
IL-17 Cytokines and Chronic Lung Diseases

 , , and
, , and {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Asthma
2.1. IL-17A and IL-17F in Asthma
2.2. IL-17E (IL-25) in Asthma
3. COPD
3.1. IL-17A in Stable COPD
3.2. IL-17A during AECOPD
3.3. IL-17C and COPD
4. Lung Cancer
5. Cystic Fibrosis
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McGeachy, M.J.; Cua, D.J.; Gaffen, S.L. The IL-17 Family of Cytokines in Health and Disease. Immunity 2019, 50, 892–906. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; He, X.; Li, X.; Qian, Y. The roles and functional mechanisms of interleukin-17 family cytokines in mucosal immunity. Cell Mol. Immunol. 2016, 13, 418–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Eddens, T.; Trevejo-Nunez, G.; Way, E.E.; Elsegeiny, W.; Ricks, D.M.; Garg, A.V.; Erb, C.J.; Bo, M.; Wang, T.; et al. IL-17 Receptor Signaling in the Lung Epithelium Is Required for Mucosal Chemokine Gradients and Pulmonary Host Defense against K. pneumoniae. Cell Host Microbe 2016, 20, 596–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaffen, S.L. Structure and signalling in the IL-17 receptor family. Nat. Rev. Immunol. 2009, 9, 556–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amatya, N.; Garg, A.V.; Gaffen, S.L. IL-17 Signaling: The Yin and the Yang. Trends Immunol. 2017, 38, 310–322. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, N.D.; Ritchie, R.; Bayes, H.K.; Mitchell, T.J.; Evans, T.J. IL-17 can be protective or deleterious in murine pneumococcal pneumonia. PLoS Pathog. 2018, 14, e1007099. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Clarke, T.B.; Weiser, J.N. Cellular effectors mediating Th17-dependent clearance of pneumococcal colonization in mice. J. Clin. Investig. 2009, 119, 1899–1909. [Google Scholar] [CrossRef] [Green Version]
- Wonnenberg, B.; Jungnickel, C.; Honecker, A.; Wolf, L.; Voss, M.; Bischoff, M.; Tschernig, T.; Herr, C.; Bals, R.; Beisswenger, C. IL-17A attracts inflammatory cells in murine lung infection with P. aeruginosa. Innate Immun. 2016, 22, 620–625. [Google Scholar] [CrossRef] [Green Version]
- Roos, A.B.; Sethi, S.; Nikota, J.; Wrona, C.T.; Dorrington, M.G.; Sanden, C.; Bauer, C.M.; Shen, P.; Bowdish, D.; Stevenson, C.S.; et al. IL-17A and the Promotion of Neutrophilia in Acute Exacerbation of Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2015, 192, 428–437. [Google Scholar] [CrossRef]
- Muir, R.; Osbourn, M.; Dubois, A.V.; Doran, E.; Small, D.M.; Monahan, A.; O’Kane, C.M.; McAllister, K.; Fitzgerald, D.C.; Kissenpfennig, A.; et al. Innate Lymphoid Cells Are the Predominant Source of IL-17A during the Early Pathogenesis of Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2016, 193, 407–416. [Google Scholar] [CrossRef] [Green Version]
- Danahay, H.; Pessotti, A.D.; Coote, J.; Montgomery, B.E.; Xia, D.; Wilson, A.; Yang, H.; Wang, Z.; Bevan, L.; Thomas, C.; et al. Notch2 is required for inflammatory cytokine-driven goblet cell metaplasia in the lung. Cell Rep. 2015, 10, 239–252. [Google Scholar] [CrossRef] [Green Version]
- Xia, W.; Bai, J.; Wu, X.; Wei, Y.; Feng, S.; Li, L.; Zhang, J.; Xiong, G.; Fan, Y.; Shi, J.; et al. Interleukin-17A promotes MUC5AC expression and goblet cell hyperplasia in nasal polyps via the Act1-mediated pathway. PLoS ONE 2014, 9, e98915. [Google Scholar] [CrossRef]
- Gurczynski, S.J.; Moore, B.B. IL-17 in the lung: The good, the bad, and the ugly. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L6–L16. [Google Scholar] [CrossRef]
- Ramirez-Carrozzi, V.; Sambandam, A.; Luis, E.; Lin, Z.; Jeet, S.; Lesch, J.; Hackney, J.; Kim, J.; Zhou, M.; Lai, J.; et al. IL-17C regulates the innate immune function of epithelial cells in an autocrine manner. Nat. Immunol. 2011, 12, 1159–1166. [Google Scholar] [CrossRef]
- Pfeifer, P.; Voss, M.; Wonnenberg, B.; Hellberg, J.; Seiler, F.; Lepper, P.M.; Bischoff, M.; Langer, F.; Schafers, H.J.; Menger, M.D.; et al. IL-17C is a mediator of respiratory epithelial innate immune response. Am. J. Respir. Cell Mol. Biol. 2013, 48, 415–421. [Google Scholar] [CrossRef]
- Kusagaya, H.; Fujisawa, T.; Yamanaka, K.; Mori, K.; Hashimoto, D.; Enomoto, N.; Inui, N.; Nakamura, Y.; Wu, R.; Maekawa, M.; et al. Toll-like receptor-mediated airway IL-17C enhances epithelial host defense in an autocrine/paracrine manner. Am. J. Respir. Cell Mol. Biol. 2014, 50, 30–39. [Google Scholar]
- Nies, J.F.; Panzer, U. IL-17C/IL-17RE: Emergence of a Unique Axis in TH17 Biology. Front. Immunol. 2020, 11, 341. [Google Scholar] [CrossRef] [Green Version]
- Wolf, L.; Sapich, S.; Honecker, A.; Jungnickel, C.; Seiler, F.; Bischoff, M.; Wonnenberg, B.; Herr, C.; Schneider-Daum, N.; Lehr, C.M.; et al. IL-17A-mediated expression of epithelial IL-17C promotes inflammation during acute Pseudomonas aeruginosa pneumonia. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 311, L1015–L1022. [Google Scholar] [CrossRef] [Green Version]
- Steck, P.; Ritzmann, F.; Honecker, A.; Vella, G.; Herr, C.; Gaupp, R.; Bischoff, M.; Speer, T.; Tschernig, T.; Bals, R.; et al. Interleukin 17 Receptor E (IL-17RE) and IL-17C Mediate the Recruitment of Neutrophils during Acute Streptococcus pneumoniae Pneumonia. Infect. Immun. 2019, 87, e00329-19. [Google Scholar] [CrossRef] [Green Version]
- Bryche, B.; Dewaele, A.; Saint-Albin, A.; Le Poupon Schlegel, C.; Congar, P.; Meunier, N. IL-17c is involved in olfactory mucosa responses to Poly (I:C) mimicking virus presence. Brain Behav. Immun. 2019, 79, 274–283. [Google Scholar] [CrossRef]
- Jamieson, K.C.; Traves, S.L.; Kooi, C.; Wiehler, S.; Dumonceaux, C.J.; Maciejewski, B.A.; Arnason, J.W.; Michi, A.N.; Leigh, R.; Proud, D. Rhinovirus and Bacteria Synergistically Induce IL-17C Release from Human Airway Epithelial Cells to Promote Neutrophil Recruitment. J. Immunol. 2019, 202, 160–170. [Google Scholar] [CrossRef] [Green Version]
- Jamieson, K.C.; Wiehler, S.; Michi, A.N.; Proud, D. Rhinovirus Induces Basolateral Release of IL-17C in Highly Differentiated Airway Epithelial Cells. Front. Cell. Infect. Microbiol. 2020, 10, 103. [Google Scholar] [CrossRef] [Green Version]
- Jeon, Y.J.; Jo, A.; Won, J.; Lee, K.M.; Yoon, S.S.; Choi, J.Y.; Kim, H.J. Interleukin-17C protects nasal epithelium from pseudomonas aeruginosa infection. Am. J. Respir. Cell Mol. Biol. 2019, 62, 95–103. [Google Scholar] [CrossRef]
- Ualiyeva, S.; Lemire, E.; Aviles, E.C.; Wong, C.; Boyd, A.A.; Lai, J.; Liu, T.; Matsumoto, I.; Barrett, N.A.; Boyce, J.A.; et al. Tuft cell-produced cysteinyl leukotrienes and IL-25 synergistically initiate lung type 2 inflammation. Sci. Immunol. 2021, 6, eabj0474. [Google Scholar] [CrossRef]
- Guida, G.; Riccio, A.M. Immune induction of airway remodeling. Semin. Immunol. 2019, 46, 101346. [Google Scholar] [CrossRef]
- De Rose, V.; Molloy, K.; Gohy, S.; Pilette, C.; Greene, C.M. Airway Epithelium Dysfunction in Cystic Fibrosis and COPD. Mediat. Inflamm. 2018, 2018, 1309746. [Google Scholar] [CrossRef] [Green Version]
- Paudel, K.R.; Dharwal, V.; Patel, V.K.; Galvao, I.; Wadhwa, R.; Malyla, V.; Shen, S.S.; Budden, K.F.; Hansbro, N.G.; Vaughan, A.; et al. Role of Lung Microbiome in Innate Immune Response Associated with Chronic Lung Diseases. Front. Med. 2020, 7, 554. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- Diseases, G.B.D.; Injuries, C. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222. [Google Scholar]
- Reddel, H.K.; Bacharier, L.B.; Bateman, E.D.; Brightling, C.E.; Brusselle, G.G.; Buhl, R.; Cruz, A.A.; Duijts, L.; Drazen, J.M.; FitzGerald, J.M.; et al. Global Initiative for Asthma Strategy 2021: Executive summary and rationale for key changes. Eur. Respir. J. 2022, 59, 17–35. [Google Scholar] [CrossRef]
- Simpson, J.L.; Scott, R.; Boyle, M.J.; Gibson, P.G. Inflammatory subtypes in asthma: Assessment and identification using induced sputum. Respirology 2006, 11, 54–61. [Google Scholar] [CrossRef]
- Baines, K.J.; Simpson, J.L.; Bowden, N.A.; Scott, R.J.; Gibson, P.G. Differential gene expression and cytokine production from neutrophils in asthma phenotypes. Eur. Respir. J. 2010, 35, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.F.; Wenzel, S.E.; Brozek, J.L.; Bush, A.; Castro, M.; Sterk, P.J.; Adcock, I.M.; Bateman, E.D.; Bel, E.H.; Bleecker, E.R.; et al. International ERS/ATS guidelines on definition, evaluation and treatment of severe asthma. Eur. Respir. J. 2014, 43, 343–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, P.J. Targeting cytokines to treat asthma and chronic obstructive pulmonary disease. Nat. Rev. Immunol. 2018, 18, 454–466. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Abel, P.W.; Casale, T.B.; Tu, Y. TH17 cells and corticosteroid insensitivity in severe asthma. J. Allergy Clin. Immunol. 2022, 149, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Ricciardolo, F.L.M.; Sorbello, V.; Folino, A.; Gallo, F.; Massaglia, G.M.; Favata, G.; Conticello, S.; Vallese, D.; Gani, F.; Malerba, M.; et al. Identification of IL-17F/frequent exacerbator endotype in asthma. J. Allergy Clin. Immunol. 2017, 140, 395–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molet, S.; Hamid, Q.; Davoine, F.; Nutku, E.; Taha, R.; Page, N.; Olivenstein, R.; Elias, J.; Chakir, J. IL-17 is increased in asthmatic airways and induces human bronchial fibroblasts to produce cytokines. J. Allergy Clin. Immunol. 2001, 108, 430–438. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Kokubu, F.; Kuga, H.; Matsukura, S.; Hoshino, H.; Ieki, K.; Imai, T.; Adachi, M.; Huang, S.K. Modulation of bronchial epithelial cells by IL-17. J. Allergy Clin. Immunol. 2001, 108, 804–809. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Onuchic, L.F.; Li, X.D.; Essayan, D.M.; Schroeder, J.; Xiao, H.Q.; Liu, M.C.; Krishnaswamy, G.; Germino, G.; Huang, S.K. Identification of a novel cytokine, ML-1, and its expression in subjects with asthma. J. Immunol. 2001, 167, 4430–4435. [Google Scholar] [CrossRef] [Green Version]
- Chakir, J.; Shannon, J.; Molet, S.; Fukakusa, M.; Elias, J.; Laviolette, M.; Boulet, L.P.; Hamid, Q. Airway remodeling-associated mediators in moderate to severe asthma: Effect of steroids on TGF-beta, IL-11, IL-17, and type I and type III collagen expression. J. Allergy Clin. Immunol. 2003, 111, 1293–1298. [Google Scholar] [CrossRef]
- Bullens, D.M.; Truyen, E.; Coteur, L.; Dilissen, E.; Hellings, P.W.; Dupont, L.J.; Ceuppens, J.L. IL-17 mRNA in sputum of asthmatic patients: Linking T cell driven inflammation and granulocytic influx? Respir. Res. 2006, 7, 135. [Google Scholar] [CrossRef] [Green Version]
- Doe, C.; Bafadhel, M.; Siddiqui, S.; Desai, D.; Mistry, V.; Rugman, P.; McCormick, M.; Woods, J.; May, R.; Sleeman, M.A.; et al. Expression of the T helper 17-associated cytokines IL-17A and IL-17F in asthma and COPD. Chest 2010, 138, 1140–1147. [Google Scholar] [CrossRef] [Green Version]
- Zheng, R.; Wang, F.; Huang, Y.; Xiang, Q.; Dai, H.; Zhang, W. Elevated Th17 cell frequencies and Th17/Treg ratio are associated with airway hyperresponsiveness in asthmatic children. J. Asthma 2021, 58, 707–716. [Google Scholar] [CrossRef]
- Barczyk, A.; Pierzchala, W.; Sozanska, E. Interleukin-17 in sputum correlates with airway hyperresponsiveness to methacholine. Respir. Med. 2003, 97, 726–733. [Google Scholar] [CrossRef] [Green Version]
- Christenson, S.A.; van den Berge, M.; Faiz, A.; Inkamp, K.; Bhakta, N.; Bonser, L.R.; Zlock, L.T.; Barjaktarevic, I.Z.; Barr, R.G.; Bleecker, E.R.; et al. An airway epithelial IL-17A response signature identifies a steroid-unresponsive COPD patient subgroup. J. Clin. Investig. 2019, 129, 169–181. [Google Scholar] [CrossRef]
- Agache, I.; Ciobanu, C.; Agache, C.; Anghel, M. Increased serum IL-17 is an independent risk factor for severe asthma. Respir. Med. 2010, 104, 1131–1137. [Google Scholar] [CrossRef] [Green Version]
- Bullone, M.; Carriero, V.; Bertolini, F.; Folino, A.; Mannelli, A.; Di Stefano, A.; Gnemmi, I.; Torchio, R.; Ricciardolo, F.L.M. Elevated serum IgE, oral corticosteroid dependence and IL-17/22 expression in highly neutrophilic asthma. Eur. Respir. J. 2019, 54, 1900068. [Google Scholar] [CrossRef]
- Kuo, C.S.; Pavlidis, S.; Loza, M.; Baribaud, F.; Rowe, A.; Pandis, I.; Hoda, U.; Rossios, C.; Sousa, A.; Wilson, S.J.; et al. A Transcriptome-driven Analysis of Epithelial Brushings and Bronchial Biopsies to Define Asthma Phenotypes in U-BIOPRED. Am. J. Respir. Crit. Care Med. 2017, 195, 443–455. [Google Scholar] [CrossRef] [Green Version]
- Holguin, F.; Cardet, J.C.; Chung, K.F.; Diver, S.; Ferreira, D.S.; Fitzpatrick, A.; Gaga, M.; Kellermeyer, L.; Khurana, S.; Knight, S.; et al. Management of severe asthma: A European Respiratory Society/American Thoracic Society guideline. Eur. Respir. J. 2020, 55, 1900588. [Google Scholar] [CrossRef] [Green Version]
- Pelletier, M.; Maggi, L.; Micheletti, A.; Lazzeri, E.; Tamassia, N.; Costantini, C.; Cosmi, L.; Lunardi, C.; Annunziato, F.; Romagnani, S.; et al. Evidence for a cross-talk between human neutrophils and Th17 cells. Blood 2010, 115, 335–343. [Google Scholar] [CrossRef]
- Jones, C.E.; Chan, K. Interleukin-17 stimulates the expression of interleukin-8, growth-related oncogene-alpha, and granulocyte-colony-stimulating factor by human airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2002, 26, 748–753. [Google Scholar] [CrossRef] [Green Version]
- Prause, O.; Laan, M.; Lotvall, J.; Linden, A. Pharmacological modulation of interleukin-17-induced GCP-2-, GRO-alpha- and interleukin-8 release in human bronchial epithelial cells. Eur. J. Pharmacol. 2003, 462, 93–198. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Onuchic, L.F.; Huang, S.K. Activation of extracellular signal-regulated kinase (ERK)1/2, but not p38 and c-Jun N-terminal kinase, is involved in signaling of a novel cytokine, ML-1. J. Biol. Chem. 2002, 277, 15229–15232. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, M.; Kokubu, F.; Matsukura, S.; Ieki, K.; Odaka, M.; Watanabe, S.; Suzuki, S.; Adachi, M.; Huang, S.K. Induction of C-X-C chemokines, growth-related oncogene alpha expression, and epithelial cell-derived neutrophil-activating protein-78 by ML-1 (interleukin-17F) involves activation of Raf1-mitogen-activated protein kinase kinase-extracellular signal-regulated kinase 1/2 pathway. J. Pharm. Exp. 2003, 307, 1213–1220. [Google Scholar]
- Kawaguchi, M.; Kokubu, F.; Odaka, M.; Watanabe, S.; Suzuki, S.; Ieki, K.; Matsukura, S.; Kurokawa, M.; Adachi, M.; Huang, S.K. Induction of granulocyte-macrophage colony-stimulating factor by a new cytokine, ML-1 (IL-17F), via Raf I-MEK-ERK pathway. J. Allergy Clin. Immunol. 2004, 114, 444–450. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Fujita, J.; Kokubu, F.; Huang, S.K.; Homma, T.; Matsukura, S.; Adachi, M.; Hizawa, N. IL-17F-induced IL-11 release in bronchial epithelial cells via MSK1-CREB pathway. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 296, L804–L810. [Google Scholar] [CrossRef] [Green Version]
- Laan, M.; Prause, O.; Miyamoto, M.; Sjostrand, M.; Hytonen, A.M.; Kaneko, T.; Lotvall, J.; Linden, A. A role of GM-CSF in the accumulation of neutrophils in the airways caused by IL-17 and TNF-alpha. Eur. Respir. J. 2003, 21, 387–393. [Google Scholar] [CrossRef] [Green Version]
- Oda, N.; Canelos, P.B.; Essayan, D.M.; Plunkett, B.A.; Myers, A.C.; Huang, S.K. Interleukin-17F induces pulmonary neutrophilia and amplifies antigen-induced allergic response. Am. J. Respir. Crit. Care Med. 2005, 171, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Hellings, P.W.; Kasran, A.; Liu, Z.; Vandekerckhove, P.; Wuyts, A.; Overbergh, L.; Mathieu, C.; Ceuppens, J.L. Interleukin-17 orchestrates the granulocyte influx into airways after allergen inhalation in a mouse model of allergic asthma. Am. J. Respir. Cell Mol. Biol. 2003, 28, 42–50. [Google Scholar] [CrossRef] [PubMed]
- McKinley, L.; Alcorn, J.F.; Peterson, A.; Dupont, R.B.; Kapadia, S.; Logar, A.; Henry, A.; Irvin, C.G.; Piganelli, J.D.; Ray, A.; et al. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J. Immunol. 2008, 181, 4089–4097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lajoie, S.; Lewkowich, I.P.; Suzuki, Y.; Clark, J.R.; Sproles, A.A.; Dienger, K.; Budelsky, A.L.; Wills-Karp, M. Complement-mediated regulation of the IL-17A axis is a central genetic determinant of the severity of experimental allergic asthma. Nat. Immunol. 2010, 11, 928–935. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.Q.; Kalim, K.W.; Li, Y.; Zheng, Y.; Guo, F. Ablation of RhoA impairs Th17 cell differentiation and alleviates house dust mite-triggered allergic airway inflammation. J. Leukoc. Biol. 2019, 106, 1139–1151. [Google Scholar] [CrossRef]
- Chenuet, P.; Fauconnier, L.; Madouri, F.; Marchiol, T.; Rouxel, N.; Ledru, A.; Mauny, P.; Lory, R.; Uttenhove, C.; van Snick, J.; et al. Neutralization of either IL-17A or IL-17F is sufficient to inhibit house dust mite induced allergic asthma in mice. Clin. Sci. 2017, 131, 2533–2548. [Google Scholar] [CrossRef]
- Zelante, T.; Bozza, S.; De Luca, A.; D’Angelo, C.; Bonifazi, P.; Moretti, S.; Giovannini, G.; Bistoni, F.; Romani, L. Th17 cells in the setting of Aspergillus infection and pathology. Med. Mycol. 2009, 47 (Suppl. S1), S162–S169. [Google Scholar] [CrossRef] [Green Version]
- De Luca, A.; Pariano, M.; Cellini, B.; Costantini, C.; Villella, V.R.; Jose, S.S.; Palmieri, M.; Borghi, M.; Galosi, C.; Paolicelli, G.; et al. The IL-17F/IL-17RC Axis Promotes Respiratory Allergy in the Proximal Airways. Cell Rep. 2017, 20, 1667–1680. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Zhang, Q.; Chen, S.; Tang, H.; Huang, P.; Wei, S.; Liang, Z.; Chen, X.; Tao, A.; Yao, L. IL-17F, rather than IL-17A, underlies airway inflammation in a steroid-insensitive toluene diisocyanate-induced asthma model. Eur. Respir. J. 2019, 53, 1801510. [Google Scholar] [CrossRef]
- Chen, Y.; Thai, P.; Zhao, Y.H.; Ho, Y.S.; DeSouza, M.M.; Wu, R. Stimulation of airway mucin gene expression by interleukin (IL)-17 through IL-6 paracrine/autocrine loop. J. Biol. Chem. 2003, 278, 17036–17043. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, K.; Graham, B.S.; Ho, S.B.; Adler, K.B.; Collins, R.D.; Olson, S.J.; Zhou, W.; Suzutani, T.; Jones, P.W.; Goleniewska, K.; et al. Respiratory syncytial virus in allergic lung inflammation increases Muc5ac and gob-5. Am. J. Respir. Crit. Care Med. 2004, 170, 306–312. [Google Scholar] [CrossRef] [Green Version]
- Starnes, T.; Robertson, M.J.; Sledge, G.; Kelich, S.; Nakshatri, H.; Broxmeyer, H.E.; Hromas, R. Cutting edge: IL-17F, a novel cytokine selectively expressed in activated T cells and monocytes, regulates angiogenesis and endothelial cell cytokine production. J. Immunol. 2001, 167, 4137–4140. [Google Scholar] [CrossRef] [Green Version]
- Schnyder-Candrian, S.; Togbe, D.; Couillin, I.; Mercier, I.; Brombacher, F.; Quesniaux, V.; Fossiez, F.; Ryffel, B.; Schnyder, B. Interleukin-17 is a negative regulator of established allergic asthma. J. Exp. Med. 2006, 203, 2715–2725. [Google Scholar] [CrossRef]
- Newcomb, D.C.; Boswell, M.G.; Zhou, W.; Huckabee, M.M.; Goleniewska, K.; Sevin, C.M.; Hershey, G.K.; Kolls, J.K.; Peebles, R.S., Jr. Human TH17 cells express a functional IL-13 receptor and IL-13 attenuates IL-17A production. J. Allergy Clin. Immunol. 2011, 127, 1006–1013.e1–4. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, K.; Fujisawa, T.; Kusagaya, H.; Mori, K.; Niwa, M.; Furuhashi, K.; Kono, M.; Hamada, E.; Suda, T.; Maekawa, M. IL-13 regulates IL-17C expression by suppressing NF-kappaB-mediated transcriptional activation in airway epithelial cells. Biochem. Biophys. Res. Commun. 2018, 495, 1534–1540. [Google Scholar] [CrossRef] [Green Version]
- Hall, S.L.; Baker, T.; Lajoie, S.; Richgels, P.K.; Yang, Y.; McAlees, J.W.; van Lier, A.; Wills-Karp, M.; Sivaprasad, U.; Acciani, T.H.; et al. IL-17A enhances IL-13 activity by enhancing IL-13-induced signal transducer and activator of transcription 6 activation. J. Allergy Clin. Immunol. 2017, 139, 462–471.e14. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; McAlees, J.W.; Bischoff, L.J.; Kaur, D.; Houshel, L.K.; Gray, J.; Hargis, J.; Davis, X.; Dudas, P.L.; Deshmukh, H.; et al. Combined administration of anti-IL-13 and anti-IL-17A at individually sub-therapeutic doses limits asthma-like symptoms in a mouse model of Th2/Th17 high asthma. Clin. Exp. Allergy 2019, 49, 317–330. [Google Scholar] [CrossRef]
- Lunding, L.P.; Webering, S.; Vock, C.; Behrends, J.; Wagner, C.; Holscher, C.; Fehrenbach, H.; Wegmann, M. Poly(inosinic-cytidylic) acid-triggered exacerbation of experimental asthma depends on IL-17A produced by NK cells. J. Immunol. 2015, 194, 5615–5625. [Google Scholar] [CrossRef] [Green Version]
- Vella, G.; Lunding, L.; Ritzmann, F.; Honecker, A.; Herr, C.; Wegmann, M.; Bals, R.; Beisswenger, C. The IL-17 receptor IL-17RE mediates polyIC-induced exacerbation of experimental allergic asthma. Respir. Res. 2020, 21, 176. [Google Scholar] [CrossRef]
- Camargo, L.D.N.; Righetti, R.F.; Aristoteles, L.; Dos Santos, T.M.; de Souza, F.C.R.; Fukuzaki, S.; Cruz, M.M.; Alonso-Vale, M.I.C.; Saraiva-Romanholo, B.M.; Prado, C.M.; et al. Effects of Anti-IL-17 on Inflammation, Remodeling, and Oxidative Stress in an Experimental Model of Asthma Exacerbated by LPS. Front. Immunol. 2017, 8, 1835. [Google Scholar] [CrossRef]
- Busse, W.W.; Holgate, S.; Kerwin, E.; Chon, Y.; Feng, J.; Lin, J.; Lin, S.L. Randomized, double-blind, placebo-controlled study of brodalumab, a human anti-IL-17 receptor monoclonal antibody, in moderate to severe asthma. Am. J. Respir. Crit. Care Med. 2013, 188, 1294–1302. [Google Scholar] [CrossRef]
- Liu, C.; Zhu, L.; Fukuda, K.; Ouyang, S.; Chen, X.; Wang, C.; Zhang, C.J.; Martin, B.; Gu, C.; Qin, L.; et al. The flavonoid cyanidin blocks binding of the cytokine interleukin-17A to the IL-17RA subunit to alleviate inflammation in vivo. Sci. Signal. 2017, 10, eaaf8823. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Coiradas, E.; Munteanu, C.R.; Diaz-Saez, L.; Pazos, A.; Huber, K.V.M.; Loza, M.I.; Dominguez, E. Discovery of novel immunopharmacological ligands targeting the IL-17 inflammatory pathway. Int. Immunopharmacol. 2020, 89, 107026. [Google Scholar] [CrossRef]
- Sel, S.; Wegmann, M.; Dicke, T.; Sel, S.; Henke, W.; Yildirim, A.O.; Renz, H.; Garn, H. Effective prevention and therapy of experimental allergic asthma using a GATA-3-specific DNAzyme. J. Allergy Clin. Immunol. 2008, 121, 910–916.e5. [Google Scholar] [CrossRef] [PubMed]
- Krug, N.; Hohlfeld, J.M.; Kirsten, A.M.; Kornmann, O.; Beeh, K.M.; Kappeler, D.; Korn, S.; Ignatenko, S.; Timmer, W.; Rogon, C.; et al. Allergen-induced asthmatic responses modified by a GATA3-specific DNAzyme. N. Engl. J. Med. 2015, 372, 1987–1995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, S.; Yosef, N.; Yang, J.; Wang, Y.; Zhou, L.; Zhu, C.; Wu, C.; Baloglu, E.; Schmidt, D.; Ramesh, R.; et al. Small-molecule RORgammat antagonists inhibit T helper 17 cell transcriptional network by divergent mechanisms. Immunity 2014, 40, 477–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamb, D.; De Sousa, D.; Quast, K.; Fundel-Clemens, K.; Erjefalt, J.S.; Sanden, C.; Hoffmann, H.J.; Kastle, M.; Schmid, R.; Menden, K.; et al. RORgammat inhibitors block both IL-17 and IL-22 conferring a potential advantage over anti-IL-17 alone to treat severe asthma. Respir. Res. 2021, 22, 158. [Google Scholar] [CrossRef] [PubMed]
- Corrigan, C.J.; Wang, W.; Meng, Q.; Fang, C.; Eid, G.; Caballero, M.R.; Lv, Z.; An, Y.; Wang, Y.H.; Liu, Y.J.; et al. Allergen-induced expression of IL-25 and IL-25 receptor in atopic asthmatic airways and late-phase cutaneous responses. J. Allergy Clin. Immunol. 2011, 128, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Paplinska-Goryca, M.; Grabczak, E.M.; Dabrowska, M.; Hermanowicz-Salamon, J.; Proboszcz, M.; Nejman-Gryz, P.; Maskey-Warzechowska, M.; Krenke, R. Sputum interleukin-25 correlates with asthma severity: A preliminary study. Postepy Derm. Alergol. 2018, 35, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Dong, C. IL-25 in allergic inflammation. Immunol. Rev. 2017, 278, 185–191. [Google Scholar] [CrossRef]
- Tamachi, T.; Maezawa, Y.; Ikeda, K.; Kagami, S.; Hatano, M.; Seto, Y.; Suto, A.; Suzuki, K.; Watanabe, N.; Saito, Y.; et al. IL-25 enhances allergic airway inflammation by amplifying a TH2 cell-dependent pathway in mice. J. Allergy Clin. Immunol. 2006, 118, 606–614. [Google Scholar] [CrossRef]
- Gregory, L.G.; Jones, C.P.; Walker, S.A.; Sawant, D.; Gowers, K.H.; Campbell, G.A.; McKenzie, A.N.; Lloyd, C.M. IL-25 drives remodelling in allergic airways disease induced by house dust mite. Thorax 2013, 68, 82–90. [Google Scholar] [CrossRef] [Green Version]
- Stock, P.; Lombardi, V.; Kohlrautz, V.; Akbari, O. Induction of airway hyperreactivity by IL-25 is dependent on a subset of invariant NKT cells expressing IL-17RB. J. Immunol. 2009, 182, 5116–5122. [Google Scholar] [CrossRef] [Green Version]
- Klein Wolterink, R.G.; Kleinjan, A.; van Nimwegen, M.; Bergen, I.; de Bruijn, M.; Levani, Y.; Hendriks, R.W. Pulmonary innate lymphoid cells are major producers of IL-5 and IL-13 in murine models of allergic asthma. Eur. J. Immunol. 2012, 42, 1106–1116. [Google Scholar] [CrossRef] [Green Version]
- Saenz, S.A.; Siracusa, M.C.; Monticelli, L.A.; Ziegler, C.G.; Kim, B.S.; Brestoff, J.R.; Peterson, L.W.; Wherry, E.J.; Goldrath, A.W.; Bhandoola, A.; et al. IL-25 simultaneously elicits distinct populations of innate lymphoid cells and multipotent progenitor type 2 (MPPtype2) cells. J. Exp. Med. 2013, 210, 1823–1837. [Google Scholar] [CrossRef] [Green Version]
- Angkasekwinai, P.; Park, H.; Wang, Y.H.; Wang, Y.H.; Chang, S.H.; Corry, D.B.; Liu, Y.J.; Zhu, Z.; Dong, C. Interleukin 25 promotes the initiation of proallergic type 2 responses. J. Exp. Med. 2007, 204, 1509–1517. [Google Scholar] [CrossRef] [Green Version]
- Barnes, P.J. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2016, 138, 16–27. [Google Scholar] [CrossRef] [Green Version]
- Sethi, S. Infection as a comorbidity of COPD. Eur. Respir. J. 2010, 35, 1209–1215. [Google Scholar] [CrossRef] [Green Version]
- Roos, A.B.; Sanden, C.; Mori, M.; Bjermer, L.; Stampfli, M.R.; Erjefalt, J.S. IL-17A Is Elevated in End-Stage Chronic Obstructive Pulmonary Disease and Contributes to Cigarette Smoke-induced Lymphoid Neogenesis. Am. J. Respir. Crit. Care Med. 2015, 191, 1232–1241. [Google Scholar] [CrossRef]
- Roos, A.B.; Mori, M.; Gura, H.K.; Lorentz, A.; Bjermer, L.; Hoffmann, H.J.; Erjefalt, J.S.; Stampfli, M.R. Increased IL-17RA and IL-17RC in End-Stage COPD and the Contribution to Mast Cell Secretion of FGF-2 and VEGF. Respir. Res. 2017, 18, 48. [Google Scholar] [CrossRef] [Green Version]
- Eustace, A.; Smyth, L.J.C.; Mitchell, L.; Williamson, K.; Plumb, J.; Singh, D. Identification of cells expressing IL-17A and IL-17F in the lungs of patients with COPD. Chest 2011, 139, 1089–1100. [Google Scholar] [CrossRef]
- Zou, Y.; Chen, X.; Liu, J.; Zhou, D.B.; Kuang, X.; Xiao, J.; Yu, Q.; Lu, X.; Li, W.; Xie, B.; et al. Serum IL-1beta and IL-17 levels in patients with COPD: Associations with clinical parameters. Int. J. Chron. Obs. Pulmon. Dis. 2017, 12, 1247–1254. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Cheng, Z.; Liu, W.; Wu, K. Expression of interleukin (IL)-10, IL-17A and IL-22 in serum and sputum of stable chronic obstructive pulmonary disease patients. COPD 2013, 10, 459–465. [Google Scholar] [CrossRef]
- Jiang, S.; Shan, F.; Zhang, Y.; Jiang, L.; Cheng, Z. Increased serum IL-17 and decreased serum IL-10 and IL-35 levels correlate with the progression of COPD. Int. J. Chron. Obs. Pulmon. Dis. 2018, 13, 2483–2494. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Pociask, D.A.; McAleer, J.P.; Chan, Y.R.; Alcorn, J.F.; Kreindler, J.L.; Keyser, M.R.; Shapiro, S.D.; Houghton, A.M.; Kolls, J.K.; et al. IL-17RA is required for CCL2 expression, macrophage recruitment, and emphysema in response to cigarette smoke. PLoS ONE 2011, 6, e20333. [Google Scholar] [CrossRef] [Green Version]
- Shan, M.; Yuan, X.; Song, L.Z.; Roberts, L.; Zarinkamar, N.; Seryshev, A.; Zhang, Y.; Hilsenbeck, S.; Chang, S.H.; Dong, C.; et al. Cigarette smoke induction of osteopontin (SPP1) mediates T(H)17 inflammation in human and experimental emphysema. Sci. Transl. Med. 2012, 4, 117ra9. [Google Scholar] [CrossRef] [Green Version]
- Kurimoto, E.; Miyahara, N.; Kanehiro, A.; Waseda, K.; Taniguchi, A.; Ikeda, G.; Koga, H.; Nishimori, H.; Tanimoto, Y.; Kataoka, M.; et al. IL-17A is essential to the development of elastase-induced pulmonary inflammation and emphysema in mice. Respir. Res. 2013, 14, 5. [Google Scholar] [CrossRef] [Green Version]
- Fukuzaki, S.; Righetti, R.F.; Santos, T.M.D.; Camargo, L.D.N.; Aristoteles, L.; Souza, F.C.R.; Garrido, A.C.; Saraiva-Romanholo, B.M.; Leick, E.A.; Prado, C.M.; et al. Preventive and therapeutic effect of anti IL-17 in an experimental model of elastase-induced lung injury in C57Bl6 mice. Am. J. Physiol. Cell Physiol. 2021, 320, C341–C354. [Google Scholar] [CrossRef]
- Yadava, K.; Pattaroni, C.; Sichelstiel, A.K.; Trompette, A.; Gollwitzer, E.S.; Salami, O.; von Garnier, C.; Nicod, L.P.; Marsland, B.J. Microbiota Promotes Chronic Pulmonary Inflammation by Enhancing IL-17A and Autoantibodies. Am. J. Respir. Crit. Care Med. 2016, 193, 975–987. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.; Al-Alwan, L.; Audusseau, S.; Chouiali, F.; Carlevaro-Fita, J.; Iwakura, Y.; Baglole, C.J.; Eidelman, D.H.; Hamid, Q. Genetic deletion of IL-17A reduces cigarette smoke-induced inflammation and alveolar type II cell apoptosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, L132–L143. [Google Scholar] [CrossRef] [Green Version]
- Pinart, M.; Zhang, M.; Li, F.; Hussain, F.; Zhu, J.; Wiegman, C.; Ryffel, B.; Chung, K.F. IL-17A modulates oxidant stress-induced airway hyperresponsiveness but not emphysema. PLoS ONE 2013, 8, e58452. [Google Scholar] [CrossRef]
- Eich, A.; Urban, V.; Jutel, M.; Vlcek, J.; Shim, J.J.; Trofimov, V.I.; Liam, C.K.; Kuo, P.H.; Hou, Y.; Xiao, J.; et al. A Randomized, Placebo-Controlled Phase 2 Trial of CNTO 6785 in Chronic Obstructive Pulmonary Disease. COPD 2017, 14, 476–483. [Google Scholar] [CrossRef]
- King, P.T.; Lim, S.; Pick, A.; Ngui, J.; Prodanovic, Z.; Downey, W.; Choong, C.; Kelman, A.; Baranyai, E.; Francis, M.; et al. Lung T-cell responses to nontypeable Haemophilus influenzae in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2013, 131, 1314–1321.e14. [Google Scholar] [CrossRef]
- Mebratu, Y.A.; Tesfaigzi, Y. IL-17 Plays a Role in Respiratory Syncytial Virus-induced Lung Inflammation and Emphysema in Elastase and LPS-injured Mice. Am. J. Respir. Cell Mol. Biol. 2018, 58, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Rao, W.; Wang, S.; Duleba, M.; Niroula, S.; Goller, K.; Xie, J.; Mahalingam, R.; Neupane, R.; Liew, A.A.; Vincent, M.; et al. Regenerative Metaplastic Clones in COPD Lung Drive Inflammation and Fibrosis. Cell 2020, 181, 848–864.e18. [Google Scholar] [CrossRef] [PubMed]
- Vella, G.; Ritzmann, F.; Wolf, L.; Kamyschnikov, A.; Stodden, H.; Herr, C.; Slevogt, H.; Bals, R.; Beisswenger, C. IL-17C contributes to NTHi-induced inflammation and lung damage in experimental COPD and is present in sputum during acute exacerbations. PLoS ONE 2021, 16, e0243484. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Rha, K.S.; Kim, D.W.; Kim, Y.M. IL-17C expression in nasal epithelial cells of chronic rhinosinusitis with nasal polyposis. Eur. Arch. Otorhinolaryngol. 2014, 271, 1097–1105. [Google Scholar] [CrossRef]
- Tan, Z.; Xue, H.; Sun, Y.; Zhang, C.; Song, Y.; Qi, Y. The Role of Tumor Inflammatory Microenvironment in Lung Cancer. Front. Pharm. 2021, 12, 688625. [Google Scholar] [CrossRef]
- Kargl, J.; Busch, S.E.; Yang, G.H.; Kim, K.H.; Hanke, M.L.; Metz, H.E.; Hubbard, J.J.; Lee, S.M.; Madtes, D.K.; McIntosh, M.W.; et al. Neutrophils dominate the immune cell composition in non-small cell lung cancer. Nat. Commun. 2017, 8, 14381. [Google Scholar] [CrossRef] [Green Version]
- Reppert, S.; Boross, I.; Koslowski, M.; Tureci, O.; Koch, S.; Lehr, H.A.; Finotto, S. A role for T-bet-mediated tumour immune surveillance in anti-IL-17A treatment of lung cancer. Nat. Commun. 2011, 2, 600. [Google Scholar] [CrossRef]
- Bao, Z.; Lu, G.; Cui, D.; Yao, Y.; Yang, G.; Zhou, J. IL-17A-producing T cells are associated with the progression of lung adenocarcinoma. Oncol. Rep. 2016, 36, 641–650. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Wan, J.; Liu, J.; Xie, W.; Diao, X.; Xu, J.; Zhu, B.; Chen, Z. Increased IL-17-producing cells correlate with poor survival and lymphangiogenesis in NSCLC patients. Lung Cancer 2010, 69, 348–354. [Google Scholar] [CrossRef]
- Akbay, E.A.; Koyama, S.; Liu, Y.; Dries, R.; Bufe, L.E.; Silkes, M.; Alam, M.M.; Magee, D.M.; Jones, R.; Jinushi, M.; et al. Interleukin-17A Promotes Lung Tumor Progression through Neutrophil Attraction to Tumor Sites and Mediating Resistance to PD-1 Blockade. J. Thorac. Oncol. 2017, 12, 1268–1279. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.H.; Mirabolfathinejad, S.G.; Katta, H.; Cumpian, A.M.; Gong, L.; Caetano, M.S.; Moghaddam, S.J.; Dong, C. T helper 17 cells play a critical pathogenic role in lung cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 5664–5669. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Guenther, J.F.; Pociask, D.A.; Wang, Y.; Kolls, J.K.; You, Z.; Chandrasekar, B.; Shan, B.; Sullivan, D.E.; Morris, G.F. Promotion of lung tumor growth by interleukin-17. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 307, L497–L508. [Google Scholar] [CrossRef] [Green Version]
- Jin, C.; Lagoudas, G.K.; Zhao, C.; Bullman, S.; Bhutkar, A.; Hu, B.; Ameh, S.; Sandel, D.; Liang, X.S.; Mazzilli, S.; et al. Commensal Microbiota Promote Lung Cancer Development via gammadelta T Cells. Cell 2019, 176, 998–1013.e16. [Google Scholar] [CrossRef] [Green Version]
- You, R.; DeMayo, F.J.; Liu, J.; Cho, S.N.; Burt, B.M.; Creighton, C.J.; Casal, R.F.; Lazarus, D.R.; Lu, W.; Tung, H.Y.; et al. IL17A Regulates Tumor Latency and Metastasis in Lung Adeno and Squamous SQ.2b and AD.1 Cancer. Cancer Immunol. Res. 2018, 6, 645–657. [Google Scholar] [CrossRef] [Green Version]
- Coffelt, S.B.; Kersten, K.; Doornebal, C.W.; Weiden, J.; Vrijland, K.; Hau, C.S.; Verstegen, N.J.M.; Ciampricotti, M.; Hawinkels, L.; Jonkers, J.; et al. IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature 2015, 522, 345–348. [Google Scholar] [CrossRef]
- Salazar, Y.; Zheng, X.; Brunn, D.; Raifer, H.; Picard, F.; Zhang, Y.; Winter, H.; Guenther, S.; Weigert, A.; Weigmann, B.; et al. Microenvironmental Th9 and Th17 lymphocytes induce metastatic spreading in lung cancer. J. Clin. Investig. 2020, 130, 3560–3575. [Google Scholar] [CrossRef] [Green Version]
- Kryczek, I.; Wei, S.; Szeliga, W.; Vatan, L.; Zou, W. Endogenous IL-17 contributes to reduced tumor growth and metastasis. Blood 2009, 114, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Martin-Orozco, N.; Muranski, P.; Chung, Y.; Yang, X.O.; Yamazaki, T.; Lu, S.; Hwu, P.; Restifo, N.P.; Overwijk, W.W.; Dong, C. T helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity 2009, 31, 787–798. [Google Scholar] [CrossRef] [Green Version]
- Ko, F.W.; Chan, K.P.; Hui, D.S.; Goddard, J.R.; Shaw, J.G.; Reid, D.W.; Yang, I.A. Acute exacerbation of COPD. Respirology 2016, 21, 1152–1165. [Google Scholar] [CrossRef] [Green Version]
- Jungnickel, C.; Schmidt, L.H.; Bittigkoffer, L.; Wolf, L.; Wolf, A.; Ritzmann, F.; Kamyschnikow, A.; Herr, C.; Menger, M.D.; Spieker, T.; et al. IL-17C mediates the recruitment of tumor-associated neutrophils and lung tumor growth. Oncogene 2017, 36, 4182–4190. [Google Scholar] [CrossRef]
- Ritzmann, F.; Jungnickel, C.; Vella, G.; Kamyschnikow, A.; Herr, C.; Li, D.; Menger, M.M.; Angenendt, A.; Hoth, M.; Lis, A.; et al. IL-17C-mediated innate inflammation decreases the response to PD-1 blockade in a model of Kras-driven lung cancer. Sci. Rep. 2019, 9, 10353. [Google Scholar] [CrossRef]
- Jiang, Z.; Chen, J.; Du, X.; Cheng, H.; Wang, X.; Dong, C. IL-25 blockade inhibits metastasis in breast cancer. Protein Cell 2017, 8, 191–201. [Google Scholar] [CrossRef] [Green Version]
- Joerger, M.; Finn, S.P.; Cuffe, S.; Byrne, A.T.; Gray, S.G. The IL-17-Th1/Th17 pathway: An attractive target for lung cancer therapy? Expert. Opin. Targets 2016, 20, 1339–1356. [Google Scholar] [CrossRef]
- Ralhan, A.; Laval, J.; Lelis, F.; Ballbach, M.; Grund, C.; Hector, A.; Hartl, D. Current Concepts and Controversies in Innate Immunity of Cystic Fibrosis Lung Disease. J. Innate Immun. 2016, 8, 531–540. [Google Scholar] [CrossRef]
- Decraene, A.; Willems-Widyastuti, A.; Kasran, A.; De Boeck, K.; Bullens, D.M.; Dupont, L.J. Elevated expression of both mRNA and protein levels of IL-17A in sputum of stable Cystic Fibrosis patients. Respir. Res. 2010, 11, 177. [Google Scholar] [CrossRef] [Green Version]
- Tan, H.L.; Regamey, N.; Brown, S.; Bush, A.; Lloyd, C.M.; Davies, J.C. The Th17 pathway in cystic fibrosis lung disease. Am. J. Respir. Crit. Care Med. 2011, 184, 252–258. [Google Scholar] [CrossRef] [Green Version]
- Tiringer, K.; Treis, A.; Fucik, P.; Gona, M.; Gruber, S.; Renner, S.; Dehlink, E.; Nachbaur, E.; Horak, F.; Jaksch, P.; et al. A Th17- and Th2-skewed cytokine profile in cystic fibrosis lungs represents a potential risk factor for Pseudomonas aeruginosa infection. Am. J. Respir. Crit. Care Med. 2013, 187, 621–629. [Google Scholar] [CrossRef]
- McAllister, F.; Henry, A.; Kreindler, J.L.; Dubin, P.J.; Ulrich, L.; Steele, C.; Finder, J.D.; Pilewski, J.M.; Carreno, B.M.; Goldman, S.J.; et al. Role of IL-17A, IL-17F, and the IL-17 receptor in regulating growth-related oncogene-alpha and granulocyte colony-stimulating factor in bronchial epithelium: Implications for airway inflammation in cystic fibrosis. J. Immunol. 2005, 175, 404–412. [Google Scholar] [CrossRef] [Green Version]
- Frija-Masson, J.; Martin, C.; Regard, L.; Lothe, M.N.; Touqui, L.; Durand, A.; Lucas, B.; Damotte, D.; Alifano, M.; Fajac, I.; et al. Bacteria-driven peribronchial lymphoid neogenesis in bronchiectasis and cystic fibrosis. Eur. Respir. J. 2017, 49, 1601873. [Google Scholar] [CrossRef] [Green Version]
- Hagner, M.; Albrecht, M.; Guerra, M.; Braubach, P.; Halle, O.; Zhou-Suckow, Z.; Butz, S.; Jonigk, D.; Hansen, G.; Schultz, C.; et al. IL-17A from innate and adaptive lymphocytes contributes to inflammation and damage in cystic fibrosis lung disease. Eur. Respir. J. 2021, 57, 1900716. [Google Scholar] [CrossRef]
- Golebski, K.; Ros, X.R.; Nagasawa, M.; van Tol, S.; Heesters, B.A.; Aglmous, H.; Kradolfer, C.M.A.; Shikhagaie, M.M.; Seys, S.; Hellings, P.W.; et al. IL-1beta, IL-23, and TGF-beta drive plasticity of human ILC2s towards IL-17-producing ILCs in nasal inflammation. Nat. Commun. 2019, 10, 2162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lore, N.I.; Cigana, C.; Riva, C.; De Fino, I.; Nonis, A.; Spagnuolo, L.; Sipione, B.; Cariani, L.; Girelli, D.; Rossi, G.; et al. IL-17A impairs host tolerance during airway chronic infection by Pseudomonas aeruginosa. Sci. Rep. 2016, 6, 25937. [Google Scholar] [CrossRef] [PubMed]
- Dubin, P.J.; Kolls, J.K. IL-23 mediates inflammatory responses to mucoid Pseudomonas aeruginosa lung infection in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, L519–L528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayes, H.K.; Ritchie, N.D.; Evans, T.J. Interleukin-17 Is Required for Control of Chronic Lung Infection Caused by Pseudomonas aeruginosa. Infect. Immun. 2016, 84, 3507–3516. [Google Scholar] [CrossRef] [Green Version]
- Hsu, D.; Taylor, P.; Fletcher, D.; van Heeckeren, R.; Eastman, J.; van Heeckeren, A.; Davis, P.; Chmiel, J.F.; Pearlman, E.; Bonfield, T.L. Interleukin-17 Pathophysiology and Therapeutic Intervention in Cystic Fibrosis Lung Infection and Inflammation. Infect. Immun. 2016, 84, 2410–2421. [Google Scholar] [CrossRef] [Green Version]
- Rehman, T.; Karp, P.H.; Tan, P.; Goodell, B.J.; Pezzulo, A.A.; Thurman, A.L.; Thornell, I.M.; Durfey, S.L.; Duffey, M.E.; Stoltz, D.A.; et al. Inflammatory cytokines TNF-alpha and IL-17 enhance the efficacy of cystic fibrosis transmembrane conductance regulator modulators. J. Clin. Investig. 2021, 131, e150398. [Google Scholar] [CrossRef]
- Rehman, T.; Thornell, I.M.; Pezzulo, A.A.; Thurman, A.L.; Romano Ibarra, G.S.; Karp, P.H.; Tan, P.; Duffey, M.E.; Welsh, M.J. TNFalpha and IL-17 alkalinize airway surface liquid through CFTR and pendrin. Am. J. Physiol. Cell Physiol. 2020, 319, C331–C344. [Google Scholar] [CrossRef]
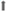 ) of mucins and antimicrobial peptides (AMP). (C) Bacteria and viruses induce the expression of IL-17C in lung epithelial cells. IL-17C enhances the expression of cytokines and chemokines in an autocrine manner, resulting in increased pulmonary inflammation.
) of mucins and antimicrobial peptides (AMP). (C) Bacteria and viruses induce the expression of IL-17C in lung epithelial cells. IL-17C enhances the expression of cytokines and chemokines in an autocrine manner, resulting in increased pulmonary inflammation.
) of mucins and antimicrobial peptides (AMP). (C) Bacteria and viruses induce the expression of IL-17C in lung epithelial cells. IL-17C enhances the expression of cytokines and chemokines in an autocrine manner, resulting in increased pulmonary inflammation.
) of mucins and antimicrobial peptides (AMP). (C) Bacteria and viruses induce the expression of IL-17C in lung epithelial cells. IL-17C enhances the expression of cytokines and chemokines in an autocrine manner, resulting in increased pulmonary inflammation.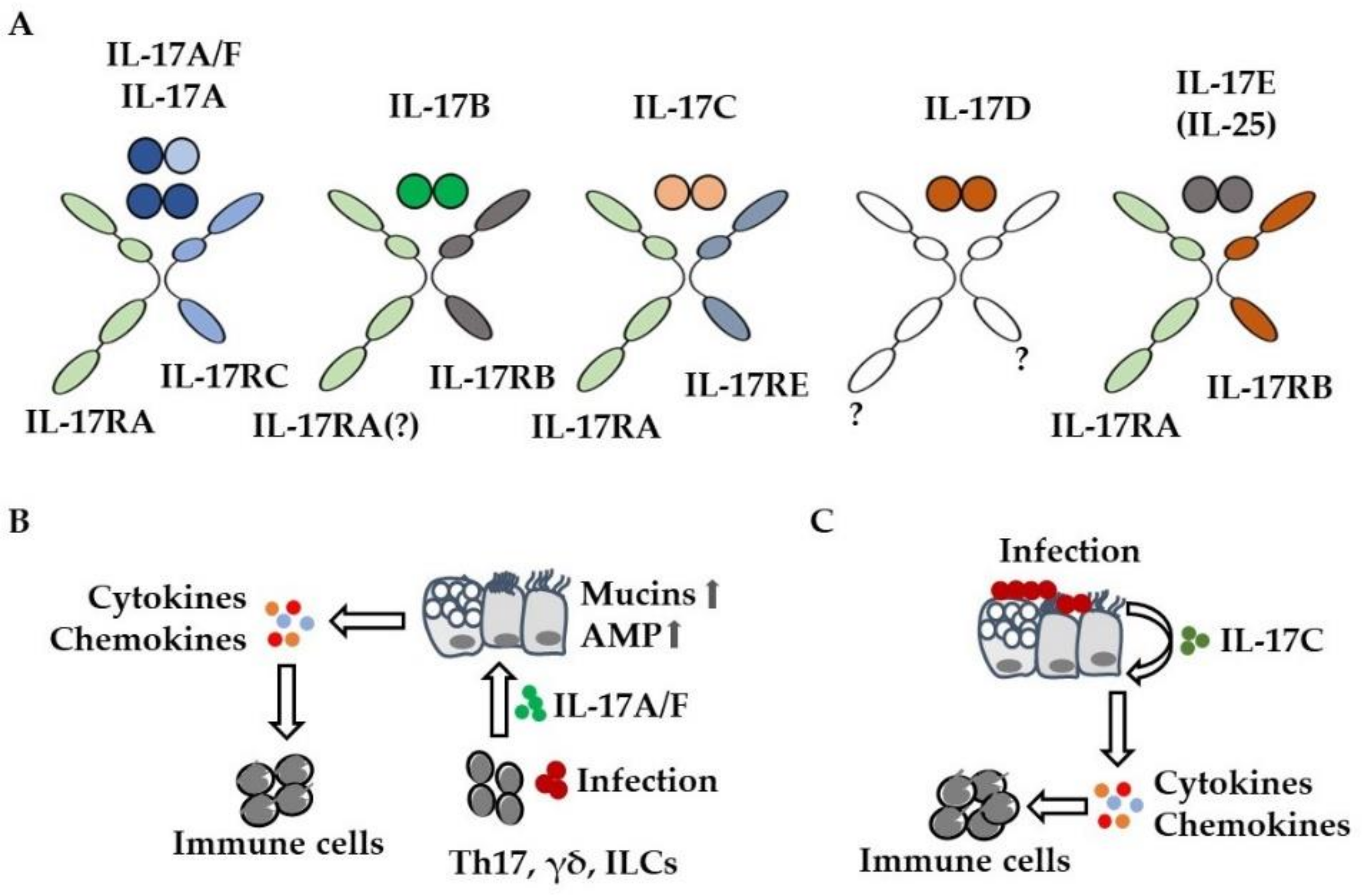

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ritzmann, F.; Lunding, L.P.; Bals, R.; Wegmann, M.; Beisswenger, C. IL-17 Cytokines and Chronic Lung Diseases. Cells 2022, 11, 2132. https://doi.org/10.3390/cells11142132
Ritzmann F, Lunding LP, Bals R, Wegmann M, Beisswenger C. IL-17 Cytokines and Chronic Lung Diseases. Cells. 2022; 11(14):2132. https://doi.org/10.3390/cells11142132
Chicago/Turabian StyleRitzmann, Felix, Lars Peter Lunding, Robert Bals, Michael Wegmann, and Christoph Beisswenger. 2022. "IL-17 Cytokines and Chronic Lung Diseases" Cells 11, no. 14: 2132. https://doi.org/10.3390/cells11142132
APA StyleRitzmann, F., Lunding, L. P., Bals, R., Wegmann, M., & Beisswenger, C. (2022). IL-17 Cytokines and Chronic Lung Diseases. Cells, 11(14), 2132. https://doi.org/10.3390/cells11142132