
Targeting Acid Ceramidase Inhibits Glioblastoma Cell Migration through Decreased AKT Signaling

 , , , , ,
, , , , ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Culture of Cell Lines and Patient-Derived Xenografts
2.3. Accession and Analysis of Publicly Available Datasets
2.4. Generation of Knockdown Cell Lines
2.5. RNA Isolation and Quantitative Real-Time PCR
2.6. Cell Growth Assays
2.7. Scratch Assays
2.8. Boyden Chamber Assays
2.9. RNA-Sequencing Analysis
2.10. Lysate Preparation and Immunoblot Analysis
2.11. Statistical Analysis
3. Results
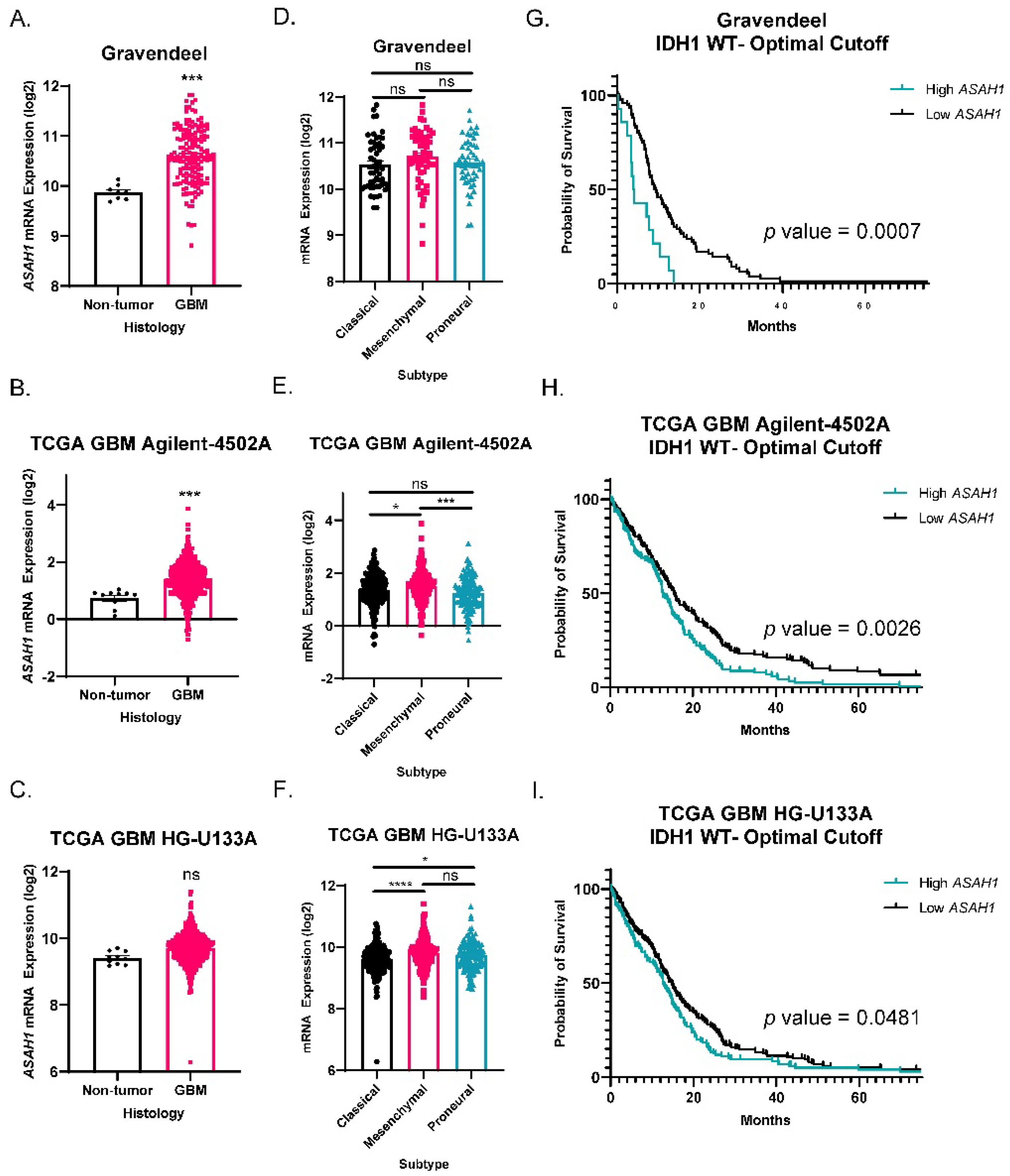
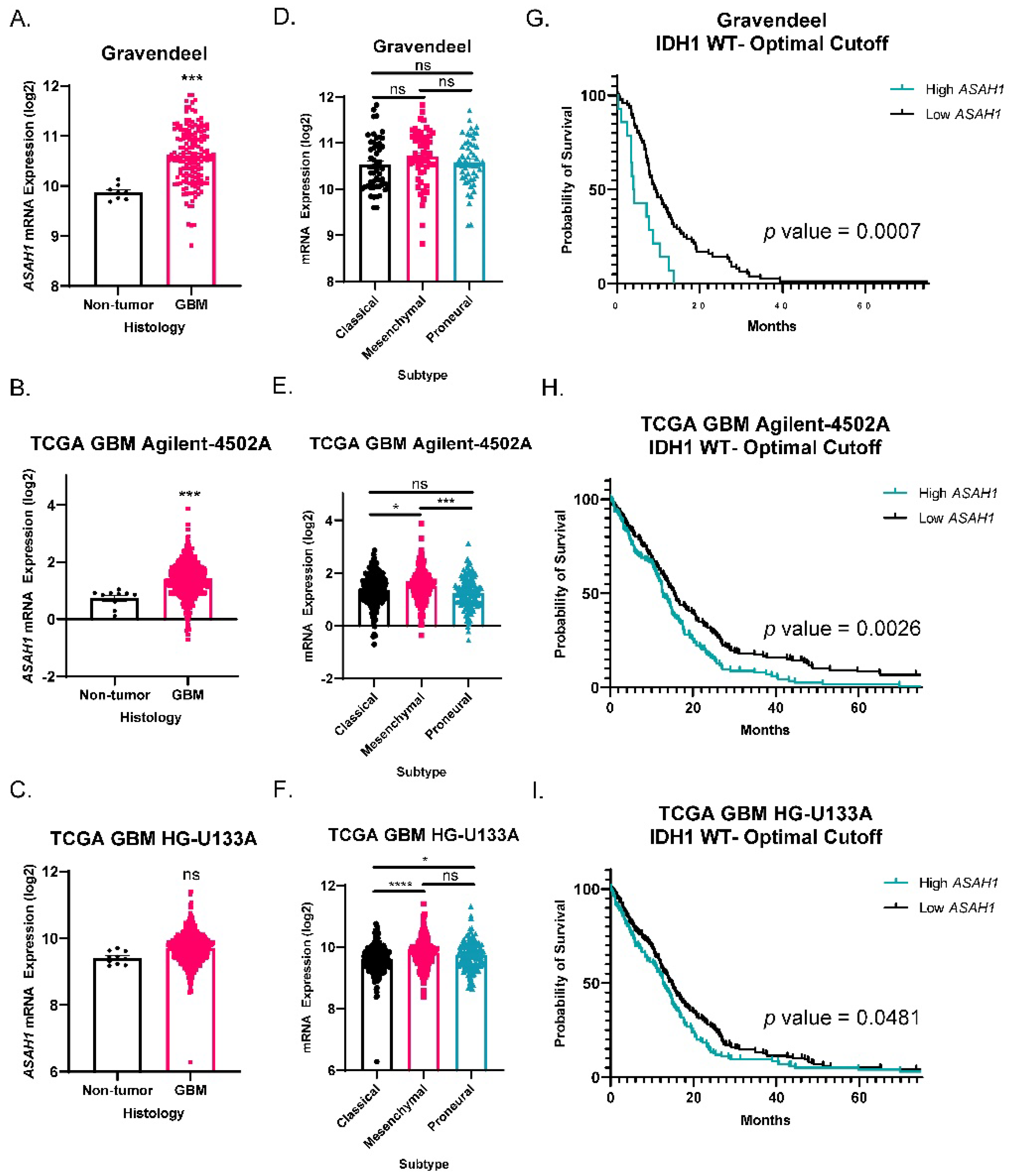
3.1. ASAH1 Expression Correlated with Worse Survival in GBM Patients
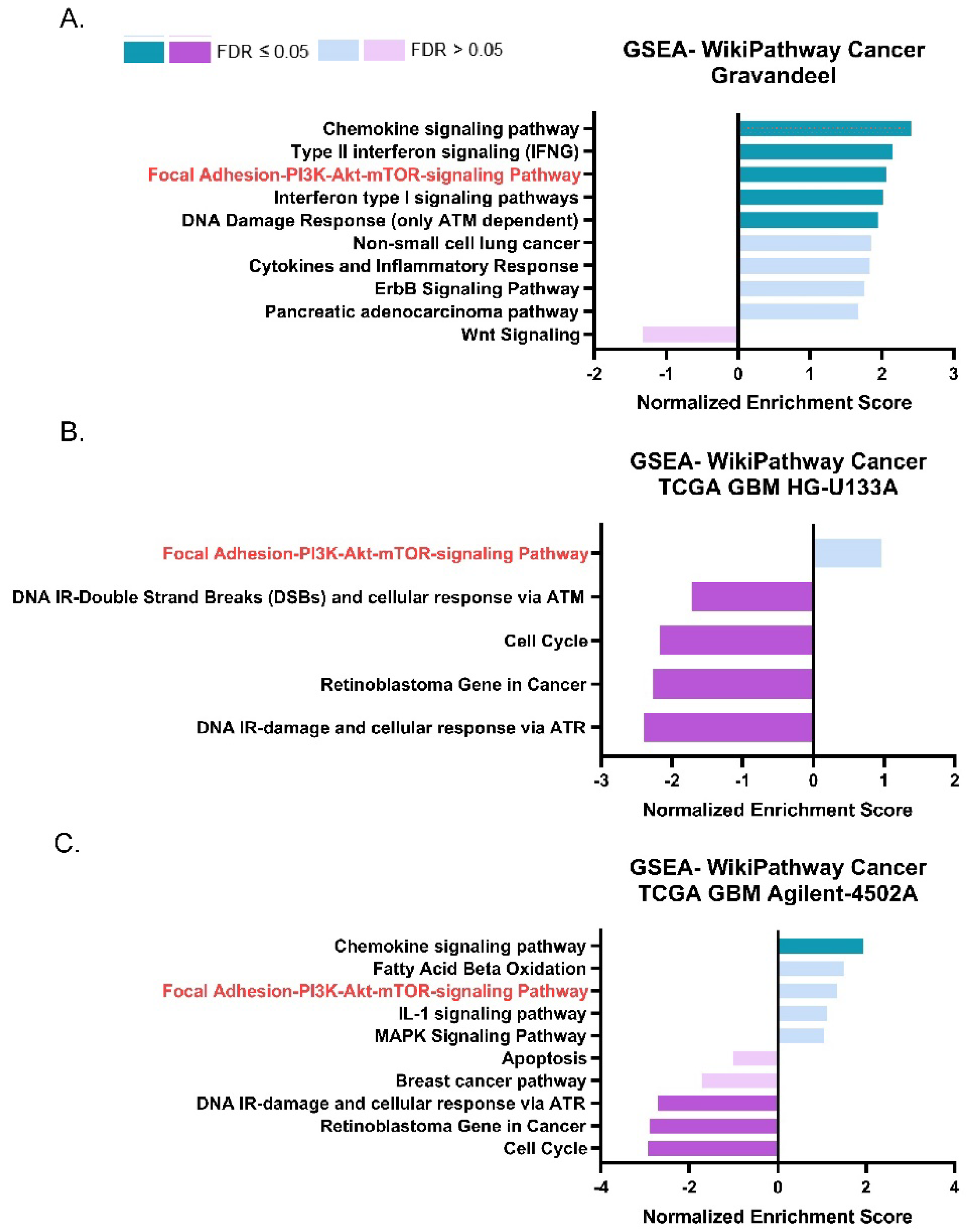
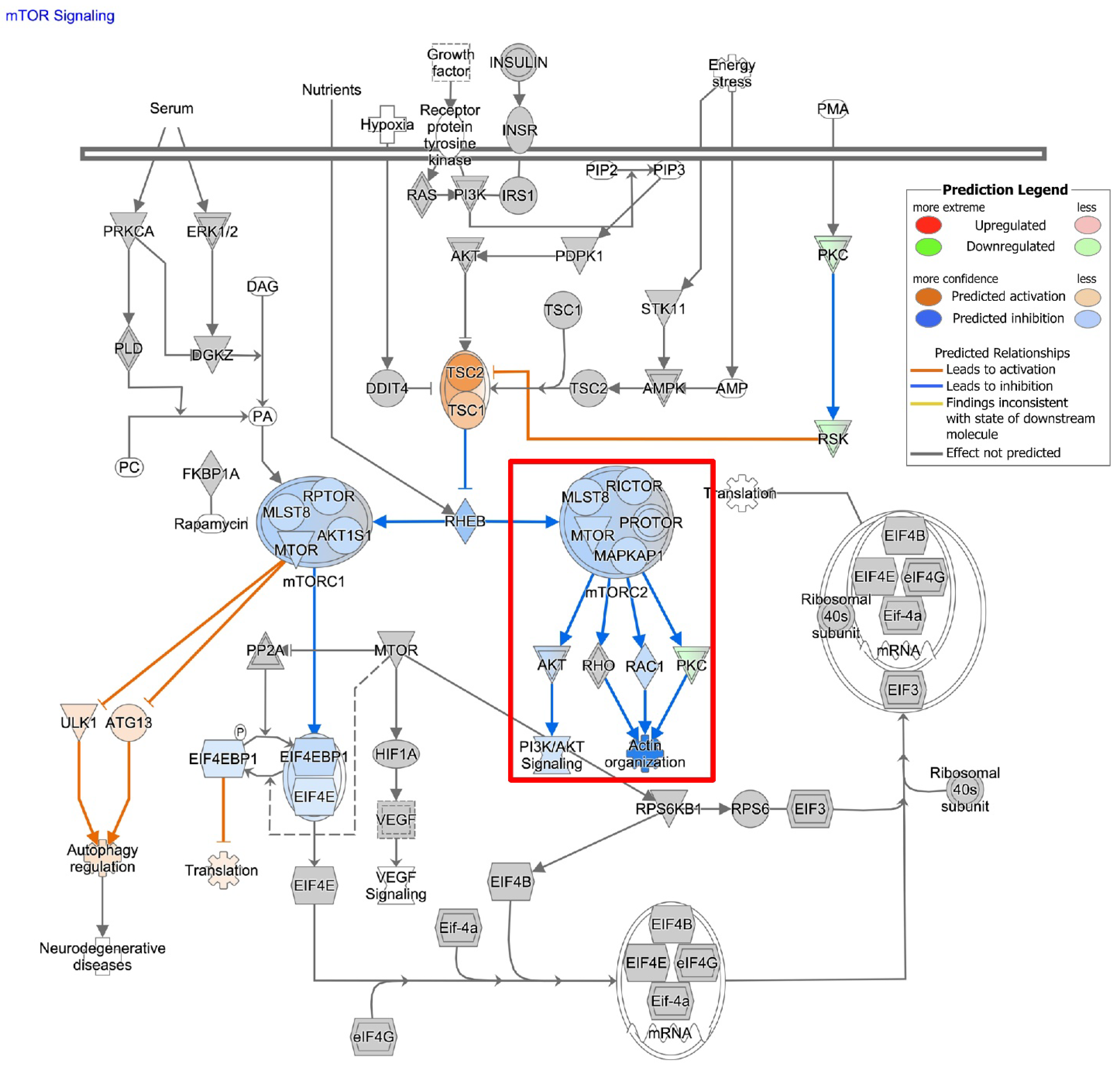
3.2. ASAH1 Expression Correlated with Genes in the Focal Adhesion-PI3K-AKT-mTOR Pathway
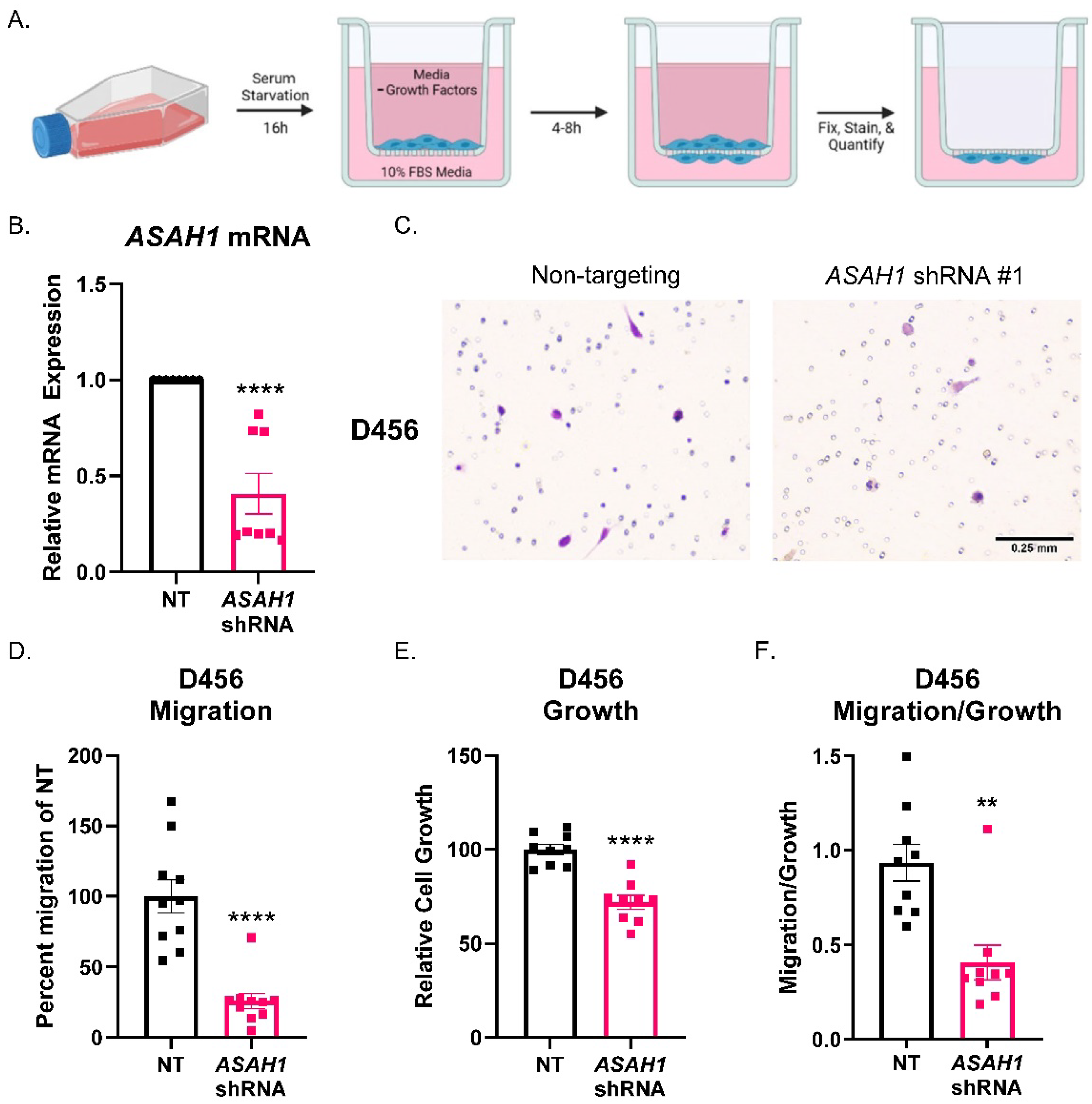
3.3. ASAH1 Knockdown Decreased Migration of GBM Cells Derived from PDXs
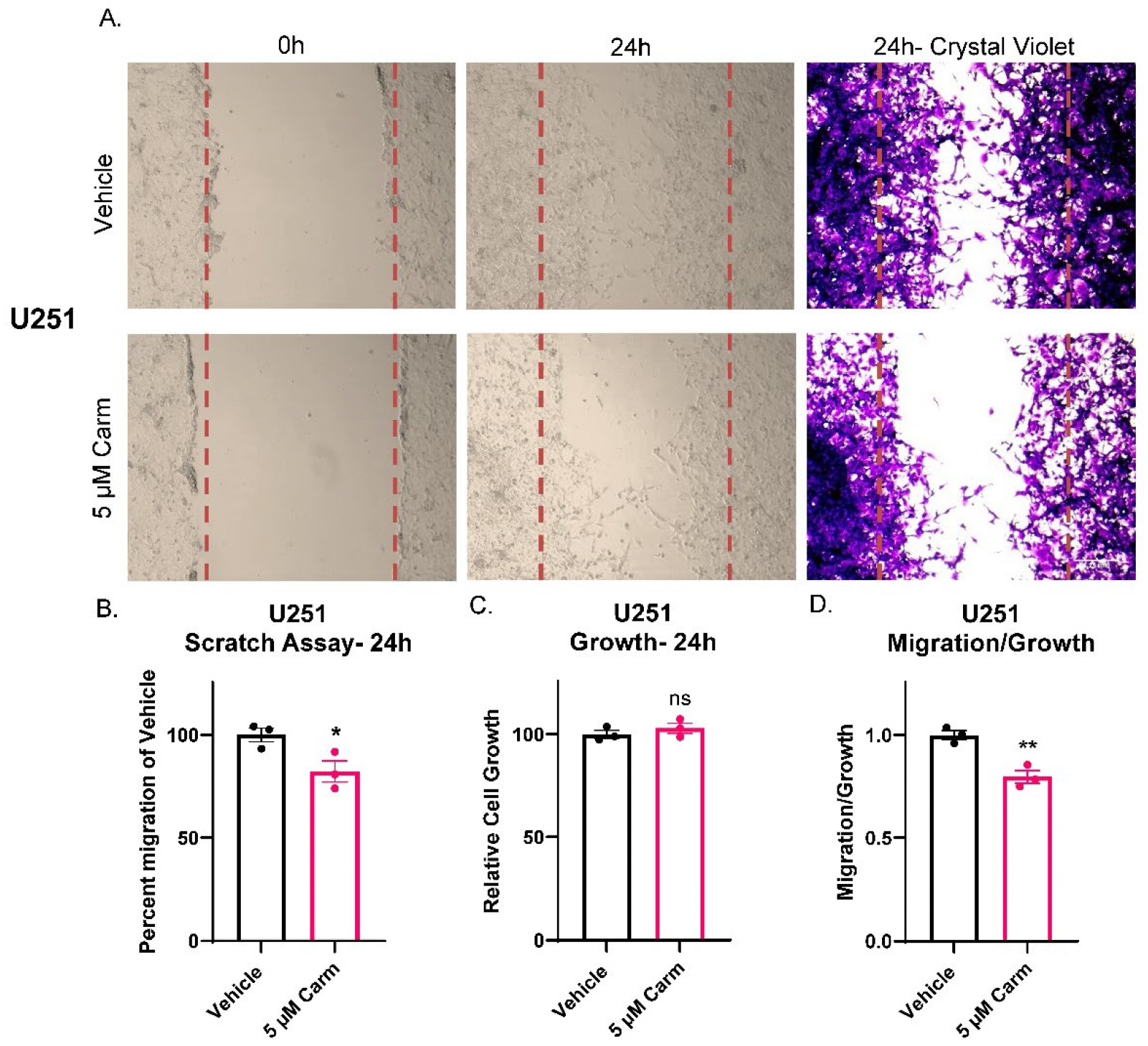
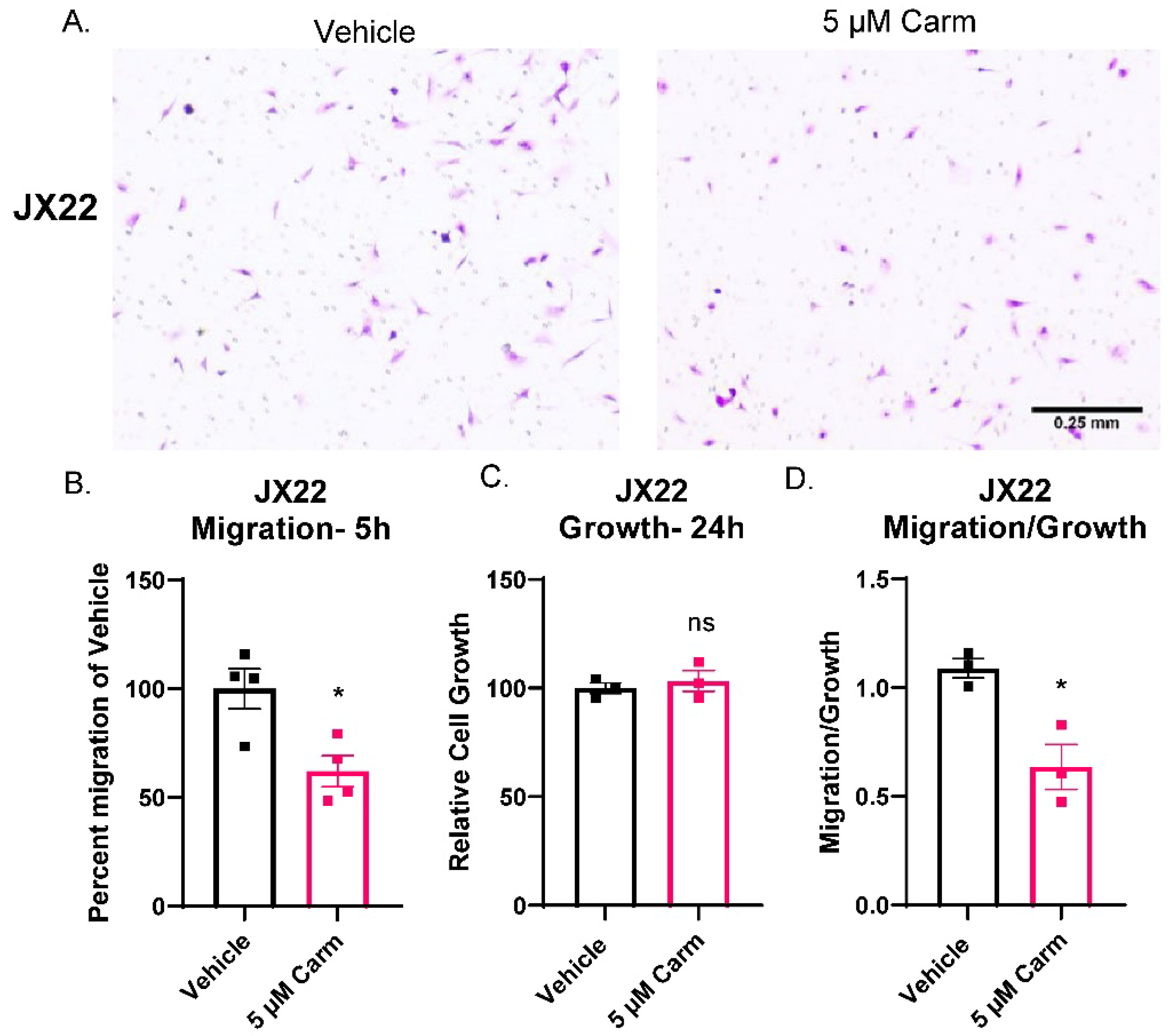
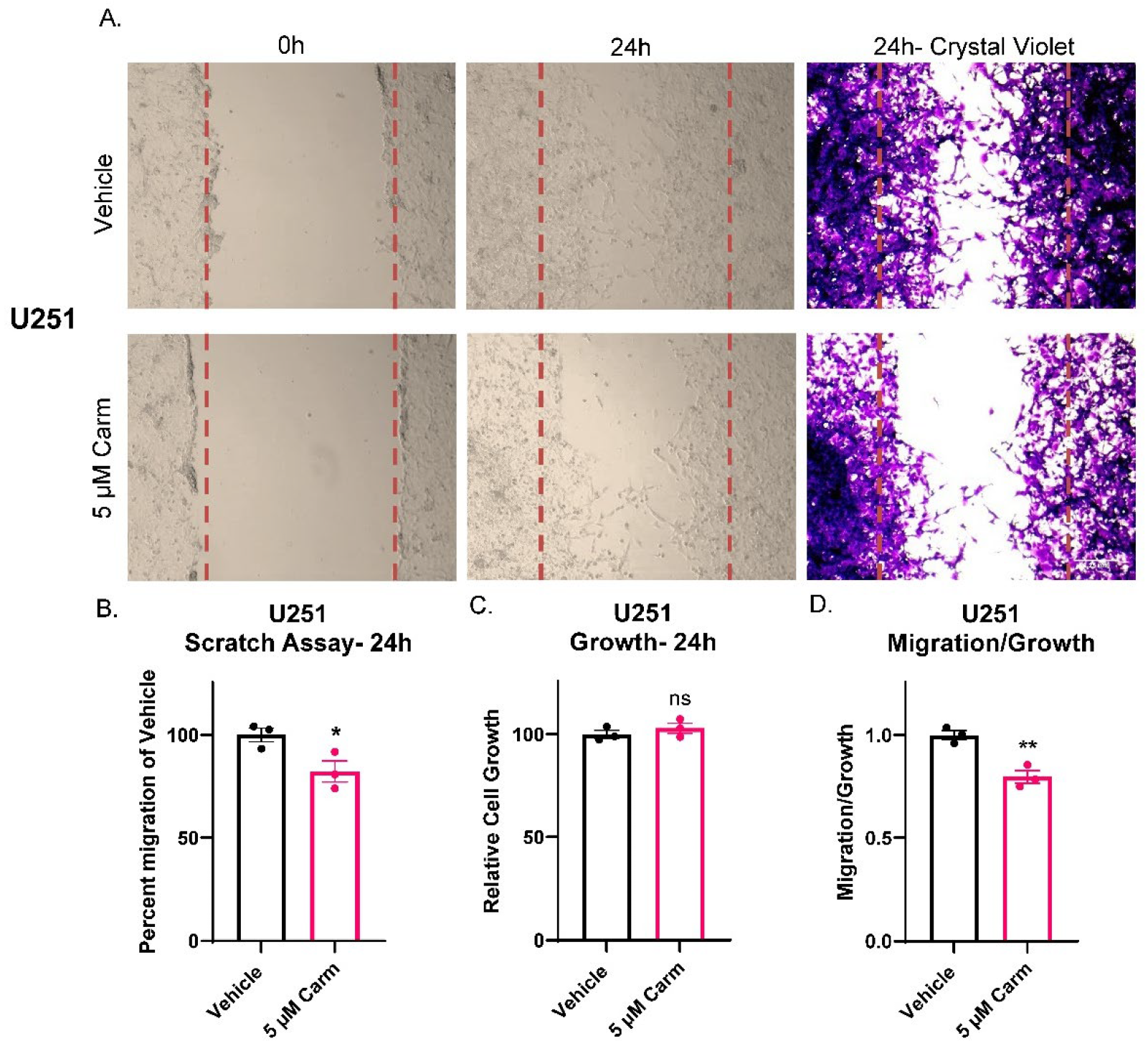
3.4. Carmofur Decreased Migration of GBM Cells
3.5. Carmofur Decreased Migration of PDX-Derived GBM Cells
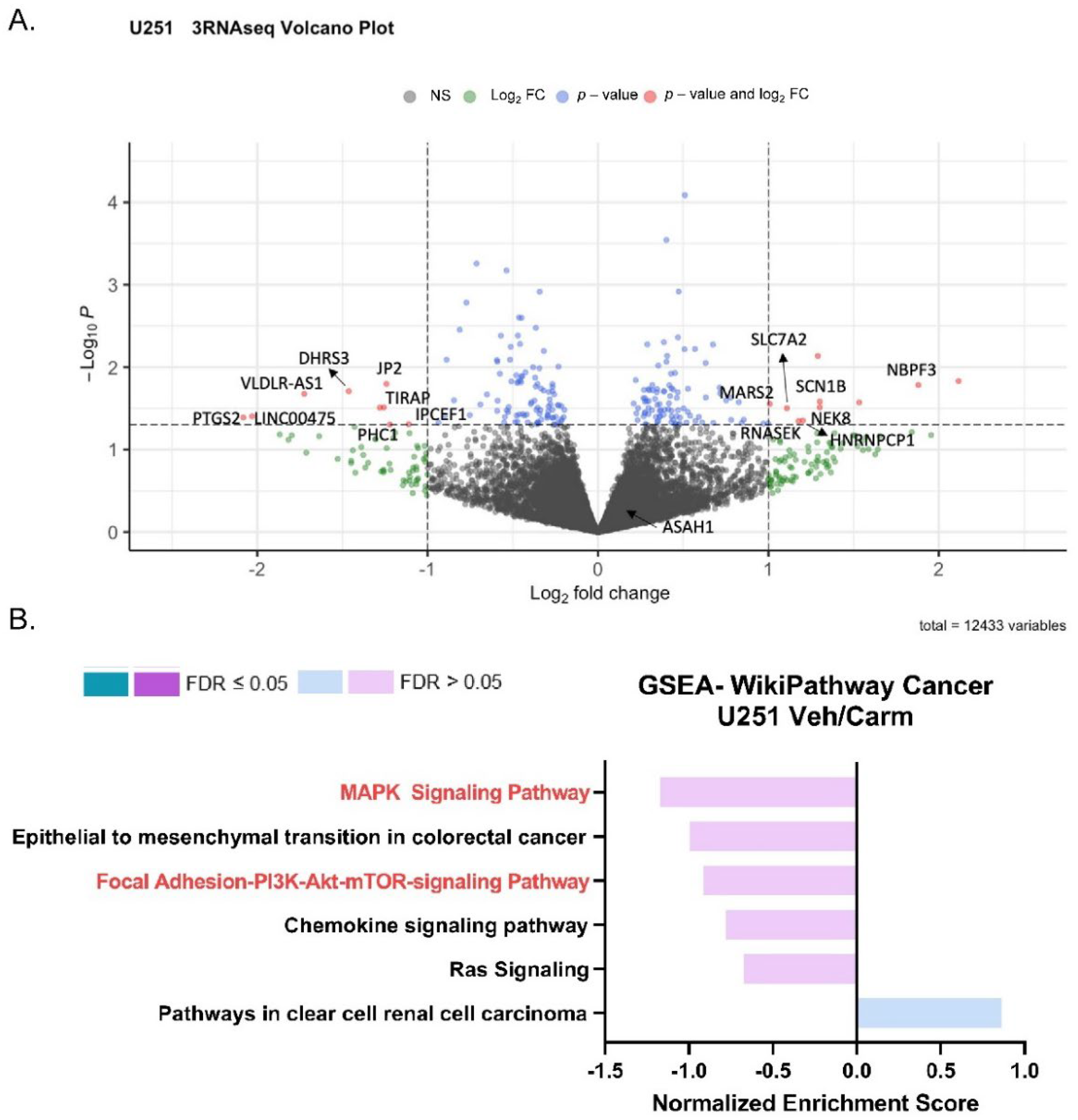
3.6. Pharmacologic Inhibition of ASAH1 Decreased Migration-Related Pathways
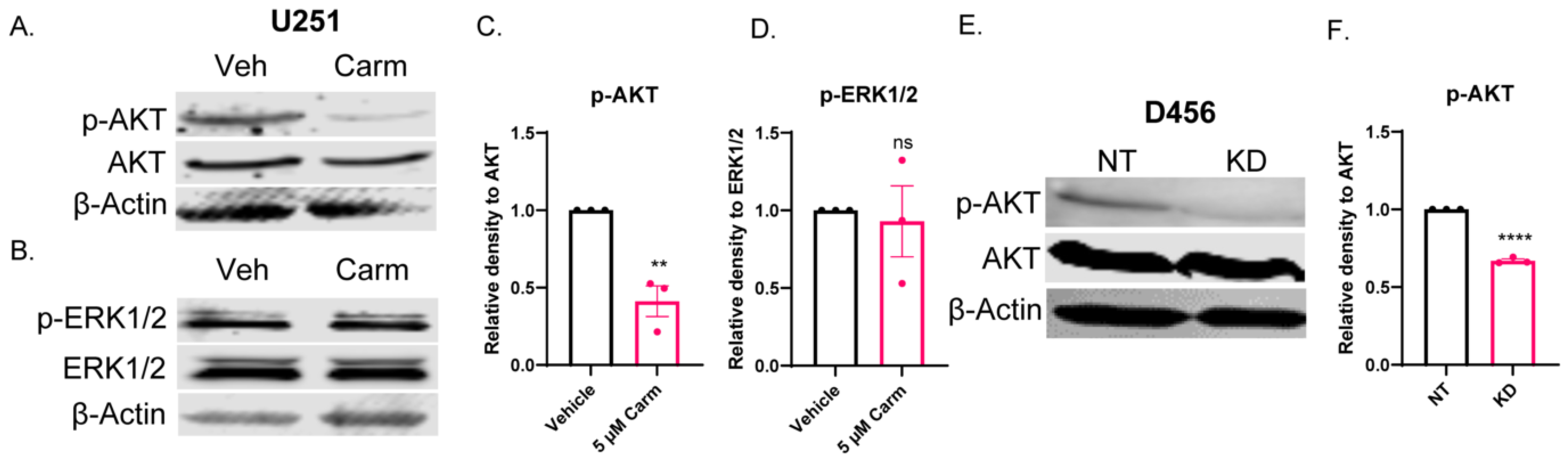
3.7. ASAH1 Inhibition Decreased pAKT, but Not pERK
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abuhusain, H.J.; Matin, A.; Qiao, Q.; Shen, H.; Kain, N.; Day, B.W.; Stringer, B.W.; Daniels, B.; Laaksonen, M.A.; Teo, C.; et al. A metabolic shift favoring sphingosine 1-phosphate at the expense of ceramide controls glioblastoma angiogenesis. J. Biol. Chem. 2013, 288, 37355–37364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, C.R.; Perry, A. Glioblastoma. Arch. Pathol. Lab. Med. 2007, 131, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Cloughesy, T.; Perry, J.R.; Wick, W. Standards of care for treatment of recurrent glioblastoma—Are we there yet? Neuro-Oncology 2013, 15, 4–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doan, N.B.; Nguyen, H.S.; Al-Gizawiy, M.M.; Mueller, W.M.; Sabbadini, R.A.; Rand, S.D.; Connelly, J.M.; Chitambar, C.R.; Schmainda, K.M.; Mirza, S.P. Acid ceramidase confers radioresistance to glioblastoma cells. Oncol. Rep. 2017, 38, 1932–1940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, C.C.; Ali, T.; Ramanadham, S.; Hjelmeland, A.B. Sphingolipid Metabolism in Glioblastoma and Metastatic Brain Tumors: A Review of Sphingomyelinases and Sphingosine-1-Phosphate. Biomolecules 2020, 10, 1357. [Google Scholar] [CrossRef]
- Van Brocklyn, J.R.; Young, N.; Roof, R. Sphingosine-1-phosphate stimulates motility and invasiveness of human glioblastoma multiforme cells. Cancer Lett. 2003, 199, 53–60. [Google Scholar] [CrossRef]
- Tea, M.N.; Poonnoose, S.I.; Pitson, S.M. Targeting the Sphingolipid System as a Therapeutic Direction for Glioblastoma. Cancers 2020, 12, 111. [Google Scholar] [CrossRef] [Green Version]
- Doan, N.B.; Alhajala, H.; Al-Gizawiy, M.M.; Mueller, W.M.; Rand, S.D.; Connelly, J.M.; Cochran, E.J.; Chitambar, C.R.; Clark, P.; Kuo, J.; et al. Acid ceramidase and its inhibitors: A de novo drug target and a new class of drugs for killing glioblastoma cancer stem cells with high efficiency. Oncotarget 2017, 8, 112662–112674. [Google Scholar] [CrossRef] [Green Version]
- Realini, N.; Solorzano, C.; Pagliuca, C.; Pizzirani, D.; Armirotti, A.; Luciani, R.; Costi, M.P.; Bandiera, T.; Piomelli, D. Discovery of highly potent acid ceramidase inhibitors with in vitro tumor chemosensitizing activity. Sci. Rep. 2013, 3, 1035. [Google Scholar] [CrossRef]
- Watanabe, M.; Kodaira, S.; Takahashi, T.; Tominaga, T.; Hojo, K.; Kato, T.; Kunitomo, K.; Isomoto, H.; Ohashi, Y.; Yasutomi, M. Randomized trial of the efficacy of adjuvant chemotherapy for colon cancer with combination therapy incorporating the oral pyrimidine 1-hexylcarbamoyl-5-fluorouracil. Langenbeck’s Arch. Surg. 2006, 391, 330–337. [Google Scholar] [CrossRef]
- Ito, K.; Yamaguchi, A.; Miura, K.; Kato, T.; Baba, S.; Matsumoto, S.; Ishii, M.; Takagi, H. Effect of oral adjuvant therapy with Carmofur (HCFU) for distant metastasis of colorectal cancer. Int. J. Clin. Oncol. 2000, 5, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, J.; Hamada, C.; Rahman, M.; Kodaira, S.; Ito, K.; Nakazato, H.; Ohashi, Y.; Yasutomi, M. An Individual Patient Data Meta-analysis of Adjuvant Therapy with Carmofur in Patients with Curatively Resected Colon Cancer. Jpn. J. Clin. Oncol. 2005, 35, 536–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dementiev, A.; Joachimiak, A.; Nguyen, H.; Gorelik, A.; Illes, K.; Shabani, S.; Gelsomino, M.; Ahn, E.-Y.E.; Nagar, B.; Doan, N. Molecular Mechanism of Inhibition of Acid Ceramidase by Carmofur. J. Med. Chem. 2019, 62, 987–992. [Google Scholar] [CrossRef] [PubMed]
- Mencarelli, C.; Martinez-Martinez, P. Ceramide function in the brain: When a slight tilt is enough. Cell. Mol. Life Sci. 2013, 70, 181–203. [Google Scholar] [CrossRef] [Green Version]
- Chalfant, C.E.; Kishikawa, K.; Mumby, M.C.; Kamibayashi, C.; Bielawska, A.; Hannun, Y.A. Long chain ceramides activate protein phosphatase-1 and protein phosphatase-2A: Activation is stereospecific and regulated by phosphatidic acid. J. Biol. Chem. 1999, 274, 20313–20317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, L.; Wu, Q.; Guryanova, O.A.; Huang, Z.; Huang, Q.; Rich, J.N.; Bao, S. Elevated invasive potential of glioblastoma stem cells. Biochem. Biophys. Res. Commun. 2011, 406, 643–648. [Google Scholar] [CrossRef] [Green Version]
- Eyler, C.E.; Foo, W.C.; LaFiura, K.M.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. Brain cancer stem cells display preferential sensitivity to Akt inhibition. Stem Cells 2008, 26, 3027–3036. [Google Scholar] [CrossRef] [Green Version]
- Lai, M.; Realini, N.; La Ferla, M.; Passalacqua, I.; Matteoli, G.; Ganesan, A.; Pistello, M.; Mazzanti, C.M.; Piomelli, D. Complete Acid Ceramidase ablation prevents cancer-initiating cell formation in melanoma cells. Sci. Rep. 2017, 7, 7411. [Google Scholar] [CrossRef]
- Boyd, N.H.; Walker, K.; Fried, J.; Hackney, J.R.; McDonald, P.C.; Benavides, G.A.; Spina, R.; Audia, A.; Scott, S.E.; Libby, C.J.; et al. Addition of carbonic anhydrase 9 inhibitor SLC-0111 to temozolomide treatment delays glioblastoma growth in vivo. JCI Insight 2017, 2, e92928. [Google Scholar] [CrossRef]
- Carlson, B.L.; Pokorny, J.L.; Schroeder, M.A.; Sarkaria, J.N. Establishment, maintenance and in vitro, and in vivo applications of primary human glioblastoma multiforme (GBM) xenograft models for translational biology studies and drug discovery. Curr. Protoc. Pharmacol. 2011, 52, 14–16. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, S.; Verreault, M.; Schmitt, C.; Guegan, J.; Guehennec, J.; Levasseur, C.; Marie, Y.; Bielle, F.; Mokhtari, K.; Hoang-Xuan, K.; et al. Multi-omics analysis of primary glioblastoma cell lines shows recapitulation of pivotal molecular features of parental tumors. Neuro-Oncology 2017, 19, 219–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and Characterization of Tumorigenic, Stem-like Neural Precursors from Human Glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aum, D.J.; Kim, D.H.; Beaumont, T.L.; Leuthardt, E.C.; Dunn, G.P.; Kim, A.H. Molecular and cellular heterogeneity: The hallmark of glioblastoma. Neurosurg. Focus 2014, 37, E11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, K.; Hjelmeland, A. Method for Efficient Transduction of Cancer Stem Cells. J. Cancer Stem Cell Res. 2014, 2, e1008. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Krämer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Behnan, J.; Finocchiaro, G.; Hanna, G. The landscape of the mesenchymal signature in brain tumours. Brain 2019, 142, 847–866. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Weigert, A.; Olesch, C.; Brüne, B. Sphingosine-1-Phosphate and Macrophage Biology-How the Sphinx Tames the Big Eater. Front. Immunol. 2019, 10, 1706. [Google Scholar] [CrossRef] [PubMed]
- Pappu, R.; Schwab, S.R.; Cornelissen, I.; Pereira, J.P.; Regard, J.B.; Xu, Y.; Camerer, E.; Zheng, Y.W.; Huang, Y.; Cyster, J.G.; et al. Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science 2007, 316, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Bourbon, N.A.; Sandirasegarane, L.; Kester, M. Ceramide-induced inhibition of Akt is mediated through protein kinase Czeta: Implications for growth arrest. J. Biol. Chem. 2002, 277, 3286–3292. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, N.; Patel, N.; Jiang, K.; Watson, J.E.; Cheng, J.; Chalfant, C.E.; Cooper, D.R. Ceramide-Activated Protein Phosphatase Involvement in Insulin Resistance via Akt, Serine/Arginine-Rich Protein 40, and Ribonucleic Acid Splicing in L6 Skeletal Muscle Cells. Endocrinology 2007, 148, 1359–1366. [Google Scholar] [CrossRef] [PubMed]
- Libby, C.J.; Gc, S.; Benavides, G.A.; Fisher, J.L.; Williford, S.E.; Zhang, S.; Tran, A.N.; Gordon, E.R.; Jones, A.B.; Tuy, K.; et al. A role for GLUT3 in glioblastoma cell invasion that is not recapitulated by GLUT1. Cell Adhes. Migr. 2021, 15, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Dolde, X.; Karreman, C.; Wiechers, M.; Schildknecht, S.; Leist, M. Profiling of Human Neural Crest Chemoattractant Activity as a Replacement of Fetal Bovine Serum for In Vitro Chemotaxis Assays. Int. J. Mol. Sci. 2021, 22, 10079. [Google Scholar] [CrossRef]
- Zhao, J.; Jiang, Y.; Chen, L.; Ma, Y.; Zhang, H.; Zhou, J.; Li, H.; Jing, Z. The EIF4A3/CASC2/RORA Feedback Loop Regulates the Aggressive Phenotype in Glioblastomas. Front. Oncol. 2021, 11, 3025. [Google Scholar] [CrossRef]
- Yang, Y.; Dodbele, S.; Park, T.; Glass, R.; Bhat, K.; Sulman, E.P.; Zhang, Y.; Abounader, R. MicroRNA-29a inhibits glioblastoma stem cells and tumor growth by regulating the PDGF pathway. J. Neuro-Oncol. 2019, 145, 23–34. [Google Scholar] [CrossRef]
- Hernández-Vega, A.M.; Del Moral-Morales, A.; Zamora-Sánchez, C.J.; Piña-Medina, A.G.; González-Arenas, A.; Camacho-Arroyo, I. Estradiol Induces Epithelial to Mesenchymal Transition of Human Glioblastoma Cells. Cells 2020, 9, 1930. [Google Scholar] [CrossRef]
- Roos, A.; Dhruv, H.D.; Peng, S.; Inge, L.J.; Tuncali, S.; Pineda, M.; Millard, N.; Mayo, Z.; Eschbacher, J.M.; Loftus, J.C.; et al. EGFRvIII—Stat5 Signaling Enhances Glioblastoma Cell Migration and Survival. Mol. Cancer Res. 2018, 16, 1185–1195. [Google Scholar] [CrossRef] [Green Version]
- Kusama, M.; Tominaga, T.; Enomoto, K.; Yoshida, M.; Koyama, H.; Sonoo, H.; Takashima, S.; Abe, R.; Nishi, T.; Yamaguchi, S.; et al. Clinical effects of carmofur (Mifurol) on advanced and recurrent breast cancer in a cooperative study. Research association for re-evaluation of direct effects of Mifurol on breast cancer. Gan Kagaku Ryoho. Cancer Chem. 1995, 22, 467–475. [Google Scholar]
- Bai, A.; Szulc, Z.M.; Bielawski, J.; Pierce, J.S.; Rembiesa, B.; Terzieva, S.; Mao, C.; Xu, R.; Wu, B.; Clarke, C.J.; et al. Targeting (cellular) lysosomal acid ceramidase by B13: Design, synthesis and evaluation of novel DMG-B13 ester prodrugs. Bioorg. Med. Chem. 2014, 22, 6933–6944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puchalski, R.B.; Shah, N.; Miller, J.; Dalley, R.; Nomura, S.R.; Yoon, J.G.; Smith, K.A.; Lankerovich, M.; Bertagnolli, D.; Bickley, K.; et al. An anatomic transcriptional atlas of human glioblastoma. Science 2018, 360, 660–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLean, G.W.; Carragher, N.O.; Avizienyte, E.; Evans, J.; Brunton, V.G.; Frame, M.C. The role of focal-adhesion kinase in cancer —A new therapeutic opportunity. Nat. Rev. Cancer 2005, 5, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Machado, J.; Merlo, A. Loss of Focal Adhesion Kinase (FAK) Inhibits Epidermal Growth Factor Receptor-dependent Migration and Induces Aggregation of NH2-Terminal FAK in the Nuclei of Apoptotic Glioblastoma Cells. Cancer Res. 2001, 61, 4978–4981. [Google Scholar] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Chin, Y.R.; Toker, A. Function of Akt/PKB signaling to cell motility, invasion and the tumor stroma in cancer. Cell Signal. 2009, 21, 470–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, G.; Hemmings, B.A. PKB/Akt-Dependent Regulation of Cell Motility. JNCI J. Natl. Cancer Inst. 2013, 105, 393–404. [Google Scholar] [CrossRef] [Green Version]
- Enomoto, A.; Murakami, H.; Asai, N.; Morone, N.; Watanabe, T.; Kawai, K.; Murakumo, Y.; Usukura, J.; Kaibuchi, K.; Takahashi, M. Akt/PKB regulates actin organization and cell motility via Girdin/APE. Dev. Cell 2005, 9, 389–402. [Google Scholar] [CrossRef] [Green Version]
- Ho, Y.P.; Kuo, C.W.; Hsu, Y.T.; Huang, Y.S.; Yew, L.P.; Huang, W.F.; Lin, K.C.; Hsu, J.H. β-Actin is a downstream effector of the PI3K/AKT signaling pathway in myeloma cells. Mol. Cell. Biochem. 2011, 348, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Dohoon, K.; Sunhong, K.; Hyongjong, K.; Sang-Oh, Y.; An-Sik, C.; Sang, C.K.; JongKyeong, C. Akt/PKB promotes cancer cell invasion via increased motility and metalloproteinase production. FASEB J. 2001, 15, 1953–1962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohn, A.D.; Takeuchi, F.; Roth, R.A. Akt, a pleckstrin homology domain containing kinase, is activated primarily by phosphorylation. J. Biol. Chem. 1996, 271, 21920–21926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinci, M.; Box, C.; Zimmermann, M.; Eccles, S.A. Tumor spheroid-based migration assays for evaluation of therapeutic agents. In Target Identification and Validation in Drug Discovery; Humana Press: Totowa, NJ, USA, 2013; Volume 986, pp. 253–266. [Google Scholar] [CrossRef]
- Li, X.; Wu, C.; Chen, N.; Gu, H.; Yen, A.; Cao, L.; Wang, E.; Wang, L. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget 2016, 7, 22440–33450. [Google Scholar] [CrossRef] [Green Version]
- Kaley, T.J.; Panageas, K.S.; Mellinghoff, I.K.; Nolan, C.; Gavrilovic, I.T.; DeAngelis, L.M.; Abrey, L.E.; Holland, E.C.; Lassman, A.B. Phase II trial of an AKT inhibitor (perifosine) for recurrent glioblastoma. J. Neuro-Oncol. 2019, 144, 403–407. [Google Scholar] [CrossRef]
- Vethakanraj, H.S.; Sesurajan, B.P.; Padmanaban, V.P.; Jayaprakasam, M.; Murali, S.; Sekar, A.K. Anticancer effect of acid ceramidase inhibitor ceranib-2 in human breast cancer cell lines MCF-7, MDA MB-231 by the activation of SAPK/JNK, p38 MAPK apoptotic pathways, inhibition of the Akt pathway, downregulation of ERα. Anti-Cancer Drugs 2018, 29, 50–60. [Google Scholar] [CrossRef]
- Mazhar, S.; Taylor, S.E.; Sangodkar, J.; Narla, G. Targeting PP2A in cancer: Combination therapies. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 51–63. [Google Scholar] [CrossRef]
- Leclerc, J.; Garandeau, D.; Pandiani, C.; Gaudel, C.; Bille, K.; Nottet, N.; Garcia, V.; Colosetti, P.; Pagnotta, S.; Bahadoran, P.; et al. Lysosomal acid ceramidase ASAH1 controls the transition between invasive and proliferative phenotype in melanoma cells. Oncogene 2019, 38, 1282–1295. [Google Scholar] [CrossRef]
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Elsamadicy, A.A.; Dechant, C.A.; Kemeny, H.R.; et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2018, 24, 1459–1468. [Google Scholar] [CrossRef]
- Newton, J.; Lima, S.; Maceyka, M.; Spiegel, S. Revisiting the sphingolipid rheostat: Evolving concepts in cancer therapy. Exp. Cell Res. 2015, 333, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Bektas, M.; Johnson, S.P.; Poe, W.E.; Bigner, D.D.; Friedman, H.S. A sphingosine kinase inhibitor induces cell death in temozolomide resistant glioblastoma cells. Cancer Chemother. Pharmacol. 2009, 64, 1053–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogretmen, B. Sphingolipid metabolism in cancer signalling and therapy. Nat. Rev. Cancer 2018, 18, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Sordillo, L.A.; Sordillo, P.P.; Helson, L. Sphingosine Kinase Inhibitors as Maintenance Therapy of Glioblastoma After Ceramide-Induced Response. Anti-Cancer Res. 2016, 36, 2085–2095. [Google Scholar]
- Miller, C.R.; Williams, C.R.; Buchsbaum, D.J.; Gillespie, G.Y. Intratumoral 5-Fluorouracil Produced by Cytosine Deaminase/5-Fluorocytosine Gene Therapy Is Effective for Experimental Human Glioblastomas. Cancer Res. 2002, 62, 773. [Google Scholar]
- White-Gilbertson, S.; Lu, P.; Jones, C.M.; Chiodini, S.; Hurley, D.; Das, A.; Delaney, J.R.; Norris, J.S.; Voelkel-Johnson, C. Tamoxifen is a candidate first-in-class inhibitor of acid ceramidase that reduces amitotic division in polyploid giant cancer cells—Unrecognized players in tumorigenesis. Cancer Med. 2020, 9, 3142–3152. [Google Scholar] [CrossRef] [Green Version]
- Graf, A.H.; Buchberger, W.; Langmayr, H.; Schmid, K.W. Site preference of metastatic tumours of the brain. Virchows Arch. A 1988, 412, 493–498. [Google Scholar] [CrossRef]
- Nussbaum, E.S.; Djalilian, H.R.; Cho, K.H.; Hall, W.A. Brain metastases. Histology, multiplicity, surgery, and survival. Cancer 1996, 78, 1781–1788. [Google Scholar] [CrossRef]
- Wanleenuwat, P.; Iwanowski, P. Metastases to the central nervous system: Molecular basis and clinical considerations. J. Neurol. Sci. 2020, 412, 116755. [Google Scholar] [CrossRef]
- Zakrzewski, J.; Geraghty, L.N.; Rose, A.E.; Christos, P.J.; Mazumdar, M.; Polsky, D.; Shapiro, R.; Berman, R.; Darvishian, F.; Hernando, E.; et al. Clinical variables and primary tumor characteristics predictive of the development of melanoma brain metastases and post-brain metastases survival. Cancer 2011, 117, 1711–1720. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Set | Gene Set | ES | NES | p Value | FDR |
|---|---|---|---|---|---|
| Gravandeel | Focal Adhesion-PI3K-Akt-mTOR-signaling pathway | 0.2472 | 2.0613 | 0.00351 | 0.03479 |
| TCGA GBM HG-U133A | 0.1867 | 0.9670 | 0.49793 | 0.48293 | |
| TCGA GBM Agilent-4502A | 0.2265 | 1.3467 | 0.14067 | 0.32006 |
| Gene Set | Description | Size | Leading Edge Number | ES | NES | p Value | FDR | DEGs |
|---|---|---|---|---|---|---|---|---|
| WP382 | MAPK Signaling Pathway | 4 | 4 | −0.5072 | −1.1697 | 0.2591 | 1 | MAPK12, PLA2G4A, PDGFB, DUSP7 |
| WP4239 | Epithelial to mesenchymal transition in colorectal cancer | 3 | 2 | −0.4992 | −0.9973 | 0.4734 | 1 | COL4A3, MAPK12 |
| WP3932 | Focal Adhesion-PI3K-Akt-mTOR-signaling pathway | 5 | 5 | −0.3613 | −0.9129 | 0.5568 | 0.9598 | ITGA7, ANGPT1, SREBF1, GNG10, PDGFB |
| WP3929 | Chemokine signaling pathway | 3 | 3 | −0.3822 | −0.7803 | 0.7407 | 0.9453 | CCL26, GRK5, GNG10 |
| WP4223 | Ras Signaling | 3 | 3 | −0.3362 | −0.6739 | 0.9022 | 0.876 | KSR1, PLA2G4A, GNG10 |
| WP4018 | Pathways in clear cell renal cell carcinoma | 4 | 4 | 0.3862 | 0.8617 | 0.6248 | 0.6455 | KSR1, PDGFB, PSAT1, PHGDH |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hawkins, C.C.; Jones, A.B.; Gordon, E.R.; Williford, S.E.; Harsh, Y.; Ziebro, J.K.; Landis, C.J.; Gc, S.; Crossman, D.K.; Cooper, S.J.; et al. Targeting Acid Ceramidase Inhibits Glioblastoma Cell Migration through Decreased AKT Signaling. Cells 2022, 11, 1873. https://doi.org/10.3390/cells11121873
Hawkins CC, Jones AB, Gordon ER, Williford SE, Harsh Y, Ziebro JK, Landis CJ, Gc S, Crossman DK, Cooper SJ, et al. Targeting Acid Ceramidase Inhibits Glioblastoma Cell Migration through Decreased AKT Signaling. Cells. 2022; 11(12):1873. https://doi.org/10.3390/cells11121873
Chicago/Turabian StyleHawkins, Cyntanna C., Amber B. Jones, Emily R. Gordon, Sarah E. Williford, Yuvika Harsh, Julia K. Ziebro, Catherine J. Landis, Sajina Gc, David K. Crossman, Sara J. Cooper, and et al. 2022. "Targeting Acid Ceramidase Inhibits Glioblastoma Cell Migration through Decreased AKT Signaling" Cells 11, no. 12: 1873. https://doi.org/10.3390/cells11121873
APA StyleHawkins, C. C., Jones, A. B., Gordon, E. R., Williford, S. E., Harsh, Y., Ziebro, J. K., Landis, C. J., Gc, S., Crossman, D. K., Cooper, S. J., Ramanadham, S., Doan, N., & Hjelmeland, A. B. (2022). Targeting Acid Ceramidase Inhibits Glioblastoma Cell Migration through Decreased AKT Signaling. Cells, 11(12), 1873. https://doi.org/10.3390/cells11121873