
ProB-Site: Protein Binding Site Prediction Using Local Features




Abstract
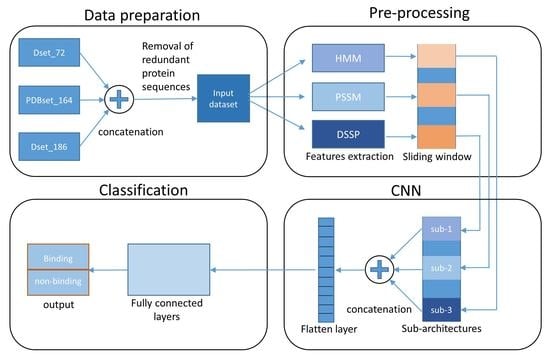
:
1. Introduction
2. Materials and Methods
2.1. Datasets
2.2. Protein Features
2.2.1. Structural Information
2.2.2. Evolutionary Features
2.3. Proposed Model
3. Results and Discussion
3.1. Evaluation Metrics
3.2. Window Size Selection
3.3. Evaluation of Features
3.4. Performance Comparison with Other Techniques
4. Webserver
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Han, J.D.J.; Bertin, N.; Hao, T.; Goldberg, D.S.; Berriz, G.F.; Zhang, L.V.; Dupuy, D.; Walhout, A.J.; Cusick, M.E.; Roth, F.P.; et al. Evidence for dynamically organized modularity in the yeast protein–protein interaction network. Nature 2004, 430, 88–93. [Google Scholar] [CrossRef] [PubMed]
- De Las Rivas, J.; Fontanillo, C. Protein–protein interactions essentials: Key concepts to building and analyzing interactome networks. PLoS Comput. Biol. 2010, 6, e1000807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Li, W.; Zeng, M.; Zheng, R.; Li, M. Network-based methods for predicting essential genes or proteins: A survey. Briefings Bioinform. 2020, 21, 566–583. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Fei, Z.; Zeng, M.; Wu, F.X.; Li, Y.; Pan, Y.; Wang, J. Automated ICD-9 coding via a deep learning approach. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018, 16, 1193–1202. [Google Scholar] [CrossRef]
- Li, M.; Gao, H.; Wang, J.; Wu, F.X. Control principles for complex biological networks. Briefings Bioinform. 2019, 20, 2253–2266. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.B.; Aloy, P. Targeting and tinkering with interaction networks. Nat. Chem. Biol. 2008, 4, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Terentiev, A.; Moldogazieva, N.; Shaitan, K. Dynamic proteomics in modeling of the living cell. Protein–protein interactions. Biochemistry 2009, 74, 1586–1607. [Google Scholar] [CrossRef]
- Wodak, S.J.; Vlasblom, J.; Turinsky, A.L.; Pu, S. Protein–protein interaction networks: The puzzling riches. Curr. Opin. Struct. Biol. 2013, 23, 941–953. [Google Scholar] [CrossRef]
- Brettner, L.M.; Masel, J. Protein stickiness, rather than number of functional protein–protein interactions, predicts expression noise and plasticity in yeast. BMC Syst. Biol. 2012, 6, 128. [Google Scholar] [CrossRef] [Green Version]
- Rehman, M.U.; Akhtar, S.; Zakwan, M.; Mahmood, M.H. Novel architecture with selected feature vector for effective classification of mitotic and non-mitotic cells in breast cancer histology images. Biomed. Signal Process. Control 2022, 71, 103212. [Google Scholar] [CrossRef]
- Abbas, Z.; Tayara, H.; Chong, K. ZayyuNet A unified deep learning model for the identification of epigenetic modifications using raw genomic sequences. IEEE/ACM Trans. Comput. Biol. Bioinform. 2021. [Google Scholar] [CrossRef] [PubMed]
- Rehman, M.U.; Cho, S.; Kim, J.; Chong, K.T. BrainSeg-net: Brain tumor MR image segmentation via enhanced encoder–decoder network. Diagnostics 2021, 11, 169. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Shujaat, M.; Tayara, H. iProm-Zea: A two-layer model to identify plant promoters and their types using convolutional neural network. Genomics 2022, 114, 110384. [Google Scholar] [CrossRef]
- Rehman, M.U.; Tayara, H.; Chong, K.T. DCNN-4mC: Densely connected neural network based N4-methylcytosine site prediction in multiple species. Comput. Struct. Biotechnol. J. 2021, 19, 6009–6019. [Google Scholar] [CrossRef] [PubMed]
- Hou, Q.; Lensink, M.F.; Heringa, J.; Feenstra, K.A. Club-martini: Selecting favourable interactions amongst available candidates, a coarse-grained simulation approach to scoring docking decoys. PLoS ONE 2016, 11, e0155251. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhou, H.X. Prediction of interface residues in protein–protein complexes by a consensus neural network method: Test against NMR data. Proteins Struct. Funct. Bioinform. 2005, 61, 21–35. [Google Scholar] [CrossRef]
- Fariselli, P.; Pazos, F.; Valencia, A.; Casadio, R. Prediction of protein–protein interaction sites in heterocomplexes with neural networks. Eur. J. Biochem. 2002, 269, 1356–1361. [Google Scholar] [CrossRef] [Green Version]
- Ofran, Y.; Rost, B. Predicted protein–protein interaction sites from local sequence information. FEBS Lett. 2003, 544, 236–239. [Google Scholar] [CrossRef] [Green Version]
- Porollo, A.; Meller, J. Prediction-based fingerprints of protein–protein interactions. Proteins Struct. Funct. Bioinform. 2007, 66, 630–645. [Google Scholar] [CrossRef]
- Northey, T.C.; Barešić, A.; Martin, A.C. IntPred: A structure-based predictor of protein–protein interaction sites. Bioinformatics 2018, 34, 223–229. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yu, B.; Ma, A.; Chen, C.; Liu, B.; Ma, Q. Protein–protein interaction sites prediction by ensemble random forests with synthetic minority oversampling technique. Bioinformatics 2019, 35, 2395–2402. [Google Scholar] [CrossRef] [PubMed]
- Hou, Q.; De Geest, P.F.; Vranken, W.F.; Heringa, J.; Feenstra, K.A. Seeing the trees through the forest: Sequence-based homo-and heteromeric protein–protein interaction sites prediction using random forest. Bioinformatics 2017, 33, 1479–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Briggs, W.J.; Padolina, J.; Timme, R.E.; Liu, W.; Linder, C.R.; Miranker, D.P. Using MoBIoS’scalable genome join to find conserved primer pair candidates between two genomes. Bioinformatics 2004, 20, i355–i362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sriwastava, B.K.; Basu, S.; Maulik, U. Protein–protein interaction site prediction in Homo sapiens and E. coli using an interaction-affinity based membership function in fuzzy SVM. J. Biosci. 2015, 40, 809–818. [Google Scholar] [CrossRef]
- Li, N.; Sun, Z.; Jiang, F. Prediction of protein–protein binding site by using core interface residue and support vector machine. BMC Bioinform. 2008, 9, 553. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Chen, X.W. Heterogeneous data integration by tree-augmented naïve B ayes for protein–protein interactions prediction. Proteomics 2013, 13, 261–268. [Google Scholar] [CrossRef]
- Li, M.H.; Lin, L.; Wang, X.L.; Liu, T. Protein–protein interaction site prediction based on conditional random fields. Bioinformatics 2007, 23, 597–604. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Guan, J.; Dong, Q.; Zhou, S. Prediction of protein–protein interaction sites using an ensemble method. BMC Bioinform. 2009, 10, 426. [Google Scholar] [CrossRef] [Green Version]
- Carl, N.; Konc, J.; Janezic, D. Protein surface conservation in binding sites. J. Chem. Inf. Model. 2008, 48, 1279–1286. [Google Scholar] [CrossRef]
- Choi, Y.S.; Yang, J.S.; Choi, Y.; Ryu, S.H.; Kim, S. Evolutionary conservation in multiple faces of protein interaction. Proteins Struct. Funct. Bioinform. 2009, 77, 14–25. [Google Scholar] [CrossRef]
- Caffrey, D.R.; Somaroo, S.; Hughes, J.D.; Mintseris, J.; Huang, E.S. Are protein–protein interfaces more conserved in sequence than the rest of the protein surface? Protein Sci. 2004, 13, 190–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.Q.; Feng, K.Y.; Chen, L.; Huang, T.; Cai, Y.D. Prediction of protein–protein interaction sites by random forest algorithm with mRMR and IFS. PLoS ONE 2012, 7, e43927. [Google Scholar] [CrossRef] [PubMed]
- Guharoy, M.; Chakrabarti, P. Secondary structure based analysis and classification of biological interfaces: Identification of binding motifs in protein–protein interactions. Bioinformatics 2007, 23, 1909–1918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Vries, S.J.; Bonvin, A.M. How proteins get in touch: Interface prediction in the study of biomolecular complexes. Curr. Protein Pept. Sci. 2008, 9, 394–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bendell, C.J.; Liu, S.; Aumentado-Armstrong, T.; Istrate, B.; Cernek, P.T.; Khan, S.; Picioreanu, S.; Zhao, M.; Murgita, R.A. Transient protein–protein interface prediction: Datasets, features, algorithms, and the RAD-T predictor. BMC Bioinform. 2014, 15, 82. [Google Scholar] [CrossRef] [Green Version]
- Hou, Q.; Dutilh, B.E.; Huynen, M.A.; Heringa, J.; Feenstra, K.A. Sequence specificity between interacting and non-interacting homologs identifies interface residues—A homodimer and monomer use case. BMC Bioinform. 2015, 16, 325. [Google Scholar] [CrossRef] [Green Version]
- Martin, J. Benchmarking protein–protein interface predictions: Why you should care about protein size. Proteins Struct. Funct. Bioinform. 2014, 82, 1444–1452. [Google Scholar] [CrossRef]
- Mihel, J.; Šikić, M.; Tomić, S.; Jeren, B.; Vlahoviček, K. PSAIA–protein structure and interaction analyzer. BMC Struct. Biol. 2008, 8, 21. [Google Scholar] [CrossRef] [Green Version]
- Yaseen, A.; Li, Y. Dinosolve: A protein disulfide bonding prediction server using context-based features to enhance prediction accuracy. BMC Bioinform. 2013, 14, S9. [Google Scholar] [CrossRef] [Green Version]
- Zeng, M.; Li, M.; Fei, Z.; Wu, F.X.; Li, Y.; Pan, Y.; Wang, J. A deep learning framework for identifying essential proteins by integrating multiple types of biological information. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 18, 296–305. [Google Scholar] [CrossRef]
- Zeng, M.; Li, M.; Fei, Z.; Yu, Y.; Pan, Y.; Wang, J. Automatic ICD-9 coding via deep transfer learning. Neurocomputing 2019, 324, 43–50. [Google Scholar] [CrossRef]
- Rao, J.; Zhou, X.; Lu, Y.; Zhao, H.; Yang, Y. Imputing single-cell RNA-seq data by combining graph convolution and autoencoder neural networks. Iscience 2021, 24, 102393. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zheng, S.; Zhao, H.; Yang, Y. Structure-aware protein solubility prediction from sequence through graph convolutional network and predicted contact map. J. Cheminformatics 2021, 13, 7. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Zheng, S.; Niu, Z.; Fu, Z.H.; Lu, Y.; Yang, Y. Communicative Representation Learning on Attributed Molecular Graphs. IJCAI 2020, 2020, 2831–2838. [Google Scholar]
- Li, Q.; Han, Z.; Wu, X.M. Deeper insights into graph convolutional networks for semi-supervised learning. In Proceedings of the Thirty-Second AAAI Conference on Artificial Intelligence, New Orleans, LA, USA, 2–7 February 2018. [Google Scholar]
- Chen, M.; Wei, Z.; Huang, Z.; Ding, B.; Li, Y. Simple and deep graph convolutional networks. In Proceedings of the International Conference on Machine Learning, PMLR, Virtual, 13–18 July 2020; pp. 1725–1735. [Google Scholar]
- Yuan, Q.; Chen, J.; Zhao, H.; Zhou, Y.; Yang, Y. Structure-aware protein–protein interaction site prediction using deep graph convolutional network. Bioinformatics 2022, 38, 125–132. [Google Scholar] [CrossRef]
- Murakami, Y.; Mizuguchi, K. Applying the Naïve Bayes classifier with kernel density estimation to the prediction of protein–protein interaction sites. Bioinformatics 2010, 26, 1841–1848. [Google Scholar] [CrossRef]
- Singh, G.; Dhole, K.; Pai, P.P.; Mondal, S. SPRINGS: Prediction of protein–protein interaction sites using artificial neural networks. PeerJ 2014. Available online: https://peerj.com/preprints/266.pdf (accessed on 19 May 2022).
- Altschul, S.F.; Gish, W.; Miller, W. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. Available online: https://www.sciencedirect.com/science/article/abs/pii/S0022283605803602 (accessed on 19 May 2022). [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolym. Orig. Res. Biomol. 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Remmert, M.; Biegert, A.; Hauser, A.; Söding, J. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 2012, 9, 173–175. [Google Scholar] [CrossRef]
- Mirdita, M.; Von Den Driesch, L.; Galiez, C.; Martin, M.J.; Söding, J.; Steinegger, M. Uniclust databases of clustered and deeply annotated protein sequences and alignments. Nucleic Acids Res. 2017, 45, D170–D176. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.; Schaffer, A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzek, B.E.; Huang, H.; McGarvey, P.; Mazumder, R.; Wu, C.H. UniRef: Comprehensive and non-redundant UniProt reference clusters. Bioinformatics 2007, 23, 1282–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, M.; Zhang, F.; Wu, F.X.; Li, Y.; Wang, J.; Li, M. Protein–protein interaction site prediction through combining local and global features with deep neural networks. Bioinformatics 2020, 36, 1114–1120. [Google Scholar] [CrossRef] [PubMed]
- Sussman, J.L.; Lin, D.; Jiang, J.; Manning, N.O.; Prilusky, J.; Ritter, O.; Abola, E.E. Protein Data Bank (PDB): Database of three-dimensional structural information of biological macromolecules. Acta Crystallogr. Sect. D Biol. Crystallogr. 1998, 54, 1078–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarap, A.F. Deep learning using rectified linear units (relu). arXiv 2018, arXiv:1803.08375. [Google Scholar]
- Light, W. Ridge functions, sigmoidal functions and neural networks. In Approximation Theory VII; Academic: Cambridge, MA, USA, 1992; pp. 163–206. [Google Scholar]
- de Boer, P.T.; Kroese, D.P.; Mannor, S.; Rubinstein, R.Y. A Tutorial on the Cross-Entropy Method. Ann. Oper. Res. 2005, 134, 19. [Google Scholar] [CrossRef]
- Zhang, J. Kurgan L. SCRIBER: Accurate and partner type-specific prediction of protein-binding residues from proteins sequences. Bioinformatics 2019, 35, 343–353. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Golding, G.B.; Ilie, L. DELPHI: Accurate deep ensemble model for protein interaction sites prediction. Bioinformatics 2021, 37, 896–904. [Google Scholar] [CrossRef]
- Qiu, J.; Bernhofer, M.; Heinzinger, M.; Kemper, S.; Norambuena, T.; Melo, F.; Rost, B. ProNA2020 predicts protein–DNA, protein–RNA, and protein–protein binding proteins and residues from sequence. J. Mol. Biol. 2020, 432, 2428–2443. [Google Scholar] [CrossRef]
- Zhang, B.; Li, J.; Quan, L.; Chen, Y.; Lü, Q. Sequence-based prediction of protein–protein interaction sites by simplified long short-term memory network. Neurocomputing 2019, 357, 86–100. [Google Scholar] [CrossRef]
- Gainza, P.; Sverrisson, F.; Monti, F.; Rodola, E.; Boscaini, D.; Bronstein, M.; Correia, B. Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning. Nat. Methods 2020, 17, 184–192. [Google Scholar] [CrossRef] [PubMed]
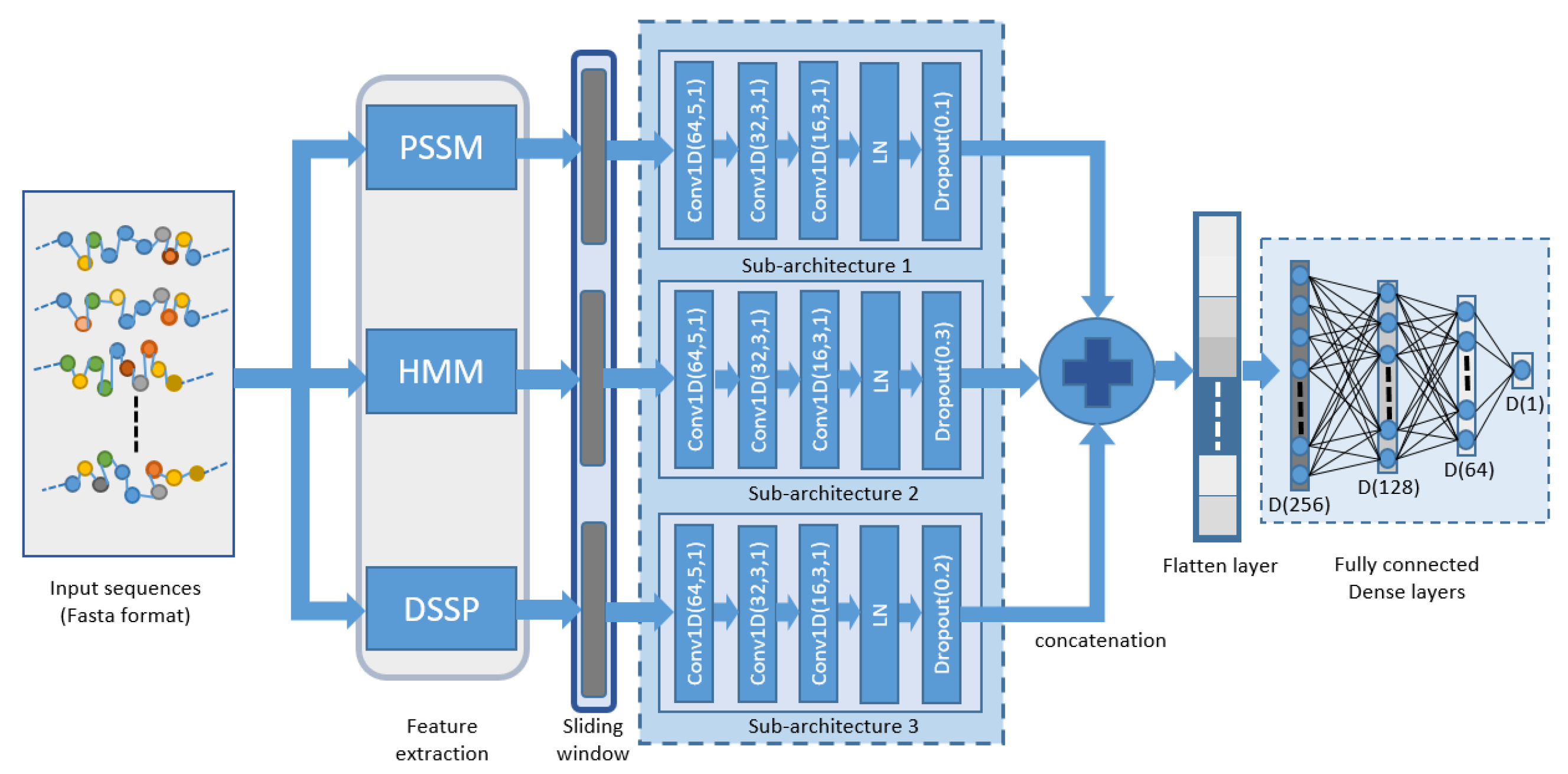


{kind=link}
{kind=link}
{kind=link}
| Layers | Parameters |
|---|---|
| Convolution layer 1 | 32, 64, 128 (Kernel Size 3, 5, 7) |
| Convolution layer 2 | 8, 16, 32, 64 (Kernel Size 3, 5) |
| Convolution layer 3 | 8, 16, 32 (Kernel Size 3, 5) |
| Dropout | 0.1, 0.15, 0.2, 0.25, 0.3, 0.35, 0.4 |
| Dense layer 1 | 256, 128, 64 |
| Dense layer 2 | 128, 64, 32 |
| Dense layer 3 | 64, 32, 16, 8 |
| Feature Set | ACC | Precision | Recall | F1 | MCC | AUROC | AUPRC |
|---|---|---|---|---|---|---|---|
| Evolutionary features | 0.651 | 0.224 | 0.675 | 0.311 | 0.149 | 0.651 | 0.243 |
| (PSSM + HMM) | |||||||
| Secondary structures | 0. 752 | 0. 354 | 0.755 | 0. 475 | 0.380 | 0.824 | 0.428 |
| (DSSP) | |||||||
| All Features | 0.799 | 0.407 | 0.612 | 0.517 | 0.368 | 0.844 | 0.467 |
| (PSSM + HMM + DSSP) |
| Method | ACC | Precision | Recall | F1 | MCC | AUROC | AUPRC |
|---|---|---|---|---|---|---|---|
| PSIVER | 0.561 | 0.188 | 0.534 | 0.278 | 0.074 | 0.573 | 0.190 |
| SCRIBER | 0.667 | 0.253 | 0.568 | 0.350 | 0.193 | 0.665 | 0.278 |
| DELPHI | 0.697 | 0.276 | 0.568 | 0.372 | 0.225 | 0.699 | 0.319 |
| ProNA2020 | 0.738 | 0.275 | 0.402 | 0.326 | 0.176 | N/A | N/A |
| DLPred | 0.682 | 0.264 | 0.565 | 0.360 | 0.208 | 0.677 | 0.294 |
| SPPIDER | 0.752 | 0.331 | 0.557 | 0.415 | 0.285 | 0.755 | 0.373 |
| DeepPPISP | 0.657 | 0.243 | 0.539 | 0.335 | 0.167 | 0.653 | 0.276 |
| MaSIF-site | 0.780 | 0.370 | 0.561 | 0.446 | 0.326 | 0.775 | 0.439 |
| GraphPPIS | 0.776 | 0.368 | 0.584 | 0.451 | 0.333 | 0.786 | 0.429 |
| ProB-site | 0.799 | 0.407 | 0.612 | 0.517 | 0.368 | 0.844 | 0.467 |
| Deep PPISP | SPPIDER | MaSIF-Site | GraphPPIS | ProB-Site | |
|---|---|---|---|---|---|
| MCC | 0.169 | 0.294 | 0.304 | 0.336 | 0.351 |
| AUPRC | 0.256 | 0.376 | 0.372 | 0.423 | 0.446 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, S.H.; Tayara, H.; Chong, K.T. ProB-Site: Protein Binding Site Prediction Using Local Features. Cells 2022, 11, 2117. https://doi.org/10.3390/cells11132117
Khan SH, Tayara H, Chong KT. ProB-Site: Protein Binding Site Prediction Using Local Features. Cells. 2022; 11(13):2117. https://doi.org/10.3390/cells11132117
Chicago/Turabian StyleKhan, Sharzil Haris, Hilal Tayara, and Kil To Chong. 2022. "ProB-Site: Protein Binding Site Prediction Using Local Features" Cells 11, no. 13: 2117. https://doi.org/10.3390/cells11132117
APA StyleKhan, S. H., Tayara, H., & Chong, K. T. (2022). ProB-Site: Protein Binding Site Prediction Using Local Features. Cells, 11(13), 2117. https://doi.org/10.3390/cells11132117