
Systemic Ablation of Camkk2 Impairs Metastatic Colonization and Improves Insulin Sensitivity in TRAMP Mice: Evidence for Cancer Cell-Extrinsic CAMKK2 Functions in Prostate Cancer


 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Experiments
2.2. GEMM Models
2.3. Immunoblot Analysis
2.4. High-Fat Diet Studies
2.5. Plasma
2.6. Cell Culture
2.7. Proliferation Assays
2.8. Syngeneic Models
2.9. Histology and Immunostaining
| Antibody | Company | Cat# | Use |
| p-HH3 (Ser10) | Millipore Sigma Burlington, MA, USA | 06-570 | IHC |
| Ki67 | Bethyl Laboratories Montgomery, TX, USA | IHC-00375-1 | IHC |
| Cleaved caspase-3 | Cell Signaling Technology Danvers, MA, USA | 9661 | IHC |
| p-S6 (Ser235/236) | Cell Signaling Technology | 2211 | IHC |
2.10. Evaluation of Metastasis
2.11. Average Cell Area and Volume
2.12. Statistical Analyses
3. Results
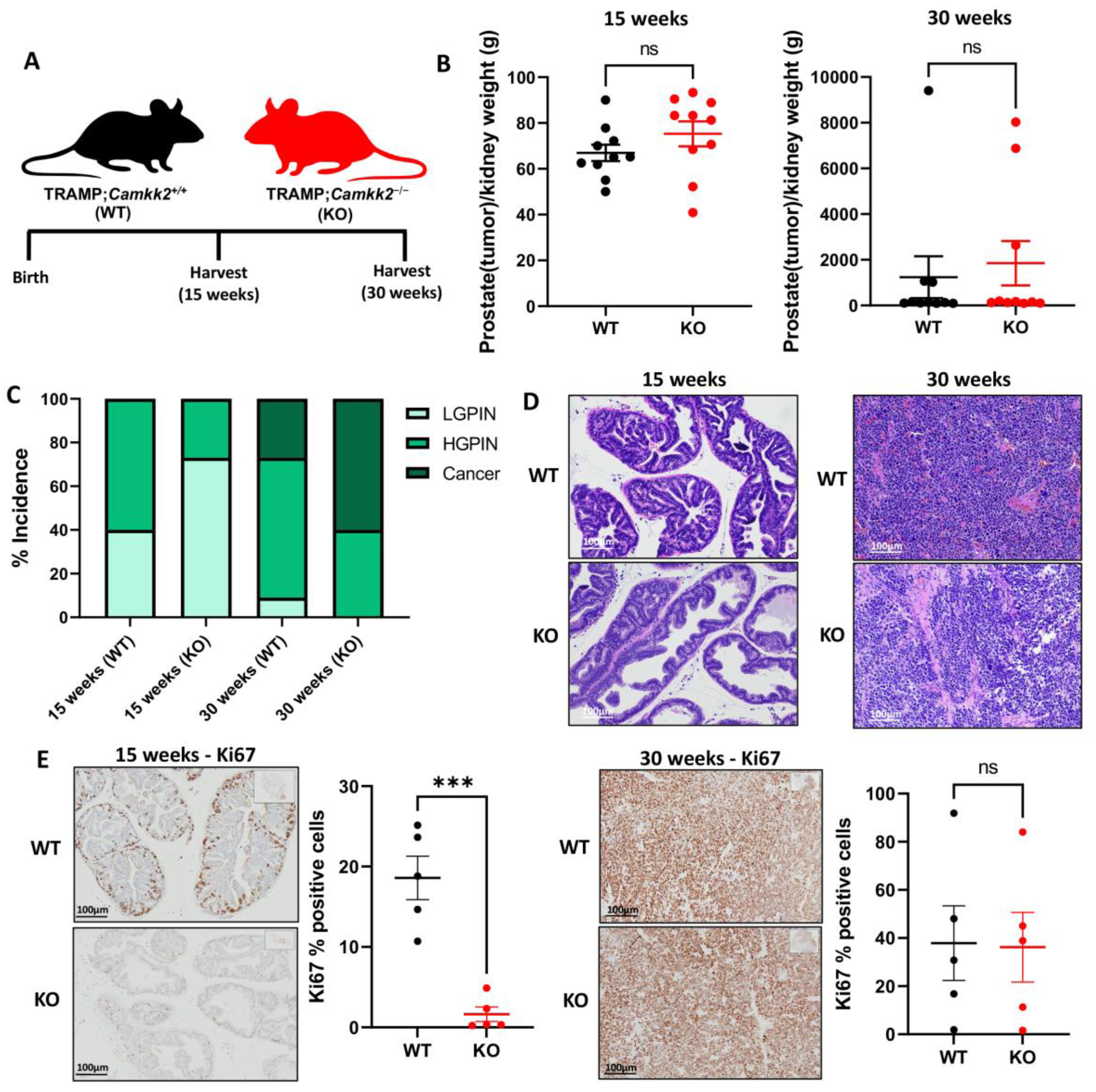
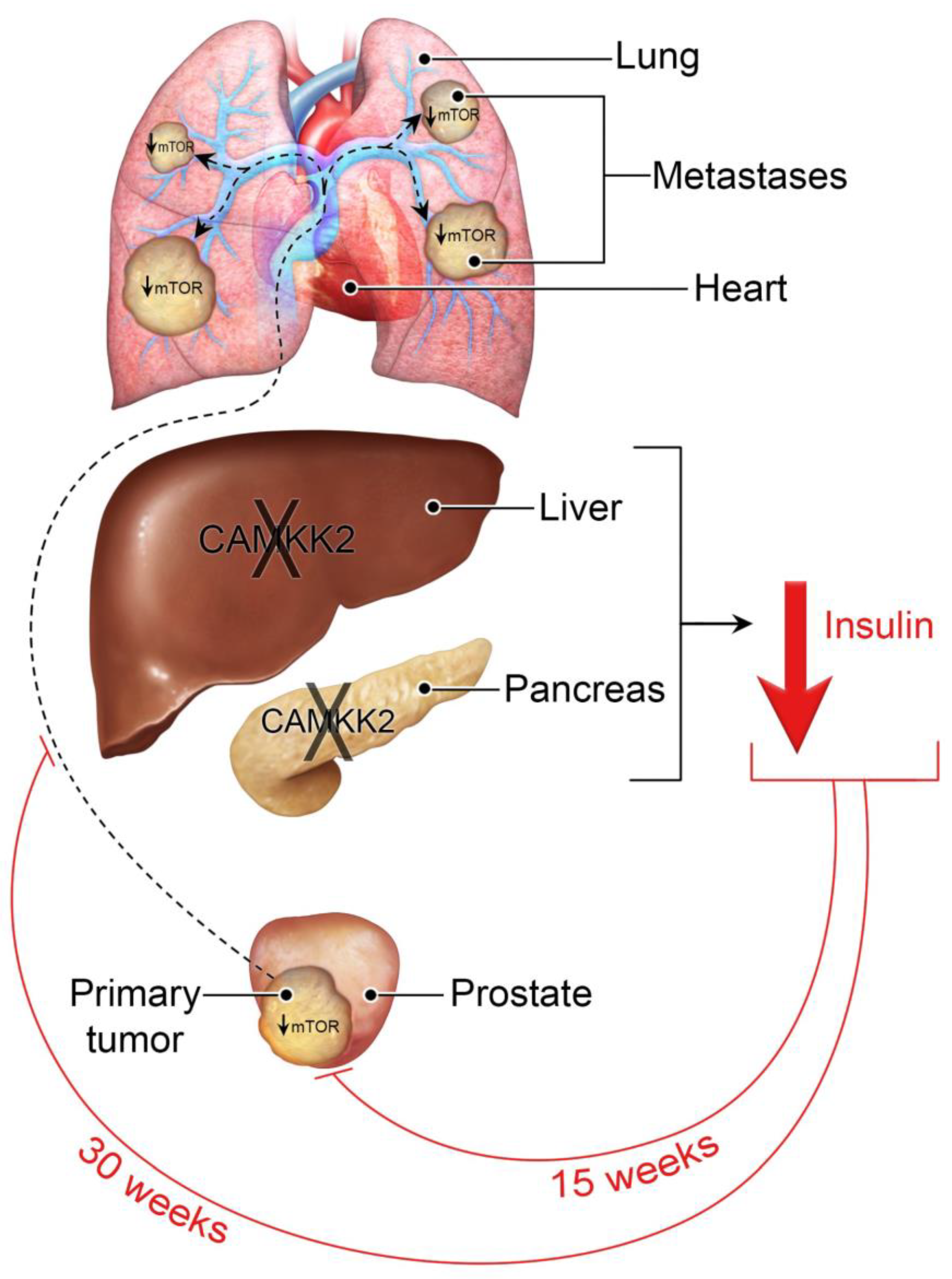
3.1. Camkk2 Deletion Initially Protects Mice from Localized Disease Progression at 15 Weeks, but Relapses at 30 Weeks in a Spontaneous Prostate Cancer Mouse Model
3.2. Camkk2 Deletion Does Not Change Cancer Progression Rates in Castrated TRAMP Mice but Does Alter Tumor Biology
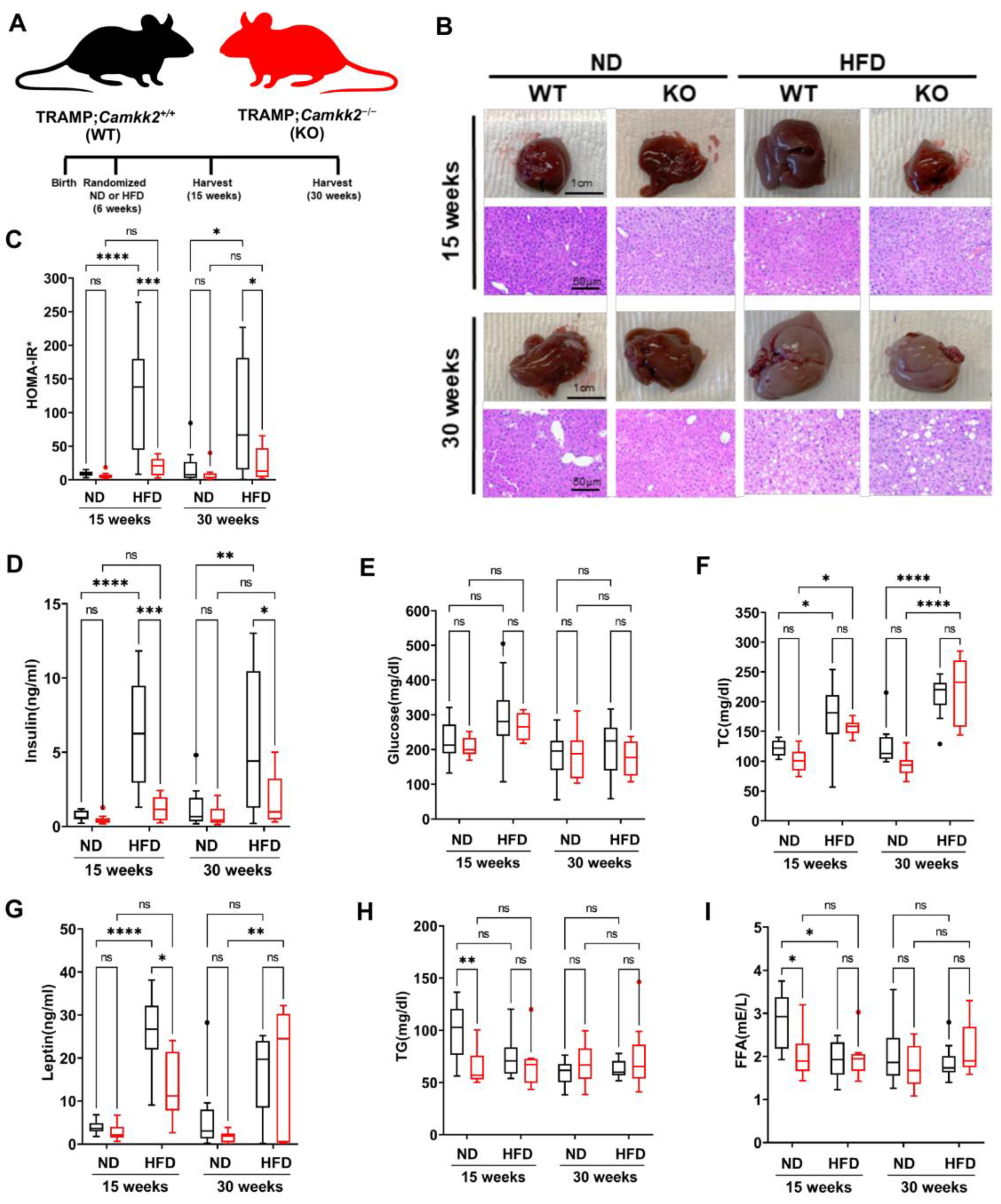
3.3. Systemic Camkk2 Deletion Protects against Metabolic Disorder in a High-Fat Diet-Induced Model of Obesity and Prostate Cancer
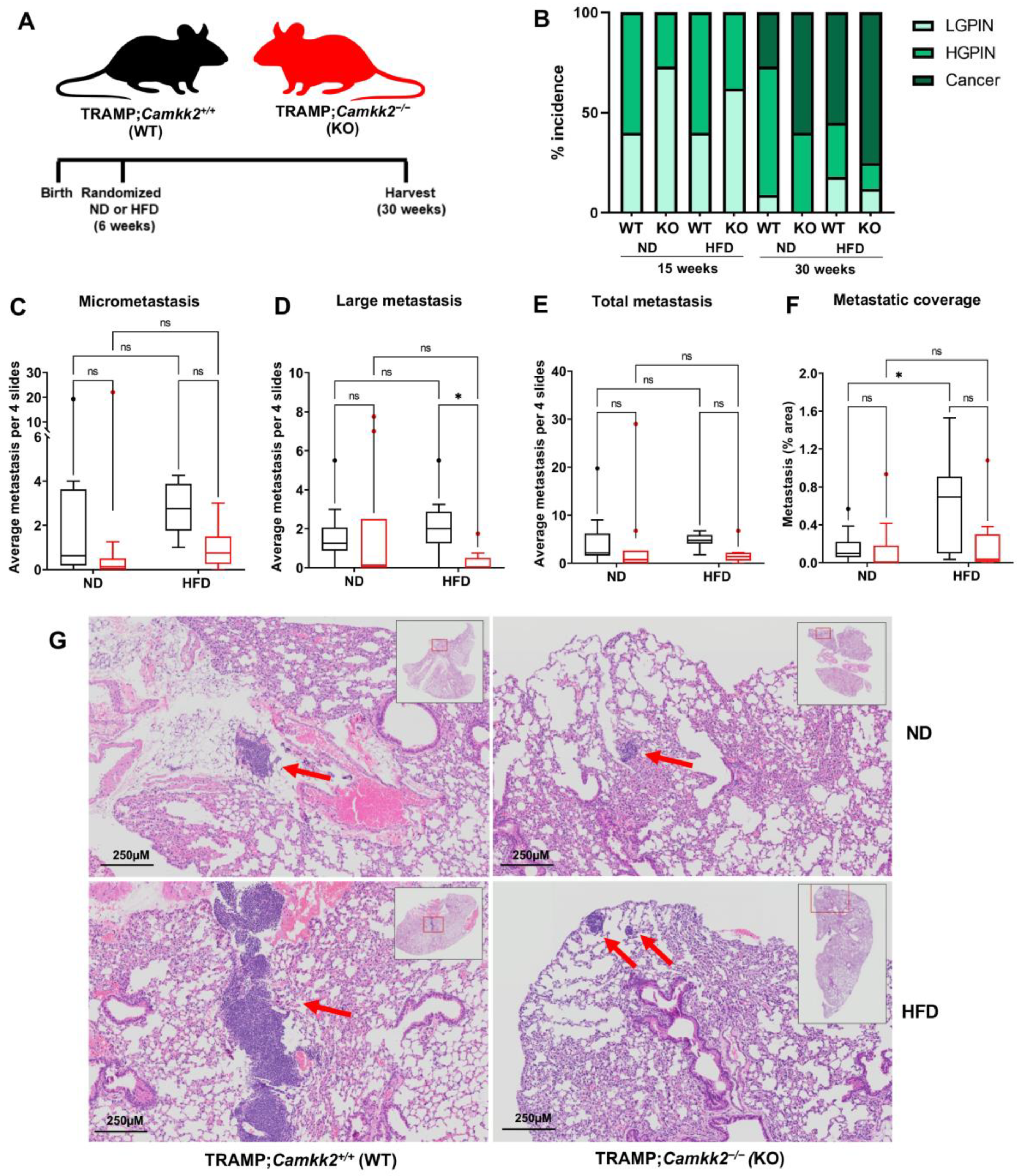
3.4. Camkk2 Deletion Impairs the Metastatic Colonization of NEPC Tumors
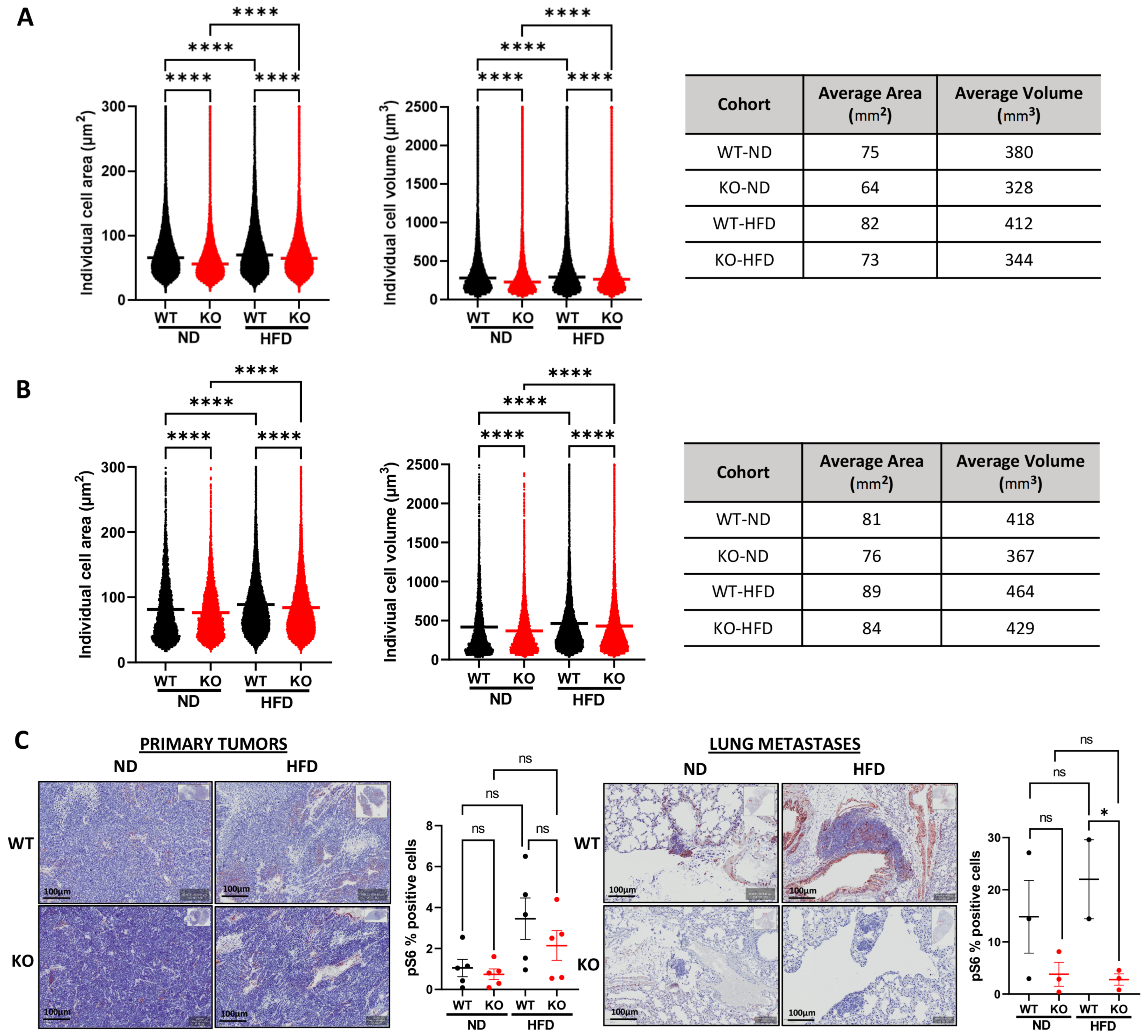
3.5. Host Camkk2 Ablation Decreases Cancer Cell Size and mTOR Signaling in the TRAMP GEMM Model of Prostate Cancer
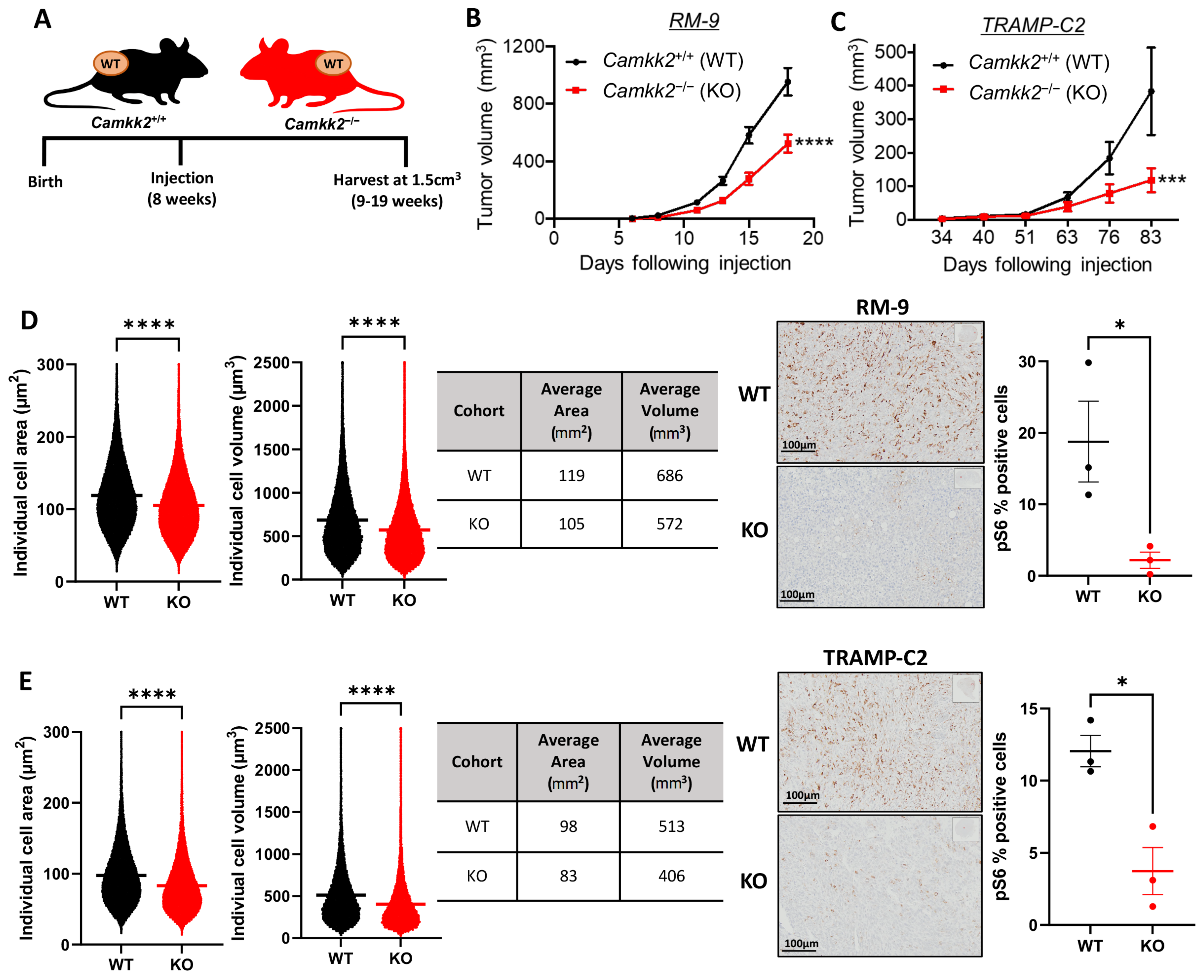
3.6. Host Camkk2 Ablation Decreases Tumor Growth and Cancer Cell Size in Syngeneic Mouse Models of Prostate Cancer: Evidence of Cancer Cell-Extrinsic Roles for CAMKK2 in Prostate Cancer
4. Discussion
Caveats
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel:, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Awad, D.; Pulliam, T.L.; Lin, C.; Wilkenfeld, S.R.; Frigo, D.E. Delineation of the androgen-regulated signaling pathways in prostate cancer facilitates the development of novel therapeutic approaches. Curr. Opin. Pharmacol. 2018, 41, 1–11. [Google Scholar] [CrossRef]
- Frigo, D.E.; Howe, M.K.; Wittmann, B.M.; Brunner, A.M.; Cushman, I.; Wang, Q.; Brown, M.; Means, A.R.; McDonnell, D.P. CaM kinase kinase beta-mediated activation of the growth regulatory kinase AMPK is required for androgen-dependent migration of prostate cancer cells. Cancer Res. 2011, 71, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Karacosta, L.G.; Foster, B.A.; Azabdaftari, G.; Feliciano, D.M.; Edelman, A.M. A regulatory feedback loop between Ca2+/calmodulin-dependent protein kinase kinase 2 (CaMKK2) and the androgen receptor in prostate cancer progression. J. Biol. Chem. 2012, 287, 24832–24843. [Google Scholar] [CrossRef] [PubMed]
- Massie, C.E.; Lynch, A.; Ramos-Montoya, A.; Boren, J.; Stark, R.; Fazli, L.; Warren, A.; Scott, H.; Madhu, B.; Sharma, N.; et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011, 30, 2719–2733. [Google Scholar] [CrossRef]
- Dadwal, U.C.; Chang, E.S.; Sankar, U. Androgen Receptor-CaMKK2 Axis in Prostate Cancer and Bone Microenvironment. Front. Endocrinol. 2018, 9, 335. [Google Scholar] [CrossRef] [PubMed]
- Marcelo, K.L.; Means, A.R.; York, B. The Ca(2+)/Calmodulin/CaMKK2 Axis: Nature’s Metabolic CaMshaft. Trends Endocrinol. Metab. 2016, 27, 706–718. [Google Scholar] [CrossRef]
- Racioppi, L. CaMKK2: A novel target for shaping the androgen-regulated tumor ecosystem. Trends Mol. Med. 2013, 19, 83–88. [Google Scholar] [CrossRef]
- Pulliam, T.L.; Goli, P.; Awad, D.; Lin, C.; Wilkenfeld, S.R.; Frigo, D.E. Regulation and role of CAMKK2 in prostate cancer. Nat. Rev. Urol. 2022. [Google Scholar] [CrossRef]
- Khan, A.S.; Frigo, D.E. A spatiotemporal hypothesis for the regulation, role, and targeting of AMPK in prostate cancer. Nat. Rev. Urol. 2017, 14, 164–180. [Google Scholar] [CrossRef]
- Lin, C.; Blessing, A.M.; Pulliam, T.L.; Shi, Y.; Wilkenfeld, S.R.; Han, J.J.; Murray, M.M.; Pham, A.H.; Duong, K.; Brun, S.N.; et al. Inhibition of CAMKK2 impairs autophagy and castration-resistant prostate cancer via suppression of AMPK-ULK1 signaling. Oncogene 2021, 40, 1690–1705. [Google Scholar] [CrossRef] [PubMed]
- Penfold, L.; Woods, A.; Muckett, P.; Nikitin, A.Y.; Kent, T.R.; Zhang, S.; Graham, R.; Pollard, A.; Carling, D. CAMKK2 Promotes Prostate Cancer Independently of AMPK via Increased Lipogenesis. Cancer Res. 2018, 78, 6747–6761. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, A.A.; Foster, B.A.; Allison, J.P.; Greenberg, N.M.; Kwon, E.D. The TRAMP mouse as a model for prostate cancer. Curr. Protoc. Immunol. 2001, 45, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Kido, L.A.; de Almeida Lamas, C.; Marostica, M.R., Jr.; Cagnon, V.H.A. Transgenic Adenocarcinoma of the Mouse Prostate (TRAMP) model: A good alternative to study PCa progression and chemoprevention approaches. Life Sci. 2019, 217, 141–147. [Google Scholar] [CrossRef]
- Crawford, S.E.; Hyser, J.M.; Utama, B.; Estes, M.K. Autophagy hijacked through viroporin-activated calcium/calmodulin-dependent kinase kinase-beta signaling is required for rotavirus replication. Proc. Natl. Acad. Sci. USA 2012, 109, E3405–E3413. [Google Scholar] [CrossRef]
- Xu, Y.; Xu, Y.; Wang, Y.; Wang, Y.; He, L.; Jiang, Z.; Huang, Z.; Liao, H.; Li, J.; Saavedra, J.M.; et al. Telmisartan prevention of LPS-induced microglia activation involves M2 microglia polarization via CaMKKbeta-dependent AMPK activation. Brain Behav. Immun. 2015, 50, 298–313. [Google Scholar] [CrossRef]
- Racioppi, L.; Noeldner, P.K.; Lin, F.; Arvai, S.; Means, A.R. Calcium/calmodulin-dependent protein kinase kinase 2 regulates macrophage-mediated inflammatory responses. J. Biol. Chem. 2012, 287, 11579–11591. [Google Scholar] [CrossRef]
- Xie, B.; Zhao, M.; Song, D.; Wu, K.; Yi, L.; Li, W.; Li, X.; Wang, K.; Chen, J. Induction of autophagy and suppression of type I IFN secretion by CSFV. Autophagy 2021, 17, 925–947. [Google Scholar] [CrossRef]
- Pritchard, Z.J.; Cary, R.L.; Yang, C.; Novack, D.V.; Voor, M.J.; Sankar, U. Inhibition of CaMKK2 reverses age-associated decline in bone mass. Bone 2015, 75, 120–127. [Google Scholar] [CrossRef]
- York, B.; Li, F.; Lin, F.; Marcelo, K.L.; Mao, J.; Dean, A.; Gonzales, N.; Gooden, D.; Maity, S.; Coarfa, C.; et al. Pharmacological inhibition of CaMKK2 with the selective antagonist STO-609 regresses NAFLD. Sci. Rep. 2017, 7, 11793. [Google Scholar] [CrossRef]
- Marcelo, K.L.; Ribar, T.; Means, C.R.; Tsimelzon, A.; Stevens, R.D.; Ilkayeva, O.; Bain, J.R.; Hilsenbeck, S.G.; Newgard, C.B.; Means, A.R.; et al. Research Resource: Roles for Calcium/Calmodulin-Dependent Protein Kinase Kinase 2 (CaMKK2) in Systems Metabolism. Mol. Endocrinol. 2016, 30, 557–572. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.A.; Lin, F.; Ribar, T.J.; Stevens, R.D.; Muehlbauer, M.J.; Newgard, C.B.; Means, A.R. Deletion of CaMKK2 from the liver lowers blood glucose and improves whole-body glucose tolerance in the mouse. Mol. Endocrinol. 2012, 26, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Freedland, S.J.; Abrahamsson, P.A. Androgen deprivation therapy and side effects: Are GnRH antagonists safer? Asian J. Androl. 2021, 23, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Higano, C.S. Update on cardiovascular and metabolic risk profiles of hormonal agents used in managing advanced prostate cancer. Urol. Oncol. 2020, 38, 912–917. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.A.; Ribar, T.J.; Lin, F.; Noeldner, P.K.; Green, M.F.; Muehlbauer, M.J.; Witters, L.A.; Kemp, B.E.; Means, A.R. Hypothalamic CaMKK2 contributes to the regulation of energy balance. Cell Metab. 2008, 7, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Vincent, S.D.; Robertson, E.J. Highly efficient transgene-independent recombination directed by a maternally derived SOX2CRE transgene. Genesis 2003, 37, 54–56. [Google Scholar] [CrossRef] [PubMed]
- Eduful, B.J.; O’Byrne, S.N.; Temme, L.; Asquith, C.R.M.; Liang, Y.; Picado, A.; Pilotte, J.R.; Hossain, M.A.; Wells, C.I.; Zuercher, W.J.; et al. Hinge Binder Scaffold Hopping Identifies Potent Calcium/Calmodulin-Dependent Protein Kinase Kinase 2 (CAMKK2) Inhibitor Chemotypes. J. Med. Chem. 2021, 64, 10849–10877. [Google Scholar] [CrossRef]
- Shi, Y.; Han, J.J.; Tennakoon, J.B.; Mehta, F.F.; Merchant, F.A.; Burns, A.R.; Howe, M.K.; McDonnell, D.P.; Frigo, D.E. Androgens promote prostate cancer cell growth through induction of autophagy. Mol. Endocrinol. 2013, 27, 280–295. [Google Scholar] [CrossRef]
- Scott, P.H.; Brunn, G.J.; Kohn, A.D.; Roth, R.A.; Lawrence, J.C., Jr. Evidence of insulin-stimulated phosphorylation and activation of the mammalian target of rapamycin mediated by a protein kinase B signaling pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 7772–7777. [Google Scholar] [CrossRef]
- Fingar, D.C.; Salama, S.; Tsou, C.; Harlow, E.; Blenis, J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 2002, 16, 1472–1487. [Google Scholar] [CrossRef]
- Kozma, S.C.; Thomas, G. Regulation of cell size in growth, development and human disease: PI3K, PKB and S6K. Bioessays 2002, 24, 65–71. [Google Scholar] [CrossRef] [PubMed]
- White, M.A.; Tsouko, E.; Lin, C.; Rajapakshe, K.; Spencer, J.M.; Wilkenfeld, S.R.; Vakili, S.S.; Pulliam, T.L.; Awad, D.; Nikolos, F.; et al. GLUT12 promotes prostate cancer cell growth and is regulated by androgens and CaMKK2 signaling. Endocr. Relat. Cancer 2018, 25, 453–469. [Google Scholar] [CrossRef] [PubMed]
- Berchuck, J.E.; Viscuse, P.V.; Beltran, H.; Aparicio, A. Clinical considerations for the management of androgen indifferent prostate cancer. Prostate Cancer Prostatic Dis. 2021, 24, 623–637. [Google Scholar] [CrossRef]
- Sharma, N.L.; Massie, C.E.; Ramos-Montoya, A.; Zecchini, V.; Scott, H.E.; Lamb, A.D.; MacArthur, S.; Stark, R.; Warren, A.Y.; Mills, I.G.; et al. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell 2013, 23, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; He, H.C.; Han, Z.D.; Wan, Y.P.; Luo, H.W.; Huang, Y.Q.; Cai, C.; Liang, Y.X.; Dai, Q.S.; Jiang, F.N.; et al. MicroRNA-224 and its target CAMKK2 synergistically influence tumor progression and patient prognosis in prostate cancer. Tumour. Biol. 2015, 36, 1983–1991. [Google Scholar] [CrossRef] [PubMed]
- Asquith, C.R.M.; Godoi, P.H.; Counago, R.M.; Laitinen, T.; Scott, J.W.; Langendorf, C.G.; Oakhill, J.S.; Drewry, D.H.; Zuercher, W.J.; Koutentis, P.A.; et al. 1,2,6-Thiadiazinones as Novel Narrow Spectrum Calcium/Calmodulin-Dependent Protein Kinase Kinase 2 (CaMKK2) Inhibitors. Molecules 2018, 23, 1221. [Google Scholar] [CrossRef]
- O’Byrne, S.N.; Scott, J.W.; Pilotte, J.R.; Santiago, A.D.S.; Langendorf, C.G.; Oakhill, J.S.; Eduful, B.J.; Counago, R.M.; Wells, C.I.; Zuercher, W.J.; et al. In Depth Analysis of Kinase Cross Screening Data to Identify CAMKK2 Inhibitory Scaffolds. Molecules 2020, 25, 325. [Google Scholar] [CrossRef]
- Price, D.J.; Drewry, D.H.; Schaller, L.T.; Thompson, B.D.; Reid, P.R.; Maloney, P.R.; Liang, X.; Banker, P.; Buckholz, R.G.; Selley, P.K.; et al. An orally available, brain-penetrant CAMKK2 inhibitor reduces food intake in rodent model. Bioorg. Med. Chem. Lett. 2018, 28, 1958–1963. [Google Scholar] [CrossRef]
- Profeta, G.S.; Dos Reis, C.V.; Santiago, A.D.S.; Godoi, P.H.C.; Fala, A.M.; Wells, C.I.; Sartori, R.; Salmazo, A.P.T.; Ramos, P.Z.; Massirer, K.B.; et al. Binding and structural analyses of potent inhibitors of the human Ca(2+)/calmodulin dependent protein kinase kinase 2 (CAMKK2) identified from a collection of commercially-available kinase inhibitors. Sci. Rep. 2019, 9, 16452. [Google Scholar] [CrossRef] [PubMed]
- Takaya, D.; Niwa, H.; Mikuni, J.; Nakamura, K.; Handa, N.; Tanaka, A.; Yokoyama, S.; Honma, T. Protein ligand interaction analysis against new CaMKK2 inhibitors by use of X-ray crystallography and the fragment molecular orbital (FMO) method. J. Mol. Graph. Model. 2020, 99, 107599. [Google Scholar] [CrossRef]
- Wang, Y.; Nasiri, A.R.; Damsky, W.E.; Perry, C.J.; Zhang, X.M.; Rabin-Court, A.; Pollak, M.N.; Shulman, G.I.; Perry, R.J. Uncoupling Hepatic Oxidative Phosphorylation Reduces Tumor Growth in Two Murine Models of Colon Cancer. Cell Rep. 2018, 24, 47–55. [Google Scholar] [CrossRef]
- Perry, R.J.; Shulman, G.I. Mechanistic Links between Obesity, Insulin, and Cancer. Trends Cancer 2020, 6, 75–78. [Google Scholar] [CrossRef]
- Rabin-Court, A.; Rodrigues, M.R.; Zhang, X.M.; Perry, R.J. Obesity-associated, but not obesity-independent, tumors respond to insulin by increasing mitochondrial glucose oxidation. PLoS ONE 2019, 14, e0218126. [Google Scholar] [CrossRef]
- Siech, C.; Rutz, J.; Maxeiner, S.; Grein, T.; Sonnenburg, M.; Tsaur, I.; Chun, F.K.; Blaheta, R.A. Insulin-like Growth Factor-1 Influences Prostate Cancer Cell Growth and Invasion through an Integrin alpha3, alpha5, alphaV, and beta1 Dependent Mechanism. Cancers 2022, 14, 363. [Google Scholar] [CrossRef]
- Anzo, M.; Cobb, L.J.; Hwang, D.L.; Mehta, H.; Said, J.W.; Yakar, S.; LeRoith, D.; Cohen, P. Targeted deletion of hepatic Igf1 in TRAMP mice leads to dramatic alterations in the circulating insulin-like growth factor axis but does not reduce tumor progression. Cancer Res. 2008, 68, 3342–3349. [Google Scholar] [CrossRef]
- Racioppi, L.; Nelson, E.R.; Huang, W.; Mukherjee, D.; Lawrence, S.A.; Lento, W.; Masci, A.M.; Jiao, Y.; Park, S.; York, B.; et al. CaMKK2 in myeloid cells is a key regulator of the immune-suppressive microenvironment in breast cancer. Nat. Commun. 2019, 10, 2450. [Google Scholar] [CrossRef]
- Huang, W.; Liu, Y.; Luz, A.; Berrong, M.; Meyer, J.N.; Zou, Y.; Swann, E.; Sundaramoorthy, P.; Kang, Y.; Jauhari, S.; et al. Calcium/Calmodulin Dependent Protein Kinase Kinase 2 Regulates the Expansion of Tumor-Induced Myeloid-Derived Suppressor Cells. Front. Immunol. 2021, 12, 754083. [Google Scholar] [CrossRef]
- Grabowska, M.M.; DeGraff, D.J.; Yu, X.; Jin, R.J.; Chen, Z.; Borowsky, A.D.; Matusik, R.J. Mouse models of prostate cancer: Picking the best model for the question. Cancer Metastasis Rev. 2014, 33, 377–397. [Google Scholar] [CrossRef]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef]
- Corn, P.G.; Heath, E.I.; Zurita, A.; Ramesh, N.; Xiao, L.; Sei, E.; Li-Ning-Tapia, E.; Tu, S.M.; Subudhi, S.K.; Wang, J.; et al. Cabazitaxel plus carboplatin for the treatment of men with metastatic castration-resistant prostate cancers: A randomised, open-label, phase 1-2 trial. Lancet Oncol. 2019, 20, 1432–1443. [Google Scholar] [CrossRef]
- Ku, S.Y.; Rosario, S.; Wang, Y.; Mu, P.; Seshadri, M.; Goodrich, Z.W.; Goodrich, M.M.; Labbe, D.P.; Gomez, E.C.; Wang, J.; et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017, 355, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Mu, P.; Zhang, Z.; Benelli, M.; Karthaus, W.R.; Hoover, E.; Chen, C.C.; Wongvipat, J.; Ku, S.Y.; Gao, D.; Cao, Z.; et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017, 355, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yan, W.; Sun, Y.; Yang, C.S. High fat diet-induced hyperinsulinemia promotes the development of prostate adenocarcinoma in prostate specific Pten-/- mice. Carcinogenesis 2022, bgac013. [Google Scholar] [CrossRef] [PubMed]
- Lengyel, E.; Makowski, L.; DiGiovanni, J.; Kolonin, M.G. Cancer as a Matter of Fat: The Crosstalk between Adipose Tissue and Tumors. Trends Cancer 2018, 4, 374–384. [Google Scholar] [CrossRef]
- Watt, M.J.; Clark, A.K.; Selth, L.A.; Haynes, V.R.; Lister, N.; Rebello, R.; Porter, L.H.; Niranjan, B.; Whitby, S.T.; Lo, J.; et al. Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Butler, L.M.; Perone, Y.; Dehairs, J.; Lupien, L.E.; de Laat, V.; Talebi, A.; Loda, M.; Kinlaw, W.B.; Swinnen, J.V. Lipids and cancer: Emerging roles in pathogenesis, diagnosis and therapeutic intervention. Adv. Drug Deliv. Rev. 2020, 159, 245–293. [Google Scholar] [CrossRef]
- Lin, F.; Ribar, T.J.; Means, A.R. The Ca2+/calmodulin-dependent protein kinase kinase, CaMKK2, inhibits preadipocyte differentiation. Endocrinology 2011, 152, 3668–3679. [Google Scholar] [CrossRef]
- Patel, M.; Taskar, K.S.; Zamek-Gliszczynski, M.J. Importance of Hepatic Transporters in Clinical Disposition of Drugs and Their Metabolites. J. Clin. Pharmacol. 2016, 56 (Suppl. 7), S23–S39. [Google Scholar] [CrossRef]
- De Nunzio, C.; Aronson, W.; Freedland, S.J.; Giovannucci, E.; Parsons, J.K. The correlation between metabolic syndrome and prostatic diseases. Eur. Urol. 2012, 61, 560–570. [Google Scholar] [CrossRef]
- Csibi, A.; Blenis, J. Hippo-YAP and mTOR pathways collaborate to regulate organ size. Nat. Cell Biol. 2012, 14, 1244–1245. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pulliam, T.L.; Awad, D.; Han, J.J.; Murray, M.M.; Ackroyd, J.J.; Goli, P.; Oakhill, J.S.; Scott, J.W.; Ittmann, M.M.; Frigo, D.E. Systemic Ablation of Camkk2 Impairs Metastatic Colonization and Improves Insulin Sensitivity in TRAMP Mice: Evidence for Cancer Cell-Extrinsic CAMKK2 Functions in Prostate Cancer. Cells 2022, 11, 1890. https://doi.org/10.3390/cells11121890
Pulliam TL, Awad D, Han JJ, Murray MM, Ackroyd JJ, Goli P, Oakhill JS, Scott JW, Ittmann MM, Frigo DE. Systemic Ablation of Camkk2 Impairs Metastatic Colonization and Improves Insulin Sensitivity in TRAMP Mice: Evidence for Cancer Cell-Extrinsic CAMKK2 Functions in Prostate Cancer. Cells. 2022; 11(12):1890. https://doi.org/10.3390/cells11121890
Chicago/Turabian StylePulliam, Thomas L., Dominik Awad, Jenny J. Han, Mollianne M. Murray, Jeffrey J. Ackroyd, Pavithr Goli, Jonathan S. Oakhill, John W. Scott, Michael M. Ittmann, and Daniel E. Frigo. 2022. "Systemic Ablation of Camkk2 Impairs Metastatic Colonization and Improves Insulin Sensitivity in TRAMP Mice: Evidence for Cancer Cell-Extrinsic CAMKK2 Functions in Prostate Cancer" Cells 11, no. 12: 1890. https://doi.org/10.3390/cells11121890
APA StylePulliam, T. L., Awad, D., Han, J. J., Murray, M. M., Ackroyd, J. J., Goli, P., Oakhill, J. S., Scott, J. W., Ittmann, M. M., & Frigo, D. E. (2022). Systemic Ablation of Camkk2 Impairs Metastatic Colonization and Improves Insulin Sensitivity in TRAMP Mice: Evidence for Cancer Cell-Extrinsic CAMKK2 Functions in Prostate Cancer. Cells, 11(12), 1890. https://doi.org/10.3390/cells11121890