
Next-Generation Sequencing on Circulating Tumor DNA in Advanced Solid Cancer: Swiss Army Knife for the Molecular Tumor Board? A Review of the Literature Focused on FDA Approved Test

 , , , , , add
Show full author list
, , , , , add
Show full author list
Abstract
:1. Introduction
2. Parameters Analyzed in FDA-Approved Pan-Cancer Panels
2.1. Tumor Fraction
2.2. Tumor Mutation Burden
2.3. Microsatellite Instability Detection (MSI)
2.4. Single-Nucleotide Variation
2.5. Copy Number Variations
2.6. Fusion/Rearrangement
3. Early Detection of Complex Drug-Resistance Mechanisms
4. Incidental Germline Variant Identification
5. Occult Malignancy Identification
6. Clonal Hematopoiesis
7. Points of Attention
8. Guidelines for Interpretation of cfDNA Clinical Reports
- -
- Tumor Fraction: this is the first parameter to look at for panels offering this information, keeping in mind the risk of overestimation if germline mutations have not been filtered out. If not calculated, results must be interpreted carefully and tumor content should be discussed. An estimation can also be given focusing on VAF of commonly identified “tracker” alterations in the histology, for example, APC mutations in sporadic colorectal cancer [68] or TP53 mutations in high-grade serous ovarian cancer [69].
- -
- -
- bTMB: High risk of overestimation exists if germline or CH mutations are not filtered out. A context of multiclonal resistance can also be a factor of overestimation. We recommend elevating the cut-off to estimate which patients will benefit from immunotherapy to 16 mutations/Mb, to interpret it in the context of other variants and to confirm the result on tissue if needed.
- -
- SNV: Mutations identified must be interpreted taking into account VAF and tumor fraction. The absence of any putative tumor variants should lead to consider the result as noncontributive, especially without the tumor fraction information. Furthermore, a SNV identified at very low VAF should be confirmed on tissue.
- -
- CNV: If up to now no literature exists on the risk of overestimating the number of copies, in our experience, reported amplifications must be interpreted with caution and to be confirmed on tissue with FISH or IHC as in case of polysomic samples, the number of copies will be artifactually overestimated with a cancer gene panel.
- -
- Fusion/Rearrangement: Currently, FDA-approved tests use DNA for fusion identification. It can represent an issue in case of large intronic regions and does not give information about expression. For these reasons, we recommend using an RNA-based technique as a confirmation tool if needed on tissue.
- -
- Germline alterations: Not all the mutations identified with VAF between 40 and 60% are germline mutations. Results must be interpreted taking into account the tumor fraction and VAF of known alterations in the histology. Suspected germline alteration has to be confirmed on whole blood by a validated technique and a geneticist has to be involved in MTB to plan a genetic counseling if needed.
- -
- Occult malignancies: In our experience, identification of other malignancies based on liquid biopsy results is a possible situation, based on the presence of recurrent drivers that are specific for certain tumor types. For this reason, we recommend including an oncologist with different specialty and a hematologist in the MTB. The exploration of a second cancer can be proposed to clinicians if the suspicion is high.
- -
- CH: The hematologic malignant potential of identified mutations with high VAF has to be discussed with a hematologist. A hematologic consultation can be planned if this risk is high or if abnormalities are seen on the complete blood count. Furthermore, suspected CH mutations occurring in genes that allow the inclusion in clinical trials must be confirmed.
9. Conclusions
Funding
Conflicts of Interest
References
- Prud’homme, C.; Deschamps, F.; Allorant, A.; Massard, C.; Hollebecque, A.; Yevich, S.; Ngo-Camus, M.; Gravel, G.; Nicotra, C.; Michiels, S.; et al. Image-guided tumour biopsies in a prospective molecular triage study (MOSCATO-01): What are the real risks? Eur. J. Cancer 2018, 103, 108–119. [Google Scholar] [CrossRef]
- Tacher, V.; Le Deley, M.-C.; Hollebecque, A.; Deschamps, F.; Vielh, P.; Hakime, A.; Ileana, E.; Abedi-Ardekani, B.; Charpy, C.; Massard, C.; et al. Factors associated with success of image-guided tumour biopsies: Results from a prospective molecular triage study (MOSCATO-01). Eur. J. Cancer 2016, 59, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Russano, M.; Napolitano, A.; Ribelli, G.; Iuliani, M.; Simonetti, S.; Citarella, F.; Pantano, F.; Dell’Aquila, E.; Anesi, C.; Silvestris, N.; et al. Liquid biopsy and tumor heterogeneity in metastatic solid tumors: The potentiality of blood samples. J. Exp. Clin. Cancer Res. 2020, 39, 95. [Google Scholar] [CrossRef] [PubMed]
- Ignatiadis, M.; Sledge, G.W.; Jeffrey, S.S. Liquid biopsy enters the clinic—Implementation issues and future challenges. Nat. Rev. Clin. Oncol. 2021, 18, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Alix-Panabières, C.; Pantel, K. Liquid Biopsy: From Discovery to Clinical Application. Cancer Discov. 2021, 11, 858–873. [Google Scholar] [CrossRef]
- Fiala, C.; Diamandis, E.P. New approaches for detecting cancer with circulating cell-free DNA. BMC Med. 2019, 17, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Garcia, D.; Hills, A.; Page, K.; Hastings, R.K.; Toghill, B.; Goddard, K.S.; Ion, C.; Ogle, O.; Boydell, A.R.; Gleason, K.; et al. Plasma cell-free DNA (cfDNA) as a predictive and prognostic marker in patients with metastatic breast cancer. Breast Cancer Res. 2019, 21, 149. [Google Scholar] [CrossRef] [PubMed]
- Iwama, E.; Sakai, K.; Azuma, K.; Harada, T.; Harada, D.; Nosaki, K.; Hotta, K.; Ohyanagi, F.; Kurata, T.; Fukuhara, T.; et al. Monitoring of somatic mutations in circulating cell-free DNA by digital PCR and next-generation sequencing during afatinib treatment in patients with lung adenocarcinoma positive for EGFR activating mutations. Ann. Oncol. 2017, 28, 136–141. [Google Scholar] [CrossRef] [PubMed]
- McCoach, C.E.; Blakely, C.M.; Banks, K.C.; Levy, B.; Chue, B.M.; Raymond, V.M.; Le, A.T.; Lee, C.E.; Diaz, J.; Waqar, S.N.; et al. Clinical Utility of Cell-Free DNA for the Detection of ALK Fusions and Genomic Mechanisms of ALK Inhibitor Resistance in Non–Small Cell Lung Cancer. Clin. Cancer Res. 2018, 24, 2758–2770. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.M.; Shah, R.H.; Pentsova, E.I.; Pourmaleki, M.; Briggs, S.; Distefano, N.; Zheng, Y.; Skakodub, A.; Mehta, S.A.; Campos, C.; et al. Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature 2019, 565, 654–658. [Google Scholar] [CrossRef] [PubMed]
- André, F.; Ciruelos, E.M.; Juric, D.; Loibl, S.; Campone, M.; Mayer, I.A.; Rubovszky, G.; Yamashita, T.; Kaufman, B.; Lu, Y.-S.; et al. Alpelisib plus fulvestrant for PIK3CA-mutated, hormone receptor-positive, human epidermal growth factor receptor-2—Negative advanced breast cancer: Final overall survival results from SOLAR-1. Ann. Oncol. 2021, 32, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Taniguchi, H.; Ikeda, M.; Bando, H.; Kato, K.; Morizane, C.; Esaki, T.; Komatsu, Y.; Kawamoto, Y.; Takahashi, N.; et al. Clinical utility of circulating tumor DNA sequencing in advanced gastrointestinal cancer: SCRUM-Japan GI-SCREEN and GOZILA studies. Nat. Med. 2020, 26, 1859–1864. [Google Scholar] [CrossRef] [PubMed]
- Fiala, C.; Diamandis, E.P. Can Grail find the trail to early cancer detection? Clin. Chem. Lab. Med. CCLM 2019, 57, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Odegaard, J.I.; Vincent, J.J.; Mortimer, S.; Vowles, J.V.; Ulrich, B.C.; Banks, K.C.; Fairclough, S.R.; Zill, O.A.; Sikora, M.; Mokhtari, R.; et al. Validation of a Plasma-Based Comprehensive Cancer Genotyping Assay Utilizing Orthogonal Tissue- and Plasma-Based Methodologies. Clin. Cancer Res. 2018, 24, 3539–3549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leighl, N.B.; Page, R.D.; Raymond, V.M.; Daniel, D.B.; Divers, S.G.; Reckamp, K.L.; Villalona-Calero, M.A.; Dix, D.; Odegaard, J.I.; Lanman, R.B.; et al. Clinical Utility of Comprehensive Cell-free DNA Analysis to Identify Genomic Biomarkers in Patients with Newly Diagnosed Metastatic Non–small Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 4691–4700. [Google Scholar] [CrossRef] [Green Version]
- Woodhouse, R.; Li, M.; Hughes, J.; Delfosse, D.; Skoletsky, J.; Ma, P.; Meng, W.; Dewal, N.; Milbury, C.; Clark, T.; et al. Clinical and analytical validation of FoundationOne Liquid CDx, a novel 324-Gene cfDNA-based comprehensive genomic profiling assay for cancers of solid tumor origin. PLoS ONE 2020, 15, e0237802. [Google Scholar] [CrossRef]
- Lee, J.K.; Hazar-Rethinam, M.; Decker, B.; Gjoerup, O.; Madison, R.W.; Lieber, D.S.; Chung, J.H.; Schrock, A.B.; Creeden, J.; Venstrom, J.M.; et al. The pan-tumor landscape of targetable kinase fusions in circulating tumor DNA. Clin. Cancer Res. 2022, 28, 728–737. [Google Scholar] [CrossRef]
- Merino, D.M.; McShane, L.M.; Fabrizio, D.; Funari, V.; Chen, S.-J.; White, J.R.; Wenz, P.; Baden, J.; Barrett, J.C.; Chaudhary, R.; et al. Establishing guidelines to harmonize tumor mutational burden (TMB): In silico assessment of variation in TMB quantification across diagnostic platforms: Phase I of the Friends of Cancer Research TMB Harmonization Project. J. Immunother. Cancer 2020, 8, e000147. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Dai, J.; Wang, D.; Lu, H.; Dai, H.; Ye, H.; Gu, J.; Chen, S.; Huang, B. Assessment of tumor mutation burden calculation from gene panel sequencing data. OncoTargets Ther. 2019, 12, 3401–3409. [Google Scholar] [CrossRef] [Green Version]
- Endris, V.; Buchhalter, I.; Allgäuer, M.; Rempel, E.; Lier, A.; Volckmar, A.; Kirchner, M.; Winterfeld, M.; Leichsenring, J.; Neumann, O.; et al. Measurement of tumor mutational burden (TMB) in routine molecular diagnostics: In silico and real-life analysis of three larger gene panels. Int. J. Cancer 2019, 144, 2303–2312. [Google Scholar] [CrossRef]
- Sturgill, E.; Misch, A.; Jones, C.; Luckett, D.; Fu, X.; Jones, S.F.; Burris, H.A., III; Spigel, D.R.; McKenzie, A.J. Concordance of blood and tissue TMB from NGS testing in real-world settings and their ability to predict response to immunotherapy. J. Clin. Oncol. 2021, 39 (Suppl. S15), 2540. [Google Scholar] [CrossRef]
- Gandara, D.R.; Paul, S.M.; Kowanetz, M.; Schleifman, E.; Zou, W.; Li, Y.; Rittmeyer, A.; Fehrenbacher, L.; Otto, G.; Malboeuf, C.; et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat. Med. 2018, 24, 1441–1448. [Google Scholar] [CrossRef] [PubMed]
- Büttner, R.; Longshore, J.W.; López-Ríos, F.; Merkelbach-Bruse, S.; Normanno, N.; Rouleau, E.; Penault-Llorca, F. Implementing TMB measurement in clinical practice: Considerations on assay requirements. ESMO Open 2019, 4, e000442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willis, J.; Lefterova, M.I.; Artyomenko, A.; Kasi, P.M.; Nakamura, Y.; Mody, K.; Catenacci, D.V.T.; Fakih, M.; Barbacioru, C.; Zhao, J.; et al. Validation of Microsatellite Instability Detection Using a Comprehensive Plasma-Based Genotyping Panel. Clin. Cancer Res. 2019, 25, 7035–7045. [Google Scholar] [CrossRef]
- Mosele, F.; Remon, J.; Mateo, J.; Westphalen, C.B.; Barlesi, F.; Lolkema, M.P.; Normanno, N.; Scarpa, A.; Robson, M.; Meric-Bernstam, F.; et al. Recommendations for the use of next-generation sequencing (NGS) for patients with metastatic cancers: A report from the ESMO Precision Medicine Working Group. Ann. Oncol. 2020, 31, 1491–1505. [Google Scholar] [CrossRef]
- Hirotsu, Y.; Otake, S.; Ohyama, H.; Amemiya, K.; Higuchi, R.; Oyama, T.; Mochizuki, H.; Goto, T.; Omata, M. Dual-molecular barcode sequencing detects rare variants in tumor and cell free DNA in plasma. Sci. Rep. 2020, 10, 3391. [Google Scholar] [CrossRef]
- Jahangiri, L.; Hurst, T. Assessing the Concordance of Genomic Alterations between Circulating-Free DNA and Tumour Tissue in Cancer Patients. Cancers 2019, 11, 1938. [Google Scholar] [CrossRef] [Green Version]
- Takeshita, T.; Yamamoto, Y.; Yamamoto-Ibusuki, M.; Tomiguchi, M.; Sueta, A.; Murakami, K.; Omoto, Y.; Iwase, H. Comparison of ESR1 Mutations in Tumor Tissue and Matched Plasma Samples from Metastatic Breast Cancer Patients. Transl. Oncol. 2017, 10, 766–771. [Google Scholar] [CrossRef]
- Thompson, J.C.; Yee, S.S.; Troxel, A.B.; Savitch, S.L.; Fan, R.; Balli, D.; Lieberman, D.B.; Morrissette, J.D.; Evans, T.L.; Bauml, J.; et al. Detection of Therapeutically Targetable Driver and Resistance Mutations in Lung Cancer Patients by Next-Generation Sequencing of Cell-Free Circulating Tumor DNA. Clin. Cancer Res. 2016, 22, 5772–5782. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.H.; Allison, D.H.R.; Feng, Y.; Jour, G.; Park, K.; Zhou, F.; Moreira, A.L.; Shen, G.; Feng, X.; Sabari, J.; et al. Comparison of solid tissue sequencing and liquid biopsy accuracy in identification of clinically relevant gene mutations and rearrangements in lung adenocarcinomas. Mod. Pathol. 2021, 34, 2168–2174. [Google Scholar] [CrossRef]
- Bieg-Bourne, C.C.; Okamura, R.; Kurzrock, R. Concordance between TP53 alterations in blood and tissue: Impact of time interval, biopsy site, cancer type and circulating tumor DNA burden. Mol. Oncol. 2020, 14, 1242–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torga, G.; Pienta, K.J. Patient-Paired Sample Congruence between 2 Commercial Liquid Biopsy Tests. JAMA Oncol. 2018, 4, 868. [Google Scholar] [CrossRef] [PubMed]
- Aldea, M.; Hendriks, L.; Mezquita, L.; Jovelet, C.; Planchard, D.; Auclin, E.; Remon, J.; Howarth, K.; Benitez, J.C.; Gazzah, A.; et al. Circulating Tumor DNA Analysis for Patients with Oncogene-Addicted NSCLC with Isolated Central Nervous System Progression. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2020, 15, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Chicard, M.; Boyault, S.; Colmet Daage, L.; Richer, W.; Gentien, D.; Pierron, G.; Lapouble, E.; Bellini, A.; Clement, N.; Iacono, I.; et al. Genomic Copy Number Profiling Using Circulating Free Tumor DNA Highlights Heterogeneity in Neuroblastoma. Clin. Cancer Res. 2016, 22, 5564–5573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, Y.; Okamoto, W.; Kato, T.; Esaki, T.; Kato, K.; Komatsu, Y.; Yuki, S.; Masuishi, T.; Nishina, T.; Ebi, H.; et al. Circulating tumor DNA-guided treatment with pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer: A phase 2 trial. Nat. Med. 2021, 27, 1899–1903. [Google Scholar] [CrossRef]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef]
- Hechtman, J.F.; Benayed, R.; Hyman, D.M.; Drilon, A.; Zehir, A.; Frosina, D.; Arcila, M.E.; Dogan, S.; Klimstra, D.S.; Ladanyi, M.; et al. Pan-Trk Immunohistochemistry Is an Efficient and Reliable Screen for the Detection of NTRK Fusions. Am. J. Surg. Pathol. 2017, 41, 1547–1551. [Google Scholar] [CrossRef]
- Marchiò, C.; Scaltriti, M.; Ladanyi, M.; Iafrate, A.J.; Bibeau, F.; Dietel, M.; Hechtman, J.F.; Troiani, T.; López-Rios, F.; Douillard, J.-Y.; et al. ESMO recommendations on the standard methods to detect NTRK fusions in daily practice and clinical research. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2019, 30, 1417–1427. [Google Scholar] [CrossRef] [Green Version]
- Belli, C.; Penault-Llorca, F.; Ladanyi, M.; Normanno, N.; Scoazec, J.-Y.; Lacroix, L.; Reis-Filho, J.S.; Subbiah, V.; Gainor, J.F.; Endris, V.; et al. ESMO recommendations on the standard methods to detect RET fusions and mutations in daily practice and clinical research. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2021, 32, 337–350. [Google Scholar] [CrossRef]
- Benayed, R.; Offin, M.; Mullaney, K.; Sukhadia, P.; Rios, K.; Desmeules, P.; Ptashkin, R.; Won, H.; Chang, J.; Halpenny, D.; et al. High Yield of RNA Sequencing for Targetable Kinase Fusions in Lung Adenocarcinomas with No Mitogenic Driver Alteration Detected by DNA Sequencing and Low Tumor Mutation Burden. Clin. Cancer Res. 2019, 25, 4712–4722. [Google Scholar] [CrossRef]
- Jonker, D.J.; O’Callaghan, C.J.; Karapetis, C.S.; Zalcberg, J.R.; Tu, D.; Au, H.-J.; Berry, S.R.; Krahn, M.; Price, T.; Simes, R.J.; et al. Cetuximab for the Treatment of Colorectal Cancer. N. Engl. J. Med. 2007, 357, 2040–2048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerrera, L.P.; Napolitano, S.; De Falco, V.; Giunta, E.F.; Vitiello, P.P.; Gravina, A.G.; Suarato, G.; Perrone, A.; Napolitano, R.; Martinelli, E.; et al. Multiple Acquired Mutations Captured by Liquid Biopsy in the EGFR Addicted Metastatic Colorectal Cancer. Clin. Colorectal Cancer 2021, 20, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.K.; Harrell, M.I.; Oza, A.M.; Oaknin, A.; Ray-Coquard, I.; Tinker, A.V.; Helman, E.; Radke, M.R.; Say, C.; Vo, L.-T.; et al. BRCA Reversion Mutations in Circulating Tumor DNA Predict Primary and Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2019, 9, 210–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burrell, R.A.; Swanton, C. Tumour heterogeneity and the evolution of polyclonal drug resistance. Mol. Oncol. 2014, 8, 1095–1111. [Google Scholar] [CrossRef] [PubMed]
- Parikh, A.R.; Leshchiner, I.; Elagina, L.; Goyal, L.; Levovitz, C.; Siravegna, G.; Livitz, D.; Rhrissorrakrai, K.; Martin, E.E.; Van Seventer, E.E.; et al. Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers. Nat. Med. 2019, 25, 1415–1421. [Google Scholar] [CrossRef]
- Enrico, D.; Lacroix, L.; Chen, J.; Rouleau, E.; Scoazec, J.-Y.; Loriot, Y.; Tselikas, L.; Jovelet, C.; Planchard, D.; Gazzah, A.; et al. Oncogenic Fusions May Be Frequently Present at Resistance of EGFR Tyrosine Kinase Inhibitors in Patients with NSCLC: A Brief Report. JTO Clin. Res. Rep. 2020, 1, 100023. [Google Scholar] [CrossRef]
- Papadimitrakopoulou, V.A.; Wu, Y.-L.; Han, J.-Y.; Ahn, M.-J.; Ramalingam, S.S.; John, T.; Okamoto, I.; Yang, J.C.-H.; Bulusu, K.C.; Laus, G.; et al. Analysis of resistance mechanisms to osimertinib in patients with EGFR T790M advanced NSCLC from the AURA3 study. Ann. Oncol. 2018, 29, viii741. [Google Scholar] [CrossRef]
- Slavin, T.P.; Banks, K.C.; Chudova, D.; Oxnard, G.R.; Odegaard, J.I.; Nagy, R.J.; Tsang, K.W.K.; Neuhausen, S.L.; Gray, S.W.; Cristofanilli, M.; et al. Identification of Incidental Germline Mutations in Patients with Advanced Solid Tumors Who Underwent Cell-Free Circulating Tumor DNA Sequencing. J. Clin. Oncol. 2018, 36, 3459–3465. [Google Scholar] [CrossRef]
- Stout, L.A.; Kassem, N.; Hunter, C.; Philips, S.; Radovich, M.; Schneider, B.P. Identification of germline cancer predisposition variants during clinical ctDNA testing. Sci. Rep. 2021, 11, 13624. [Google Scholar] [CrossRef]
- Balmaña, J.; Balaguer, F.; Cervantes, A.; Arnold, D.; ESMO Guidelines Working Group. Familial risk-colorectal cancer: ESMO Clinical Practice Guidelines. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2013, 24 (Suppl. S6), vi73–vi80. [Google Scholar] [CrossRef]
- Paluch-Shimon, S.; Cardoso, F.; Sessa, C.; Balmana, J.; Cardoso, M.J.; Gilbert, F.; Senkus, E.; ESMO Guidelines Committee. Prevention and screening in BRCA mutation carriers and other breast/ovarian hereditary cancer syndromes: ESMO Clinical Practice Guidelines for cancer prevention and screening. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016, 27 (Suppl. S5), v103–v110. [Google Scholar] [CrossRef] [PubMed]
- Vogt, A.; Schmid, S.; Heinimann, K.; Frick, H.; Herrmann, C.; Cerny, T.; Omlin, A. Multiple primary tumours: Challenges and approaches, a review. ESMO Open 2017, 2, e000172. [Google Scholar] [CrossRef] [Green Version]
- Aldea, M.; Cerbone, L.; Bayle, A.; Parisi, C.; Sarkozy, C.; Vasseur, D.; Verlingue, L.; Blanc-Durand, F.; Mosele, F.; Sakkal, M.; et al. Detection of additional occult malignancy through profiling of ctDNA in late-stage cancer patients. Ann. Oncol. 2021, 32, 1642–1645. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Li, W.; Deng, Q.; Li, L.; Hsi, E.D.; Young, K.H.; Zhang, M.; Li, Y. MYD88 L265P Mutation in Lymphoid Malignancies. Cancer Res. 2018, 78, 2457–2462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Swanton, C.; Seiden, M.V.; Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Smith, D.; Richards, D.; et al. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann. Oncol. 2020, 31, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Chabon, J.J.; Hamilton, E.G.; Kurtz, D.M.; Esfahani, M.S.; Moding, E.J.; Stehr, H.; Schroers-Martin, J.; Nabet, B.Y.; Chen, B.; Chaudhuri, A.A.; et al. Integrating genomic features for non-invasive early lung cancer detection. Nature 2020, 580, 245–251. [Google Scholar] [CrossRef]
- Coombs, C.C.; Zehir, A.; Devlin, S.M.; Kishtagari, A.; Syed, A.; Jonsson, P.; Hyman, D.M.; Solit, D.B.; Robson, M.E.; Baselga, J.; et al. Therapy-Related Clonal Hematopoiesis in Patients with Non-hematologic Cancers Is Common and Associated with Adverse Clinical Outcomes. Cell Stem Cell 2017, 21, 374–382.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef] [PubMed]
- Vaughn, C.P.; ZoBell, S.D.; Furtado, L.V.; Baker, C.L.; Samowitz, W.S. Frequency of KRAS, BRAF, and NRAS mutations in colorectal cancer. Genes Chromosom. Cancer 2011, 50, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.; Leighl, N.B.; Tsao, M.-S.; Shepherd, F.A. KRAS Mutations as Prognostic and Predictive Markers in Non–Small Cell Lung Cancer. J. Thorac. Oncol. 2013, 8, 530–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Ulrich, B.C.; Supplee, J.; Kuang, Y.; Lizotte, P.H.; Feeney, N.B.; Guibert, N.M.; Awad, M.M.; Wong, K.-K.; Jänne, P.A.; et al. False-Positive Plasma Genotyping Due to Clonal Hematopoiesis. Clin. Cancer Res. 2018, 24, 4437–4443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, K.; Konnick, E.Q.; Schweizer, M.T.; Sokolova, A.O.; Grivas, P.; Cheng, H.H.; Klemfuss, N.M.; Beightol, M.; Yu, E.Y.; Nelson, P.S.; et al. Association of Clonal Hematopoiesis in DNA Repair Genes with Prostate Cancer Plasma Cell-free DNA Testing Interference. JAMA Oncol. 2021, 7, 107. [Google Scholar] [CrossRef]
- Tie, J.; Kinde, I.; Wang, Y.; Wong, H.L.; Roebert, J.; Christie, M.; Tacey, M.; Wong, R.; Singh, M.; Karapetis, C.S.; et al. Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Ann. Oncol. 2015, 26, 1715–1722. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yao, Y.; Xu, Y.; Li, L.; Gong, Y.; Zhang, K.; Zhang, M.; Guan, Y.; Chang, L.; Xia, X.; et al. Pan-cancer circulating tumor DNA detection in over 10,000 Chinese patients. Nat. Commun. 2021, 12, 11. [Google Scholar] [CrossRef] [PubMed]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of Circulating Tumor DNA in Early- and Late-Stage Human Malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [Green Version]
- Rolfo, C.; Manca, P.; Salgado, R.; Van Dam, P.; Dendooven, A.; Ferri Gandia, J.; Rutten, A.; Lybaert, W.; Vermeij, J.; Gevaert, T.; et al. Multidisciplinary molecular tumour board: A tool to improve clinical practice and selection accrual for clinical trials in patients with cancer. ESMO Open 2018, 3, e000398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowan, A.J.; Lamlum, H.; Ilyas, M.; Wheeler, J.; Straub, J.; Papadopoulou, A.; Bicknell, D.; Bodmer, W.F.; Tomlinson, I.P.M. APC mutations in sporadic colorectal tumors: A mutational “hotspot” and interdependence of the “two hits”. Proc. Natl. Acad. Sci. USA 2000, 97, 3352–3357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, A.J.; Dwight, T.; Gill, A.J.; Dickson, K.-A.; Zhu, Y.; Clarkson, A.; Gard, G.B.; Maidens, J.; Valmadre, S.; Clifton-Bligh, R.; et al. Assessing mutant p53 in primary high-grade serous ovarian cancer using immunohistochemistry and massively parallel sequencing. Sci. Rep. 2016, 6, 26191. [Google Scholar] [CrossRef] [PubMed]
- Berg, K.D.; Glaser, C.L.; Thompson, R.E.; Hamilton, S.R.; Griffin, C.A.; Eshleman, J.R. Detection of microsatellite instability by fluorescence multiplex polymerase chain reaction. J. Mol. Diagn. JMD 2000, 2, 20–28. [Google Scholar] [CrossRef] [Green Version]
- Shia, J. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part I. The utility of immunohistochemistry. J. Mol. Diagn. JMD 2008, 10, 293–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larson, M.H.; Pan, W.; Kim, H.J.; Mauntz, R.E.; Stuart, S.M.; Pimentel, M.; Zhou, Y.; Knudsgaard, P.; Demas, V.; Aravanis, A.M.; et al. A comprehensive characterization of the cell-free transcriptome reveals tissue- and subtype-specific biomarkers for cancer detection. Nat. Commun. 2021, 12, 2357. [Google Scholar] [CrossRef]


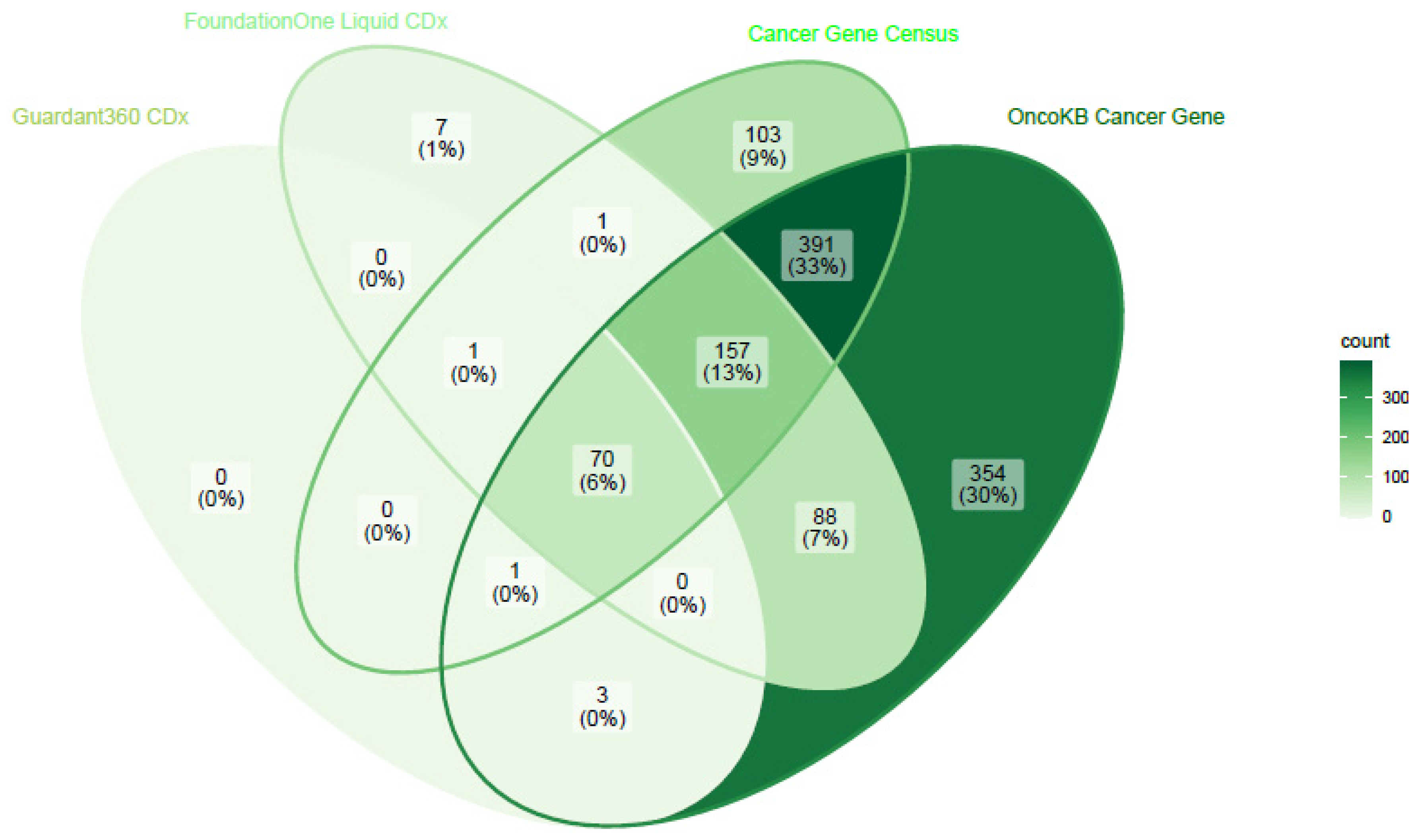{kind=link}
| Characteristics | G360 | F1LCDx | |
|---|---|---|---|
| Starting material | Whole blood | 2 × 8.5 mL | 2 × 10 mL |
| Alterations types | SNV | 73 genes | 311 genes |
| Copy number variations | 18 genes (amplification only) | 310 genes | |
| Fusions/Rearrangements | 8 genes | 324 genes | |
| Microsatellite status | Yes | Yes | |
| TMB | No | Yes | |
| Turnaround time | Announced | 7 calendar days | Less than 2 weeks |
| LoD | SNV-indels 95–100% | 0.20–0.25% | 0.4–0.82% |
| Fusions/Rearrangements | 0.20% | 0.37–0.90% | |
| Tumor fraction | No | Yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasseur, D.; Sassi, H.; Bayle, A.; Tagliamento, M.; Besse, B.; Marzac, C.; Arbab, A.; Auger, N.; Cotteret, S.; Aldea, M.; et al. Next-Generation Sequencing on Circulating Tumor DNA in Advanced Solid Cancer: Swiss Army Knife for the Molecular Tumor Board? A Review of the Literature Focused on FDA Approved Test. Cells 2022, 11, 1901. https://doi.org/10.3390/cells11121901
Vasseur D, Sassi H, Bayle A, Tagliamento M, Besse B, Marzac C, Arbab A, Auger N, Cotteret S, Aldea M, et al. Next-Generation Sequencing on Circulating Tumor DNA in Advanced Solid Cancer: Swiss Army Knife for the Molecular Tumor Board? A Review of the Literature Focused on FDA Approved Test. Cells. 2022; 11(12):1901. https://doi.org/10.3390/cells11121901
Chicago/Turabian StyleVasseur, Damien, Hela Sassi, Arnaud Bayle, Marco Tagliamento, Benjamin Besse, Christophe Marzac, Ahmadreza Arbab, Nathalie Auger, Sophie Cotteret, Mihaela Aldea, and et al. 2022. "Next-Generation Sequencing on Circulating Tumor DNA in Advanced Solid Cancer: Swiss Army Knife for the Molecular Tumor Board? A Review of the Literature Focused on FDA Approved Test" Cells 11, no. 12: 1901. https://doi.org/10.3390/cells11121901
APA StyleVasseur, D., Sassi, H., Bayle, A., Tagliamento, M., Besse, B., Marzac, C., Arbab, A., Auger, N., Cotteret, S., Aldea, M., Blanc-Durand, F., Géraud, A., Gazzah, A., Loriot, Y., Hollebecque, A., Martín-Romano, P., Ngo-Camus, M., Nicotra, C., Ponce, S., ... Lacroix, L. (2022). Next-Generation Sequencing on Circulating Tumor DNA in Advanced Solid Cancer: Swiss Army Knife for the Molecular Tumor Board? A Review of the Literature Focused on FDA Approved Test. Cells, 11(12), 1901. https://doi.org/10.3390/cells11121901