
Immune Checkpoint Proteins, Metabolism and Adhesion Molecules: Overlooked Determinants of CAR T-Cell Migration?


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
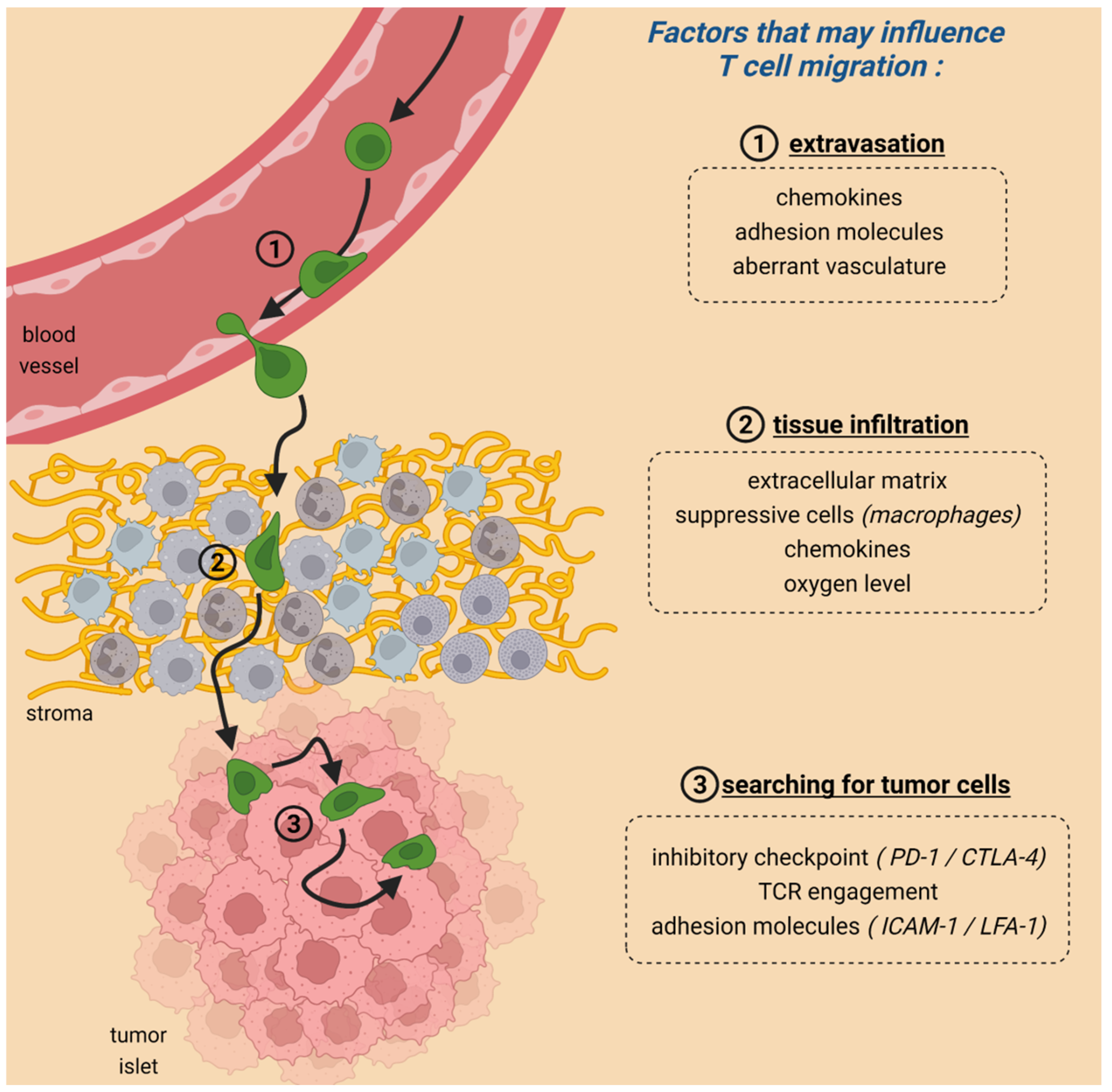
2. Migration Is Crucial for CAR T-Cell Anti-Tumoral Activities
3. The Role of Immune Checkpoint Proteins in T-Cell Migration within Tumors
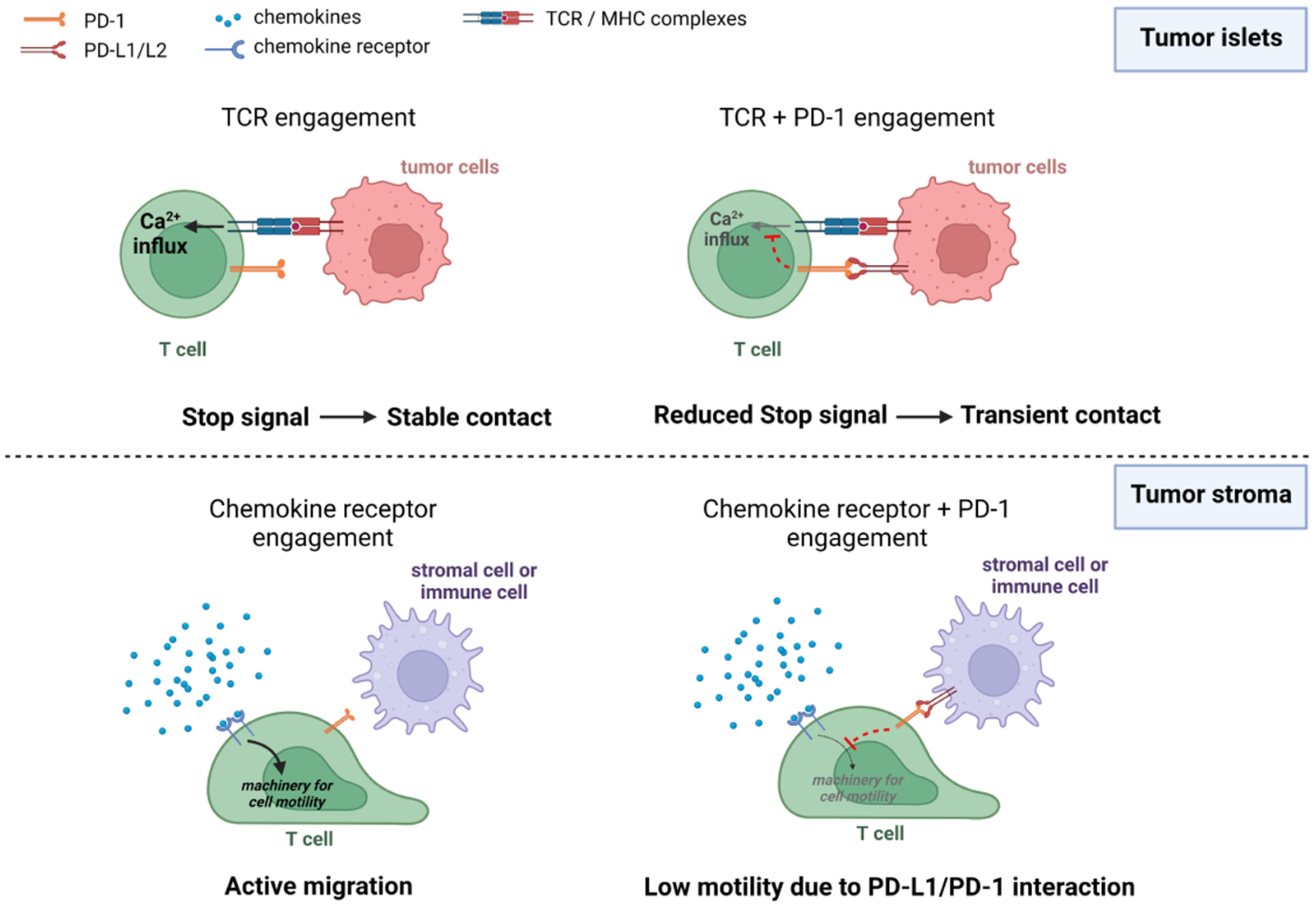
3.1. CTLA-4 and PD-1 Interfere with the Ability of T Cells to Form Stable Conjugates with Their Targets
3.2. Blocking CTLA-4 and PD-1 Produces Variable Effects on T-Cell Motility in Tumors
4. Metabolism and T-Cell Migration within Tumors
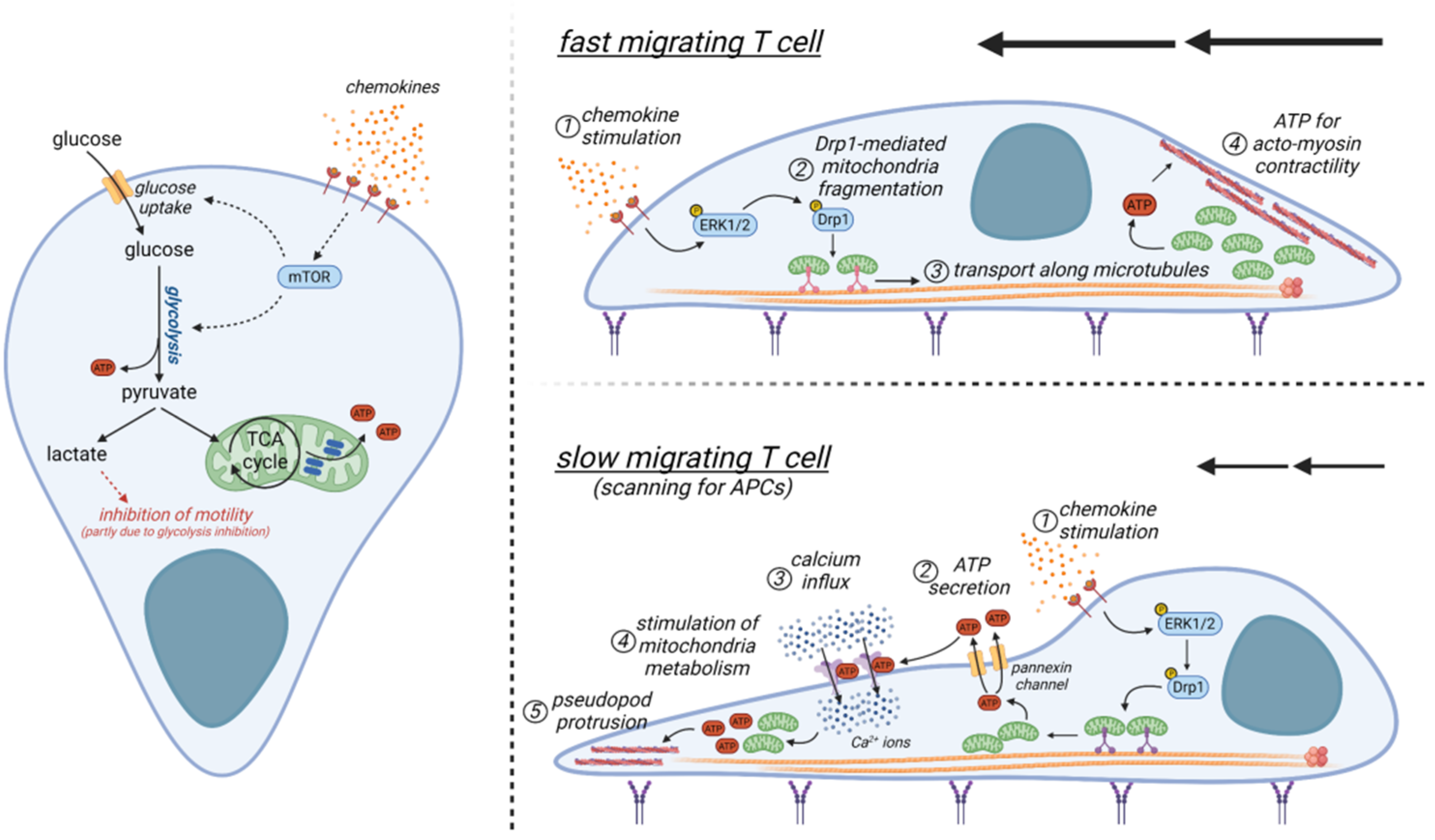
4.1. A Brief Overview of T-Cell Metabolism
4.2. Metabolic Determinants Controlling the Motility of T Cells and Comparison with Those of Other Cells
4.3. Metabolic Alterations within the Tumor Microenvironment and Their Impact on T-Cell Motility
5. Adhesion Molecules Controlling the Contact between CAR T Cells and Their Targets
6. Restoring Defective CAR T Cell Migration by Targeting Immune Checkpoint Proteins, Metabolic Factors, and Adhesion Molecules
6.1. Promoting CAR T-Cell Interaction with Tumor Cells via Targeting of Adhesion Molecules
6.1.1. Increasing ICAM-1 Expression by Cancer Cells
6.1.2. Increasing Integrin Activity and Expression on CAR T Cells
6.2. Promoting CAR T-Cell Migration in Tumors via Targeting of Metabolism
6.3. Promoting CAR T-Cell Migration in Tumors via Targeting Immune Checkpoint Proteins
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Milone, M.C.; Xu, J.; Chen, S.-J.; Collins, M.A.; Zhou, J.; Powell, D.J., Jr.; Melenhorst, J.J. Engineering enhanced CAR T-cells for improved cancer therapy. Nat. Cancer 2021, 2, 780–793. [Google Scholar] [CrossRef]
- Guedan, S.; Ruella, M.; June, C.H. Emerging Cellular Therapies for Cancer. Annu. Rev. Immunol. 2019, 37, 145–171. [Google Scholar] [CrossRef]
- Lesch, S.; Benmebarek, M.-R.; Cadilha, B.L.; Stoiber, S.; Subklewe, M.; Endres, S.; Kobold, S. Determinants of response and resistance to CAR T cell therapy. Semin. Cancer Biol. 2020, 65, 80–90. [Google Scholar] [CrossRef] [PubMed]
- White, L.G.; Goy, H.E.; Rose, A.J.; McLellan, A.D. Controlling Cell Trafficking: Addressing Failures in CAR T and NK Cell Therapy of Solid Tumours. Cancers 2022, 14, 978. [Google Scholar] [CrossRef] [PubMed]
- Donnadieu, E.; Dupre, L.; Pinho, L.G.; Cotta-de-Almeida, V. Surmounting the obstacles that impede effective CAR T cell trafficking to solid tumors. J. Leukoc. Biol. 2020, 108, 1067–1079. [Google Scholar] [CrossRef] [PubMed]
- Nourshargh, S.; Alon, R. Leukocyte migration into inflamed tissues. Immunity 2014, 41, 694–707. [Google Scholar] [CrossRef]
- Lanitis, E.; Dangaj, D.; Irving, M.; Coukos, G. Mechanisms regulating T-cell infiltration and activity in solid tumors. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, xii18–xii32. [Google Scholar] [CrossRef]
- Bernhard, H.; Neudorfer, J.; Gebhard, K.; Conrad, H.; Hermann, C.; Nährig, J.; Fend, F.; Weber, W.; Busch, D.H.; Peschel, C. Adoptive transfer of autologous, HER2-specific, cytotoxic T lymphocytes for the treatment of HER2-overexpressing breast cancer. Cancer Immunol. Immunother. CII 2008, 57, 271–280. [Google Scholar] [CrossRef]
- Majzner, R.G.; Ramakrishna, S.; Yeom, K.W.; Patel, S.; Chinnasamy, H.; Schultz, L.M.; Richards, R.M.; Jiang, L.; Barsan, V.; Mancusi, R.; et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature 2022, 603, 934–941. [Google Scholar] [CrossRef]
- Adusumilli, P.S.; Cherkassky, L.; Villena-Vargas, J.; Colovos, C.; Servais, E.; Plotkin, J.; Jones, D.R.; Sadelain, M. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci. Transl. Med. 2014, 6, 261ra151. [Google Scholar] [CrossRef]
- Dirkx, A.E.; Oude Egbrink, M.G.; Kuijpers, M.J.; van der Niet, S.T.; Heijnen, V.V.; Bouma-ter Steege, J.C.; Wagstaff, J.; Griffioen, A.W. Tumor angiogenesis modulates leukocyte-vessel wall interactions in vivo by reducing endothelial adhesion molecule expression. Cancer Res. 2003, 63, 2322–2329. [Google Scholar] [PubMed]
- Lesch, S.; Blumenberg, V.; Stoiber, S.; Gottschlich, A.; Ogonek, J.; Cadilha, B.L.; Dantes, Z.; Rataj, F.; Dorman, K.; Lutz, J.; et al. T cells armed with C-X-C chemokine receptor type 6 enhance adoptive cell therapy for pancreatic tumours. Nat. Biomed. Eng. 2021, 5, 1246–1260. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef]
- Salmon, H.; Franciszkiewicz, K.; Damotte, D.; Dieu-Nosjean, M.C.; Validire, P.; Trautmann, A.; Mami-Chouaib, F.; Donnadieu, E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Investig. 2012, 122, 899–910. [Google Scholar] [CrossRef]
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guerin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Powell, D.; Tauzin, S.; Hind, L.E.; Deng, Q.; Beebe, D.J.; Huttenlocher, A. Chemokine Signaling and the Regulation of Bidirectional Leukocyte Migration in Interstitial Tissues. Cell Rep. 2017, 19, 1572–1585. [Google Scholar] [CrossRef]
- Wang, Z.; Moresco, P.; Yan, R.; Li, J.; Gao, Y.; Biasci, D.; Yao, M.; Pearson, J.; Hechtman, J.F.; Janowitz, T.; et al. Carcinomas assemble a filamentous CXCL12-keratin-19 coating that suppresses T cell-mediated immune attack. Proc. Natl. Acad. Sci. USA 2022, 119, e2119463119. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef]
- Guruprasad, P.; Lee, Y.G.; Kim, K.H.; Ruella, M. The current landscape of single-cell transcriptomics for cancer immunotherapy. J. Exp. Med. 2021, 218, e2119463119. [Google Scholar] [CrossRef]
- Kishton, R.J.; Vodnala, S.K.; Vizcardo, R.; Restifo, N.P. Next generation immunotherapy: Enhancing stemness of polyclonal T cells to improve anti-tumor activity. Curr. Opin. Immunol. 2021, 74, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; Han, G.; Puebla-Osorio, N.; Ma, M.C.J.; Strati, P.; Chasen, B.; Dai, E.; Dang, M.; Jain, N.; Yang, H.; et al. Characteristics of anti-CD19 CAR T cell infusion products associated with efficacy and toxicity in patients with large B cell lymphomas. Nat. Med. 2020, 26, 1878–1887. [Google Scholar] [CrossRef] [PubMed]
- Mami-Chouaib, F.; Blanc, C.; Corgnac, S.; Hans, S.; Malenica, I.; Granier, C.; Tihy, I.; Tartour, E. Resident memory T cells, critical components in tumor immunology. J. Immunother. Cancer 2018, 6, 87. [Google Scholar] [CrossRef]
- Jansen, C.S.; Prokhnevska, N.; Master, V.A.; Sanda, M.G.; Carlisle, J.W.; Bilen, M.A.; Cardenas, M.; Wilkinson, S.; Lake, R.; Sowalsky, A.G.; et al. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature 2019, 576, 465–470. [Google Scholar] [CrossRef]
- Di Pilato, M.; Kfuri-Rubens, R.; Pruessmann, J.N.; Ozga, A.J.; Messemaker, M.; Cadilha, B.L.; Sivakumar, R.; Cianciaruso, C.; Warner, R.D.; Marangoni, F.; et al. CXCR6 positions cytotoxic T cells to receive critical survival signals in the tumor microenvironment. Cell 2021, 184, 4512–4530.e22. [Google Scholar] [CrossRef]
- Van Braeckel-Budimir, N.; Dolina, J.S.; Wei, J.; Wang, X.; Chen, S.H.; Santiago, P.; Tu, G.; Micci, L.; Al-Khami, A.A.; Pfister, S.; et al. Combinatorial immunotherapy induces tumor-infiltrating CD8+ T cells with distinct functional, migratory, and stem-like properties. J. Immunother. Cancer 2021, 9, e003614. [Google Scholar] [CrossRef]
- You, R.; Artichoker, J.; Fries, A.; Edwards, A.W.; Combes, A.J.; Reeder, G.C.; Samad, B.; Krummel, M.F. Active surveillance characterizes human intratumoral T cell exhaustion. J. Clin. Investig. 2021, 131, e144353. [Google Scholar] [CrossRef]
- Miller, B.C.; Sen, D.R.; Al Abosy, R.; Bi, K.; Virkud, Y.V.; LaFleur, M.W.; Yates, K.B.; Lako, A.; Felt, K.; Naik, G.S.; et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol. 2019, 20, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Bhakta, N.R.; Oh, D.Y.; Lewis, R.S. Calcium oscillations regulate thymocyte motility during positive selection in the three-dimensional thymic environment. Nat. Immunol. 2005, 6, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Donnadieu, E.; Bismuth, G.; Trautmann, A. Antigen recognition by helper T cells elicits a sequence of distinct changes of their shape and intracellular calcium. Curr. Biol. 1994, 4, 584–595. [Google Scholar] [CrossRef]
- Negulescu, P.A.; Krasieva, T.B.; Khan, A.; Kerschbaum, H.H.; Cahalan, M.D. Polarity of T cell shape, motility, and sensitivity to antigen. Immunity 1996, 4, 421–430. [Google Scholar] [CrossRef]
- Springer, T.A.; Dustin, M.L. Integrin inside-out signaling and the immunological synapse. Curr. Opin. Cell Biol. 2012, 24, 107–115. [Google Scholar] [CrossRef]
- Deguine, J.; Breart, B.; Lemaitre, F.; Di Santo, J.P.; Bousso, P. Intravital imaging reveals distinct dynamics for natural killer and CD8+ T cells during tumor regression. Immunity 2010, 33, 632–644. [Google Scholar] [CrossRef]
- Davenport, A.J.; Cross, R.S.; Watson, K.A.; Liao, Y.; Shi, W.; Prince, H.M.; Beavis, P.A.; Trapani, J.A.; Kershaw, M.H.; Ritchie, D.S.; et al. Chimeric antigen receptor T cells form nonclassical and potent immune synapses driving rapid cytotoxicity. Proc. Natl. Acad. Sci. USA 2018, 115, E2068–E2076. [Google Scholar] [CrossRef]
- Davenport, A.J.; Jenkins, M.R.; Cross, R.S.; Yong, C.S.; Prince, H.M.; Ritchie, D.S.; Trapani, J.A.; Kershaw, M.H.; Darcy, P.K.; Neeson, P.J. CAR-T Cells Inflict Sequential Killing of Multiple Tumor Target Cells. Cancer Immunol. Res. 2015, 3, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Wei, Q.; Brzostek, J.; Gascoigne, N.R.J. Signaling from T cell receptors (TCRs) and chimeric antigen receptors (CARs) on T cells. Cell Mol. Immunol. 2020, 17, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Fife, B.T.; Pauken, K.E.; Eagar, T.N.; Obu, T.; Wu, J.; Tang, Q.; Azuma, M.; Krummel, M.F.; Bluestone, J.A. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat. Immunol. 2009, 10, 1185–1192. [Google Scholar] [CrossRef]
- Schneider, H.; Downey, J.; Smith, A.; Zinselmeyer, B.H.; Rush, C.; Brewer, J.M.; Wei, B.; Hogg, N.; Garside, P.; Rudd, C.E. Reversal of the TCR stop signal by CTLA-4. Science 2006, 313, 1972–1975. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Yuan, Z.; Lyu, J.; Ahn, E.; Davis, S.J.; Ahmed, R.; Zhu, C. PD-1 suppresses TCR-CD8 cooperativity during T-cell antigen recognition. Nat. Commun. 2021, 12, 2746. [Google Scholar] [CrossRef]
- Brunner-Weinzierl, M.C.; Rudd, C.E. CTLA-4 and PD-1 Control of T-Cell Motility and Migration: Implications for Tumor Immunotherapy. Front. Immunol. 2018, 9, 2737. [Google Scholar] [CrossRef] [PubMed]
- Yokosuka, T.; Takamatsu, M.; Kobayashi-Imanishi, W.; Hashimoto-Tane, A.; Azuma, M.; Saito, T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med. 2012, 209, 1201–1217. [Google Scholar] [CrossRef]
- Honda, T.; Egen, J.G.; Lammermann, T.; Kastenmuller, W.; Torabi-Parizi, P.; Germain, R.N. Tuning of antigen sensitivity by T cell receptor-dependent negative feedback controls T cell effector function in inflamed tissues. Immunity 2014, 40, 235–247. [Google Scholar] [CrossRef]
- Ruocco, M.G.; Pilones, K.A.; Kawashima, N.; Cammer, M.; Huang, J.; Babb, J.S.; Liu, M.; Formenti, S.C.; Dustin, M.L.; Demaria, S. Suppressing T cell motility induced by anti-CTLA-4 monotherapy improves antitumor effects. J. Clin. Investig. 2012, 122, 3718–3730. [Google Scholar] [CrossRef]
- Lau, D.; Garcon, F.; Chandra, A.; Lechermann, L.M.; Aloj, L.; Chilvers, E.R.; Corrie, P.G.; Okkenhaug, K.; Gallagher, F.A. Intravital Imaging of Adoptive T-Cell Morphology, Mobility and Trafficking Following Immune Checkpoint Inhibition in a Mouse Melanoma Model. Front. Immunol. 2020, 11, 1514. [Google Scholar] [CrossRef] [PubMed]
- Pentcheva-Hoang, T.; Simpson, T.R.; Montalvo-Ortiz, W.; Allison, J.P. Cytotoxic T lymphocyte antigen-4 blockade enhances antitumor immunity by stimulating melanoma-specific T-cell motility. Cancer Immunol. Res. 2014, 2, 970–980. [Google Scholar] [CrossRef]
- Zinselmeyer, B.H.; Heydari, S.; Sacristan, C.; Nayak, D.; Cammer, M.; Herz, J.; Cheng, X.; Davis, S.J.; Dustin, M.L.; McGavern, D.B. PD-1 promotes immune exhaustion by inducing antiviral T cell motility paralysis. J. Exp. Med. 2013, 210, 757–774. [Google Scholar] [CrossRef] [PubMed]
- Dangaj, D.; Bruand, M.; Grimm, A.J.; Ronet, C.; Barras, D.; Duttagupta, P.A.; Lanitis, E.; Duraiswamy, J.; Tanyi, J.L.; Benencia, F.; et al. Cooperation between Constitutive and Inducible Chemokines Enables T Cell Engraftment and Immune Attack in Solid Tumors. Cancer Cell 2019, 35, 885–900.e10. [Google Scholar] [CrossRef]
- Mikucki, M.E.; Fisher, D.T.; Matsuzaki, J.; Skitzki, J.J.; Gaulin, N.B.; Muhitch, J.B.; Ku, A.W.; Frelinger, J.G.; Odunsi, K.; Gajewski, T.F.; et al. Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat. Commun. 2015, 6, 7458. [Google Scholar] [CrossRef] [PubMed]
- Kanda, Y.; Okazaki, T.; Katakai, T. Motility Dynamics of T Cells in Tumor-Draining Lymph Nodes: A Rational Indicator of Antitumor Response and Immune Checkpoint Blockade. Cancers 2021, 13, 4616. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.; Chen, J.; Gonzalez-Avalos, E.; Samaniego-Castruita, D.; Das, A.; Wang, Y.H.; Lopez-Moyado, I.F.; Georges, R.O.; Zhang, W.; Onodera, A.; et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8+ T cell exhaustion. Proc. Natl. Acad. Sci. USA 2019, 116, 12410–12415. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Park, S.; Park, S.Y.; Kim, G.; Park, S.M.; Cho, J.W.; Kim, D.H.; Park, Y.M.; Koh, Y.W.; Kim, H.R.; et al. Single-cell transcriptome analysis reveals TOX as a promoting factor for T cell exhaustion and a predictor for anti-PD-1 responses in human cancer. Genome Med. 2020, 12, 22. [Google Scholar] [CrossRef] [PubMed]
- Thommen, D.S.; Koelzer, V.H.; Herzig, P.; Roller, A.; Trefny, M.; Dimeloe, S.; Kiialainen, A.; Hanhart, J.; Schill, C.; Hess, C.; et al. A transcriptionally and functionally distinct PD-1+ CD8+ T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat. Med. 2018, 24, 994–1004. [Google Scholar] [CrossRef]
- Lindner, S.E.; Johnson, S.M.; Brown, C.E.; Wang, L.D. Chimeric antigen receptor signaling: Functional consequences and design implications. Sci. Adv. 2020, 6, eaaz3223. [Google Scholar] [CrossRef]
- Adusumilli, P.S.; Zauderer, M.G.; Riviere, I.; Solomon, S.B.; Rusch, V.W.; O’Cearbhaill, R.E.; Zhu, A.; Cheema, W.; Chintala, N.K.; Halton, E.; et al. A Phase I Trial of Regional Mesothelin-Targeted CAR T-cell Therapy in Patients with Malignant Pleural Disease, in Combination with the Anti-PD-1 Agent Pembrolizumab. Cancer Discov. 2021, 11, 2748–2763. [Google Scholar] [CrossRef]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 126, 3130–3144. [Google Scholar] [CrossRef]
- Almeida, L.; Lochner, M.; Berod, L.; Sparwasser, T. Metabolic pathways in T cell activation and lineage differentiation. Semin. Immunol. 2016, 28, 514–524. [Google Scholar] [CrossRef]
- Icard, P.; Alifano, M.; Donnadieu, E.; Simula, L. Fructose-1,6-bisphosphate promotes PI3K and glycolysis in T cells? Trends Endocrinol. Metab. 2021, 32, 540–543. [Google Scholar] [CrossRef] [PubMed]
- Tarasenko, T.N.; Pacheco, S.E.; Koenig, M.K.; Gomez-Rodriguez, J.; Kapnick, S.M.; Diaz, F.; Zerfas, P.M.; Barca, E.; Sudderth, J.; DeBerardinis, R.J.; et al. Cytochrome c Oxidase Activity Is a Metabolic Checkpoint that Regulates Cell Fate Decisions during T Cell Activation and Differentiation. Cell Metab. 2017, 25, 1254–1268.e7. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D.A.; Wang, C.R.; Schumacker, P.T.; Licht, J.D.; Perlman, H.; et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 2013, 38, 225–236. [Google Scholar] [CrossRef]
- Cluxton, D.; Petrasca, A.; Moran, B.; Fletcher, J.M. Differential Regulation of Human Treg and Th17 Cells by Fatty Acid Synthesis and Glycolysis. Front. Immunol. 2019, 10, 115. [Google Scholar] [CrossRef]
- Shin, B.; Benavides, G.A.; Geng, J.; Koralov, S.B.; Hu, H.; Darley-Usmar, V.M.; Harrington, L.E. Mitochondrial Oxidative Phosphorylation Regulates the Fate Decision between Pathogenic Th17 and Regulatory T Cells. Cell Rep. 2020, 30, 1898–1909.e4. [Google Scholar] [CrossRef]
- Chang, C.H.; Curtis, J.D.; Maggi, L.B., Jr.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.; van der Windt, G.J.; Blagih, J.; Qiu, J.; et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef]
- van der Windt, G.J.; Everts, B.; Chang, C.H.; Curtis, J.D.; Freitas, T.C.; Amiel, E.; Pearce, E.J.; Pearce, E.L. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 2012, 36, 68–78. [Google Scholar] [CrossRef]
- Bachem, A.; Makhlouf, C.; Binger, K.J.; de Souza, D.P.; Tull, D.; Hochheiser, K.; Whitney, P.G.; Fernandez-Ruiz, D.; Dahling, S.; Kastenmuller, W.; et al. Microbiota-Derived Short-Chain Fatty Acids Promote the Memory Potential of Antigen-Activated CD8+ T Cells. Immunity 2019, 51, 285–297.e5. [Google Scholar] [CrossRef]
- Bailis, W.; Shyer, J.A.; Zhao, J.; Canaveras, J.C.G.; Al Khazal, F.J.; Qu, R.; Steach, H.R.; Bielecki, P.; Khan, O.; Jackson, R.; et al. Distinct modes of mitochondrial metabolism uncouple T cell differentiation and function. Nature 2019, 571, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Norris, V.; Amar, P.; Legent, G.; Ripoll, C.; Thellier, M.; Ovadi, J. Sensor potency of the moonlighting enzyme-decorated cytoskeleton: The cytoskeleton as a metabolic sensor. BMC Biochem. 2013, 14, 3. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Burckhardt, C.J.; Lazcano, R.; Solis, L.M.; Isogai, T.; Li, L.; Chen, C.S.; Gao, B.; Minna, J.D.; Bachoo, R.; et al. Mechanical regulation of glycolysis via cytoskeleton architecture. Nature 2020, 578, 621–626. [Google Scholar] [CrossRef]
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquiere, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 2013, 154, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.C.; Ford, W.C. The role of glucose in supporting motility and capacitation in human spermatozoa. J. Androl. 2001, 22, 680–695. [Google Scholar]
- Tsujii, H.; Ohta, E.; Miah, A.G.; Hossain, S.; Salma, U. Effect of fructose on motility, acrosome reaction and in vitro fertilization capability of boar spermatozoa. Reprod. Med. Biol. 2006, 5, 255–261. [Google Scholar] [CrossRef]
- Toragall, M.M.; Satapathy, S.K.; Kadadevaru, G.G.; Hiremath, M.B. Evaluation of Seminal Fructose and Citric Acid Levels in Men with Fertility Problem. J. Hum. Reprod. Sci. 2019, 12, 199–203. [Google Scholar] [CrossRef]
- Nguyen, T.M.; Alves, S.; Grasseau, I.; Metayer-Coustard, S.; Praud, C.; Froment, P.; Blesbois, E. Central role of 5′-AMP-activated protein kinase in chicken sperm functions. Biol. Reprod. 2014, 91, 121. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.M.; Froment, P.; Combarnous, Y.; Blesbois, E. AMPK, regulator of sperm energy and functions. Med. Sci. 2016, 32, 491–496. [Google Scholar] [CrossRef][Green Version]
- Hurtado de Llera, A.; Martin-Hidalgo, D.; Gil, M.C.; Garcia-Marin, L.J.; Bragado, M.J. AMP-activated kinase AMPK is expressed in boar spermatozoa and regulates motility. PLoS ONE 2012, 7, e38840. [Google Scholar] [CrossRef]
- Yamada, K.M.; Sixt, M. Mechanisms of 3D cell migration. Nat. Rev. Mol. Cell Biol. 2019, 20, 738–752. [Google Scholar] [CrossRef]
- Reversat, A.; Gaertner, F.; Merrin, J.; Stopp, J.; Tasciyan, S.; Aguilera, J.; de Vries, I.; Hauschild, R.; Hons, M.; Piel, M.; et al. Cellular locomotion using environmental topography. Nature 2020, 582, 582–585. [Google Scholar] [CrossRef] [PubMed]
- Tabdanov, E.D.; Rodriguez-Merced, N.J.; Cartagena-Rivera, A.X.; Puram, V.V.; Callaway, M.K.; Ensminger, E.A.; Pomeroy, E.J.; Yamamoto, K.; Lahr, W.S.; Webber, B.R.; et al. Engineering T cells to enhance 3D migration through structurally and mechanically complex tumor microenvironments. Nat. Commun. 2021, 12, 2815. [Google Scholar] [CrossRef]
- Haas, R.; Smith, J.; Rocher-Ros, V.; Nadkarni, S.; Montero-Melendez, T.; D’Acquisto, F.; Bland, E.J.; Bombardieri, M.; Pitzalis, C.; Perretti, M.; et al. Lactate Regulates Metabolic and Pro-inflammatory Circuits in Control of T Cell Migration and Effector Functions. PLoS Biol. 2015, 13, e1002202. [Google Scholar] [CrossRef] [PubMed]
- Chan, O.; Burke, J.D.; Gao, D.F.; Fish, E.N. The chemokine CCL5 regulates glucose uptake and AMP kinase signaling in activated T cells to facilitate chemotaxis. J. Biol. Chem. 2012, 287, 29406–29416. [Google Scholar] [CrossRef]
- Simula, L.; Antonucci, Y.; Scarpelli, G.; Cancila, V.; Colamatteo, A.; Manni, S.; De Angelis, B.; Quintarelli, C.; Procaccini, C.; Matarese, G.; et al. PD-1-induced T cell exhaustion is controlled by a Drp1-dependent mechanism. Mol. Oncol. 2022, 16, 188–205. [Google Scholar] [CrossRef] [PubMed]
- Simula, L.; Pacella, I.; Colamatteo, A.; Procaccini, C.; Cancila, V.; Bordi, M.; Tregnago, C.; Corrado, M.; Pigazzi, M.; Barnaba, V.; et al. Drp1 Controls Effective T Cell Immune-Surveillance by Regulating T Cell Migration, Proliferation, and cMyc-Dependent Metabolic Reprogramming. Cell Rep. 2018, 25, 3059–3073.e10. [Google Scholar] [CrossRef]
- Marko, A.J.; Miller, R.A.; Kelman, A.; Frauwirth, K.A. Induction of glucose metabolism in stimulated T lymphocytes is regulated by mitogen-activated protein kinase signaling. PLoS ONE 2010, 5, e15425. [Google Scholar] [CrossRef]
- Kishore, M.; Cheung, K.C.P.; Fu, H.; Bonacina, F.; Wang, G.; Coe, D.; Ward, E.J.; Colamatteo, A.; Jangani, M.; Baragetti, A.; et al. Regulatory T Cell Migration Is Dependent on Glucokinase-Mediated Glycolysis. Immunity 2017, 47, 875–889.e10. [Google Scholar] [CrossRef]
- Campello, S.; Lacalle, R.A.; Bettella, M.; Manes, S.; Scorrano, L.; Viola, A. Orchestration of lymphocyte chemotaxis by mitochondrial dynamics. J. Exp. Med. 2006, 203, 2879–2886. [Google Scholar] [CrossRef]
- Ledderose, C.; Liu, K.; Kondo, Y.; Slubowski, C.J.; Dertnig, T.; Denicolo, S.; Arbab, M.; Hubner, J.; Konrad, K.; Fakhari, M.; et al. Purinergic P2X4 receptors and mitochondrial ATP production regulate T cell migration. J. Clin. Investig. 2018, 128, 3583–3594. [Google Scholar] [CrossRef]
- Ledderose, C.; Bromberger, S.; Slubowski, C.J.; Sueyoshi, K.; Aytan, D.; Shen, Y.; Junger, W.G. The purinergic receptor P2Y11 choreographs the polarization, mitochondrial metabolism, and migration of T lymphocytes. Sci. Signal. 2020, 13, eaba3300. [Google Scholar] [CrossRef] [PubMed]
- Bader, J.E.; Voss, K.; Rathmell, J.C. Targeting Metabolism to Improve the Tumor Microenvironment for Cancer Immunotherapy. Mol. Cell 2020, 78, 1019–1033. [Google Scholar] [CrossRef]
- van Baren, N.; Van den Eynde, B.J. Tumoral Immune Resistance Mediated by Enzymes That Degrade Tryptophan. Cancer Immunol. Res. 2015, 3, 978–985. [Google Scholar] [CrossRef]
- de la Cruz-Lopez, K.G.; Castro-Munoz, L.J.; Reyes-Hernandez, D.O.; Garcia-Carranca, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.R.; Rathmell, W.K.; Rathmell, J.C. The tumor microenvironment as a metabolic barrier to effector T cells and immunotherapy. eLife 2020, 9, e55185. [Google Scholar] [CrossRef]
- Sugiura, A.; Rathmell, J.C. Metabolic Barriers to T Cell Function in Tumors. J. Immunol. 2018, 200, 400–407. [Google Scholar] [CrossRef]
- Gemta, L.F.; Siska, P.J.; Nelson, M.E.; Gao, X.; Liu, X.; Locasale, J.W.; Yagita, H.; Slingluff, C.L., Jr.; Hoehn, K.L.; Rathmell, J.C.; et al. Impaired enolase 1 glycolytic activity restrains effector functions of tumor-infiltrating CD8+ T cells. Sci. Immunol. 2019, 4, eaap9520. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, J.M.; Chen, W.; Gookin, J.; Wu, G.Y.; Fu, Q.; Blikslager, A.T.; Rippe, R.A.; Argenzio, R.A.; Cance, W.G.; Weaver, E.M.; et al. Arginine stimulates intestinal cell migration through a focal adhesion kinase dependent mechanism. Gut 2004, 53, 514–522. [Google Scholar] [CrossRef]
- Gu, K.; Liu, G.; Wu, C.; Jia, G.; Zhao, H.; Chen, X.; Tian, G.; Cai, J.; Zhang, R.; Wang, J. Tryptophan improves porcine intestinal epithelial cell restitution through the CaSR/Rac1/PLC-gamma1 signaling pathway. Food Funct. 2021, 12, 8787–8799. [Google Scholar] [CrossRef]
- Huang, J.H.; Cardenas-Navia, L.I.; Caldwell, C.C.; Plumb, T.J.; Radu, C.G.; Rocha, P.N.; Wilder, T.; Bromberg, J.S.; Cronstein, B.N.; Sitkovsky, M.; et al. Requirements for T lymphocyte migration in explanted lymph nodes. J. Immunol. 2007, 178, 7747–7755. [Google Scholar] [CrossRef] [PubMed]
- Manaster, Y.; Shipony, Z.; Hutzler, A.; Kolesnikov, M.; Avivi, C.; Shalmon, B.; Barshack, I.; Besser, M.J.; Feferman, T.; Shakhar, G. Reduced CTL motility and activity in avascular tumor areas. Cancer Immunol. Immunother. CII 2019, 68, 1287–1301. [Google Scholar] [CrossRef] [PubMed]
- Ogando, J.; Saez, M.E.; Santos, J.; Nuevo-Tapioles, C.; Gut, M.; Esteve-Codina, A.; Heath, S.; Gonzalez-Perez, A.; Cuezva, J.M.; Lacalle, R.A.; et al. PD-1 signaling affects cristae morphology and leads to mitochondrial dysfunction in human CD8+ T lymphocytes. J. Immunother. Cancer 2019, 7, 151. [Google Scholar] [CrossRef]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef]
- Patsoukis, N.; Brown, J.; Petkova, V.; Liu, F.; Li, L.; Boussiotis, V.A. Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci. Signal. 2012, 5, ra46. [Google Scholar] [CrossRef]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Yun, S.J.; Lee, B.; Jeong, E.; Yoon, G.; Kim, K.; Park, S. Association of TIM-3 expression with glucose metabolism in Jurkat T cells. BMC Immunol. 2020, 21, 48. [Google Scholar] [CrossRef] [PubMed]
- Franciszkiewicz, K.; Le Floc’h, A.; Boutet, M.; Vergnon, I.; Schmitt, A.; Mami-Chouaib, F. CD103 or LFA-1 engagement at the immune synapse between cytotoxic T cells and tumor cells promotes maturation and regulates T-cell effector functions. Cancer Res. 2013, 73, 617–628. [Google Scholar] [CrossRef]
- Harjunpaa, H.; Llort Asens, M.; Guenther, C.; Fagerholm, S.C. Cell Adhesion Molecules and Their Roles and Regulation in the Immune and Tumor Microenvironment. Front. Immunol. 2019, 10, 1078. [Google Scholar] [CrossRef] [PubMed]
- Demetriou, P.; Abu-Shah, E.; Valvo, S.; McCuaig, S.; Mayya, V.; Kvalvaag, A.; Starkey, T.; Korobchevskaya, K.; Lee, L.Y.W.; Friedrich, M.; et al. A dynamic CD2-rich compartment at the outer edge of the immunological synapse boosts and integrates signals. Nat. Immunol. 2020, 21, 1232–1243. [Google Scholar] [CrossRef]
- Gudipati, V.; Rydzek, J.; Doel-Perez, I.; Goncalves, V.D.R.; Scharf, L.; Konigsberger, S.; Lobner, E.; Kunert, R.; Einsele, H.; Stockinger, H.; et al. Inefficient CAR-proximal signaling blunts antigen sensitivity. Nat. Immunol. 2020, 21, 848–856. [Google Scholar] [CrossRef]
- Koneru, M.; Monu, N.; Schaer, D.; Barletta, J.; Frey, A.B. Defective adhesion in tumor infiltrating CD8+ T cells. J. Immunol. 2006, 176, 6103–6111. [Google Scholar] [CrossRef]
- Kantari-Mimoun, C.; Barrin, S.; Vimeux, L.; Haghiri, S.; Gervais, C.; Joaquina, S.; Mittelstaet, J.; Mockel-Tenbrinck, N.; Kinkhabwala, A.; Damotte, D.; et al. CAR T cell entry into tumor islets is a two-step process dependent on IFN and ICAM-1. Cancer Immunol. Res. 2021, 9, 1425–1438. [Google Scholar] [CrossRef] [PubMed]
- Lotscher, J.; Marti, I.L.A.A.; Kirchhammer, N.; Cribioli, E.; Giordano Attianese, G.M.P.; Trefny, M.P.; Lenz, M.; Rothschild, S.I.; Strati, P.; Kunzli, M.; et al. Magnesium sensing via LFA-1 regulates CD8+ T cell effector function. Cell 2022, 185, 585–602.e29. [Google Scholar] [CrossRef]
- Ramkumar, P.; Abarientos, A.B.; Tian, R.; Seyler, M.; Leong, J.T.; Chen, M.; Choudhry, P.; Hechler, T.; Shah, N.; Wong, S.W.; et al. CRISPR-based screens uncover determinants of immunotherapy response in multiple myeloma. Blood Adv. 2020, 4, 2899–2911. [Google Scholar] [CrossRef] [PubMed]
- Larson, R.C.; Kann, M.C.; Bailey, S.R.; Haradhvala, N.J.; Llopis, P.M.; Bouffard, A.A.; Scarfo, I.; Leick, M.B.; Grauwet, K.; Berger, T.R.; et al. CAR T cell killing requires the IFNgammaR pathway in solid but not liquid tumours. Nature 2022, 604, 563–570. [Google Scholar] [CrossRef]
- Cappell, K.M.; Kochenderfer, J.N. A comparison of chimeric antigen receptors containing CD28 versus 4-1BB costimulatory domains. Nat. Rev. Clin. Oncol. 2021, 18, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Gocher, A.M.; Workman, C.J.; Vignali, D.A.A. Interferon-gamma: Teammate or opponent in the tumour microenvironment? Nat. Rev. Immunol. 2021, 22, 158–172. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Fisher, D.T.; Clancy, K.A.; Gauguet, J.M.; Wang, W.C.; Unger, E.; Rose-John, S.; von Andrian, U.H.; Baumann, H.; Evans, S.S. Fever-range thermal stress promotes lymphocyte trafficking across high endothelial venules via an interleukin 6 trans-signaling mechanism. Nat. Immunol. 2006, 7, 1299–1308. [Google Scholar] [CrossRef] [PubMed]
- Palena, C.; Foon, K.A.; Panicali, D.; Yafal, A.G.; Chinsangaram, J.; Hodge, J.W.; Schlom, J.; Tsang, K.Y. Potential approach to immunotherapy of chronic lymphocytic leukemia (CLL): Enhanced immunogenicity of CLL cells via infection with vectors encoding for multiple costimulatory molecules. Blood 2005, 106, 3515–3523. [Google Scholar] [CrossRef][Green Version]
- Sartor, W.M.; Kyprianou, N.; Fabian, D.F.; Lefor, A.T. Enhanced expression of ICAM-1 in a murine fibrosarcoma reduces tumor growth rate. J. Surg. Res. 1995, 59, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Lefor, A.T.; Fabian, D.F. Enhanced cytolytic activity of tumor infiltrating lymphocytes (TILs) derived from an ICAM-1 transfected tumor in a murine model. J. Surg. Res. 1998, 75, 49–53. [Google Scholar] [CrossRef]
- Lokugamage, N.; Chowdhury, I.H.; Biediger, R.J.; Market, R.V.; Khounlo, S.; Warier, N.D.; Hwang, S.A.; Actor, J.K.; Woodside, D.G.; Marathi, U.; et al. Use of a small molecule integrin activator as a systemically administered vaccine adjuvant in controlling Chagas disease. NPJ Vaccines 2021, 6, 114. [Google Scholar] [CrossRef]
- Kochl, R.; Thelen, F.; Vanes, L.; Brazao, T.F.; Fountain, K.; Xie, J.; Huang, C.L.; Lyck, R.; Stein, J.V.; Tybulewicz, V.L. WNK1 kinase balances T cell adhesion versus migration in vivo. Nat. Immunol. 2016, 17, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, S.M.; Kjaergaard, J.; Lukashev, D.; Schreiber, T.H.; Belikoff, B.; Abbott, R.; Sethumadhavan, S.; Philbrook, P.; Ko, K.; Cannici, R.; et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci. Transl. Med. 2015, 7, 277ra230. [Google Scholar] [CrossRef] [PubMed]
- Jayaprakash, P.; Ai, M.; Liu, A.; Budhani, P.; Bartkowiak, T.; Sheng, J.; Ager, C.; Nicholas, C.; Jaiswal, A.R.; Sun, Y.; et al. Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J. Clin. Investig. 2018, 128, 5137–5149. [Google Scholar] [CrossRef]
- Amitrano, A.M.; Berry, B.J.; Lim, K.; Kim, K.D.; Waugh, R.E.; Wojtovich, A.P.; Kim, M. Optical Control of CD8+ T Cell Metabolism and Effector Functions. Front. Immunol. 2021, 12, 666231. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simula, L.; Ollivier, E.; Icard, P.; Donnadieu, E. Immune Checkpoint Proteins, Metabolism and Adhesion Molecules: Overlooked Determinants of CAR T-Cell Migration? Cells 2022, 11, 1854. https://doi.org/10.3390/cells11111854
Simula L, Ollivier E, Icard P, Donnadieu E. Immune Checkpoint Proteins, Metabolism and Adhesion Molecules: Overlooked Determinants of CAR T-Cell Migration? Cells. 2022; 11(11):1854. https://doi.org/10.3390/cells11111854
Chicago/Turabian StyleSimula, Luca, Emma Ollivier, Philippe Icard, and Emmanuel Donnadieu. 2022. "Immune Checkpoint Proteins, Metabolism and Adhesion Molecules: Overlooked Determinants of CAR T-Cell Migration?" Cells 11, no. 11: 1854. https://doi.org/10.3390/cells11111854
APA StyleSimula, L., Ollivier, E., Icard, P., & Donnadieu, E. (2022). Immune Checkpoint Proteins, Metabolism and Adhesion Molecules: Overlooked Determinants of CAR T-Cell Migration? Cells, 11(11), 1854. https://doi.org/10.3390/cells11111854