1. Introduction
The CRISPR/Cas9 system is derived from the type II CRISPR/Cas system, which provides adaptive immunity to viruses and plasmids for bacteria [
1,
2,
3]. The full name of CRISPR is Clustered Regularly Interspaced Short Palindromic Repeats [
4], and these repetitive sequences are separated by distinct non-repetitive sequences called spacers. When an exogenous virus invades the host, the viral DNA is processed by Cas nuclease, into small DNA fragments, which are then integrated into the CRISPR of the host genome as spacers. The spacers are used as the transcriptional templates to produce crRNA, and the mature crRNA with tracrRNA form a special RNA structure (gRNA), through base complementary pairing, which guides the cas9 protein to recognize the specific site (PAM motif) [
5,
6] of the target DNA, causes double-strand breaks, and enables targeted editing of the genome [
7]. Since cells cannot survive long with their DNA cleavage, their alarms go off, as soon as the DNA is broken. The steps to repair the break begin quickly.
DNA double-strand breaks are, primarily, repaired by one of two pathways: non-homologous end-joining (NHEJ) and homology-directed repair (HDR) [
8,
9]. In addition to this, there is evidence for microhomology-mediated end-joining (MMEJ). HDR [
10] is complex and precise, can only occur in the
G2 and
S phase of the cells, and repairs double-strand breaks (DSBs), using homologous template sequences, but it is inefficient. In contrast, NHEJ [
11] is rapid and imprecise, directly rejoining broken ends, usually in the form of short insertions or deletions (indels), which occur throughout the cell cycle. Unlike classical NHEJ, MMEJ [
12] relies on regions of microhomology, to repair breaks. MMEJ repair of broken DNA, often, results in the deletion of micro-homologous fragments at the junction or, even, leads to gene rearrangements, so it is an error-prone repair method. Recent studies have shown that the repair outcomes of NHEJ and MMEJ are determined by the sequence characteristics of the target DNA [
13].
Owing to the fact that mutational outcomes vary with cell lines and Cas9 modifications [
14], accurate prediction of template-free CRISPR/Cas9 editing outcomes is, undoubtedly, a challenging bioinformatics problem. Thanks to rapid advances in machine learning, researchers better conducted CRISPR/Cas9 studies. Various machine-learning-based prediction tools have been developed and put into use (
Table 1).
inDelphi [
15] introduced different sequence features, such as microhomology length, built three interconnected modules based on the deep neural network or k-Nearest Neighbor algorithm, and, then, predicted MH deletions, MH-less deletions, and 1 bp insertions, respectively. SPROUT [
16] took the 20 nucleotides of the spacer sequences plus the PAM, as inputs, and built a Gradient Boosting Decision Tree model. At each target site, the model predicted nine statistics, such as the average deletion length. CROTON [
17] is a deep learning framework based on the Convolutional Neural Network (CNN) and Neural Architecture Search (NAS), which predicted 1 bp insertion and deletion probability as well as deletion and frameshift frequency, from raw one-hot encoded DNA sequences. Several models mentioned above predicted only a few fixed repair labels during model building, which cannot cover all mutational outcomes and have insufficient prediction accuracy. FORECasT [
14] generated candidate mutations for each gRNA and derived features for them based on local sequence characteristics, training a multi-class Logistic Regression model designed to predict repair outcomes. Lindel [
18] defined binary features to characterize the target sequences and used one-hot encoding to convert the sequences to matrixes as model inputs, so three components were trained independently using different Logistic Regression models aiming to predict the ratio of insertions to deletions, the distribution of deletion events, and the distribution of insertion events. Although the above two models can predict the vast majority of mutational outcomes, the inherent mechanism of CRISPR gene editing technology is, still, unclear, and both the artificial construction of a set of binary features describing the sequences or mutational information alone and the conversion of the sequences into sparse matrixes using unique one-hot encoding as model inputs may, negatively, affect the prediction performance.
Here, we developed a model named Apindel (a deep learning framework based on the Attention mechanism and Positional Encoding, for predicting CRISPR/Cas9 repair outcomes) to address the problem of predicting DNA mutations, using contextual sequences surrounding the cleavage sites. First, through introducing the GloVe embedding model, the cooccurrence matrixes were constructed to extract the global statistical information of the input sequences; second, the target sequences were converted into dense matrixes, by combining the Positional Encoding; finally, the model was combined with the deep neural network model, adding an attention layer, which can reveal that the bases at different sequence loci have varying degrees of effect on predicting DNA repair outcomes. Apindel can, accurately, predict the mutational outcomes for a given target sequence. We demonstrated that Apindel, which was trained on the FORECasT dataset, outperformed existing approaches on the most prediction tasks. Therefore, it is expected to be a potential tool to aid research on CRISPR systems.
3. Results
3.1. Apindel Architecture
Based on the previous research findings, that “repair outcomes can be predicted by the local context of the sequences” [
19,
21,
24], we constructed the Apindel model (
Figure 2), which could accurately predict the outcomes and repair probabilities, based on the sequences near the cleavage sites. Considering the efficiency and accuracy of the model, we truncated 60 bp genomic sequences as model inputs, and we described the mutational outcomes as model outputs, with a total of 557 labels considered.
The first layer of the model was the embedding layer. The k-mer corpus was obtained, by setting the sliding window size Kmer to 2 and the sliding step length Kstep to 1, so the size of the k-mer vocabulary was, thus, , and the indexes corresponded to the set of positive integers . We transformed the input target sequences into the k-mer sequences of length , resulting in a location information vocabulary of size , with indexes corresponding to the set of positive integers . The k-mer were combined with the position information, to obtain a vocabulary of size , and the indexes corresponded to the set of positive integers . The vector-conversion dictionary was trained through the GloVe model and Positional Encoding, respectively, the corresponding GloVe word vector and the positional vector were added according to the index, and, subsequently, the individual k-mer was transformed into a vector, so the whole k-mer sequence was converted into the matrix, as the input to the deep learning model. We called the Python extension package mittens to train the GloVe model, based on our own code implementation of the GloVe cooccurrence matrix. Regarding the hyperparameters of the GloVe word-vector-generation model, the dimension of the embedding vector was set to 150, the size of the window diameter for calculating the cooccurrence matrix was set to 5, the truncation value in the auxiliary function was set to 10,000, and the maximum number of iterations was set to 20,000. We called the torch to build the PE model, and the dimension of the embedding vector was set, as the same as the GloVe model: dropout parameter (p = 0).
The second layer was the BiLSTM network, which was, mainly, used to extract the contextual features of the input information. The number of neurons (units) was set to 50, the activation function was ReLU, and L1 regularization was added to this layer. The output of the layer was used for the computation of the next layer and the final result.
The third layer was the Attention layer, and the computational process of this layer was divided into three steps. First, we calculated the weight matrix, multiplied it with the feature matrix output from the previous layer, and chose the activation function as tanh. Then, we calculated the weight vector, multiplied it with the matrix output by the previous step, selected the activation function as SoftMax, and obtained the attention vector a. Finally, the attention vector was multiplied by the feature-matrix input from this layer, and, then, the result was fed into a multilayer perception (MLP) network, followed by the SoftMax activation function. We added L1 regularization to this layer. The prediction results of the model were obtained.
During the training process, we used the Adam optimizer [
25] with a learning rate of 0.0001, to optimize the loss function. In order to avoid the occurrence of overfitting, we set the batch size to 64 and used the categorical cross-entropy loss. The factor of the callback function ReduceLROnPlateau was set to 0.2, training proceeded for a maximum of 100 epochs, with a “patience” of 1 and a “patience” of 3, meaning that training shrunk the learning rate after one epoch and stopped after three epochs, with no improvement in validation-set performance. The detailed description of the model structure is in the
Supplementary Material.
3.2. Model Selection
We evaluated the predictive performance of Apindel against other pre-selected models (
Table 2). The structure of the benchmark model Apindel was described in the “Apindel architecture” section. Apindel_NoPE was obtained, by removing the Positional Encoding part from the data encoding of the Apindel model; Apindel_Noattention was the Apindel model, encoded without the Attention mechanism. First, all models were trained on the Lindel dataset and were evaluated on the test set using Mean Square Error (MSE). Second, all models were trained on the FORECasT dataset and were evaluated on the test set using Area Under Curve (AUC) and Pearson correlation coefficients; the relevant data descriptions are described in the “Data Sources” section.
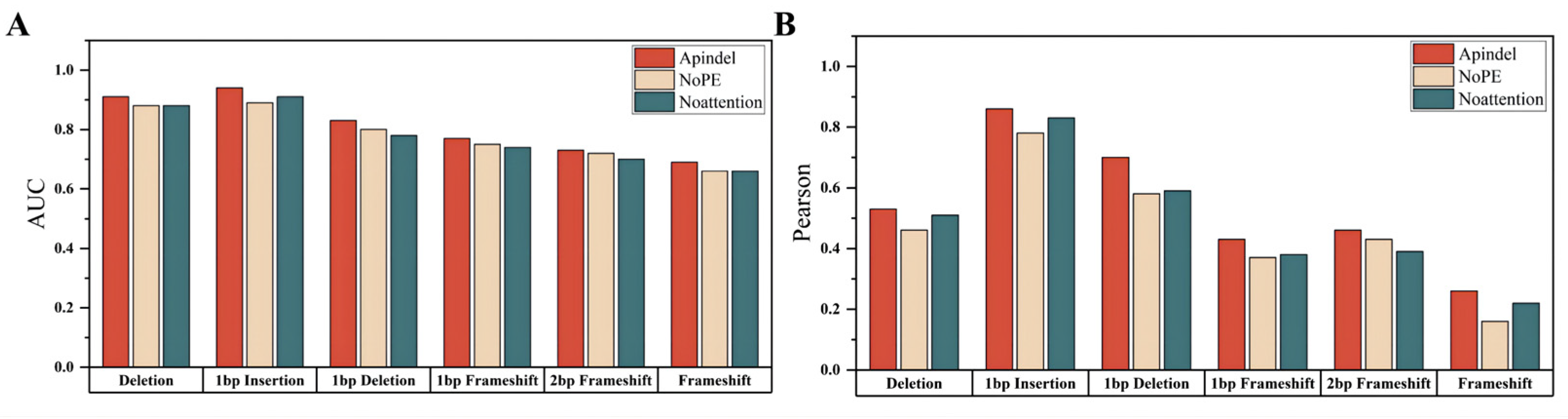
Compared with the Apindel_NoPE model and the Apindel_Noattention model (
Table 3), we can clearly see that Apindel achieved better prediction performance, on the Lindel test set.
Compared with the benchmark model, we can see that the Apindel model predicted the six prediction tasks used for model comparison, with much better accuracy than the Apindel_NoPE model and the Apindel_Noattention model (
Figure 3), The AUC values for predicting Deletion frequency and 1 bp Insertion frequency were both greater than 90% (
Table S2). In summary, the position information was very important to our model, and assigning different weights to different sequence loci, through the Attention mechanism, was helpful to improve the prediction of the model.
3.3. Model Comparison
Recently, five machine-learning models have been developed to predict the outcome of DNA sequence repair, namely SPROUT [
16], inDelphi [
15], CROTON [
17], FORECasT [
14], and Lindel [
18]. SPROUT was a Gradient Boosting Decision Tree model that predicted nine repair types, including average deletion length, and provided an independent dataset of CRISPR/Cas9 editing results. CROTON utilized NAS, to automatically create a multi-task deep Convolutional Neural Network framework, for prediction of CRISPR/Cas9 editing outcomes, which can predict six repair types, including deletion frequencies. inDelphi built three interconnected modules, based on neural networks and k-Nearest Neighbors, aiming to predict microhomology (MH) deletions, MH-less deletions, and 1 bp insertions. FORECasT built a multi-class Logistic Regression model, capable of predicting all possible unique repair outcomes, and it provided one of the largest datasets of CRISPR/Cas9 editing results, relative to inDelphi, SPROUT, and Lindel, for the establishment of Apindel. Lindel was a Logistic Regression model; unlike the above models, it provided an exact category criterion for predicting repair outcomes, including insertion and deletion sites, insertion nucleotide types, and deletion length.
For a more comprehensive comparison with the five models mentioned above, we applied the Apindel model, trained on the Lindel dataset, to the Lindel test set and the FORECasT test set, and evaluated the predictive performance of the model using the mean squared error (mse). The Apindel model trained on the FORECasT dataset was applied to the randomly selected FORECasT test set, the SPROUT test set, and the Lindel test set, and the predictive performance of the models was evaluated using the Kendall’s tau rank correlation coefficient, Pearson correlation coefficient, or AUC between predicted and observed values (
Table S3).
3.3.1. Evaluation of Predictive Performance on 557 Predictive Labels
First, since Lindel was the only model in the published papers, so far, that predicted 557 repair outcomes, we compared the Apindel model trained on the Lindel data with Lindel. We trained on the Lindel dataset and predicted the frequency of 557 classes of indels. Comparing the results of our model and Lindel on the test set, our model worked better (MSE = 0.000164 and 0.000172 for our model and Lindel, respectively). The above results illustrated that our model had better performance, in predicting 557 repair outcomes. To validate the generalization ability of Apindel, we analyzed the prediction performance of Apindel on the independent FORECasT dataset. Predicting 557 classes of indels on the FORECasT dataset did not work as well as on the Lindel test set (MSE = 0.000197). Based on the previous research findings, that “the overall distribution of repair outcome types was not the same across the different cell lines” [
14], we speculated that this problem may be caused by differences in repair outcomes between different cell lines.
3.3.2. Evaluation of Predictive Performance on Six Classes of Prediction Tasks
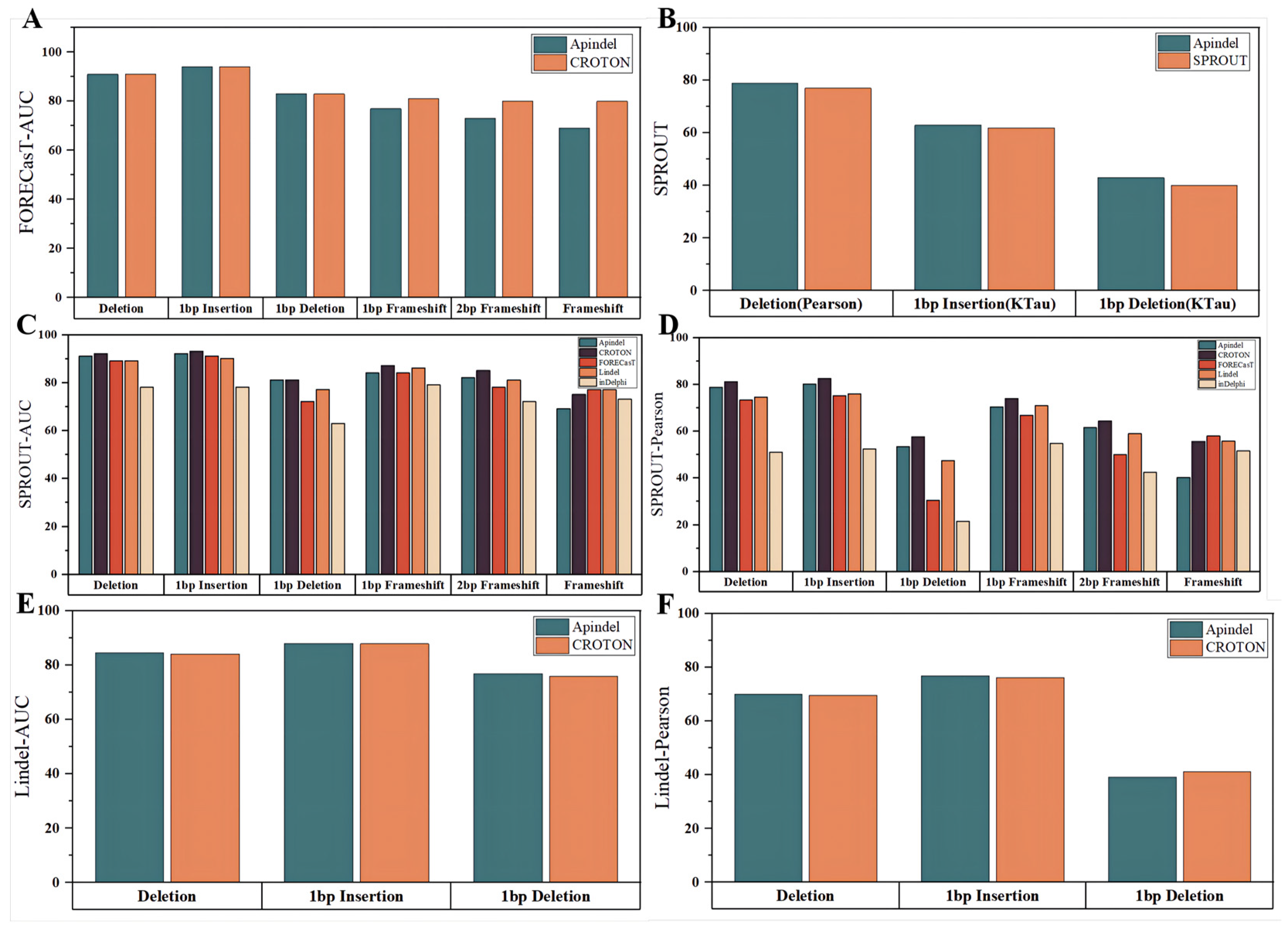
In the next step, Apindel was compared with other models on the CROTON test set (described in detail in the “Data sources” section). First of all, since CROTON was built based on the FORECasT dataset, we considered a comparison of the predicted edit outcomes, with the CROTON model on the FORECasT data. Apindel achieved AUCs greater than 90%, for both the deletion frequency and 1 bp insertion-frequency tasks, and AUCs of 91%,94%, and 83% for the deletion frequency,1 bp Insertion frequency, and 1 bp deletion frequency, respectively, which were the same as those of CROTON, and slightly inferior to the other models, in terms of frameshift frequency prediction (
Figure 4A).
Next, in order to test the accuracy and generalization ability of Apindel in predicting DNA repair outcomes, we applied Apindel to the SPROUT dataset and the Lindel dataset, which was compared with existing machine-learning-based predictors of CRISPR/Cas9 editing results: SPROUT, inDelphi, CROTON, FORECasT, and Lindel. Given that the inDelphi, FORECasT, and Lindel models predicted more detailed repair labels, while the CROTON model predicted only six repair outcomes, including deletion frequency, we would compare model performance from the following two aspects.
On the one hand, the repair outcomes predicted by Apindel, inDelphi, FORECasT, and Lindel were organized into six repair tasks, for comparison (
Figure 4C,D). Apindel outperformed other models, for most of the prediction tasks (inferior to inDelphi, FORECasT, and Lindel, for frameshift frequency). Especially in deletion frequency and 1 bp insertion frequency, Apindel achieved AUC values greater than 90% and a Pearson correlation of 80% for 1 bp insertion frequency, indicating that Apindel achieved better prediction ability.
On the other hand, for CROTON, we could, intuitively, see that on the SPROUT dataset, the prediction performance of Apindel was slightly inferior to CROTON. However, on the Lindel test set, Apindel was better than CROTON (
Figure 4E,F), indicating that the prediction performance of Apindel was comparable to CROTON. It was worth noting that Apindel could predict 557 repair labels, while CROTON could predict only 6 repair labels, including deletion frequency. Apindel was more practical because it could predict the repair outcomes in a more detailed and comprehensive way. For SPROUT, the performance of Apindel was similar to or even better than SPROUT, for the three metrics (
Figure 4B) thatwere trained on the SPROUT dataset. In conclusion, Apindel ran robustly on different experimental datasets, with high accuracy and generalization ability.
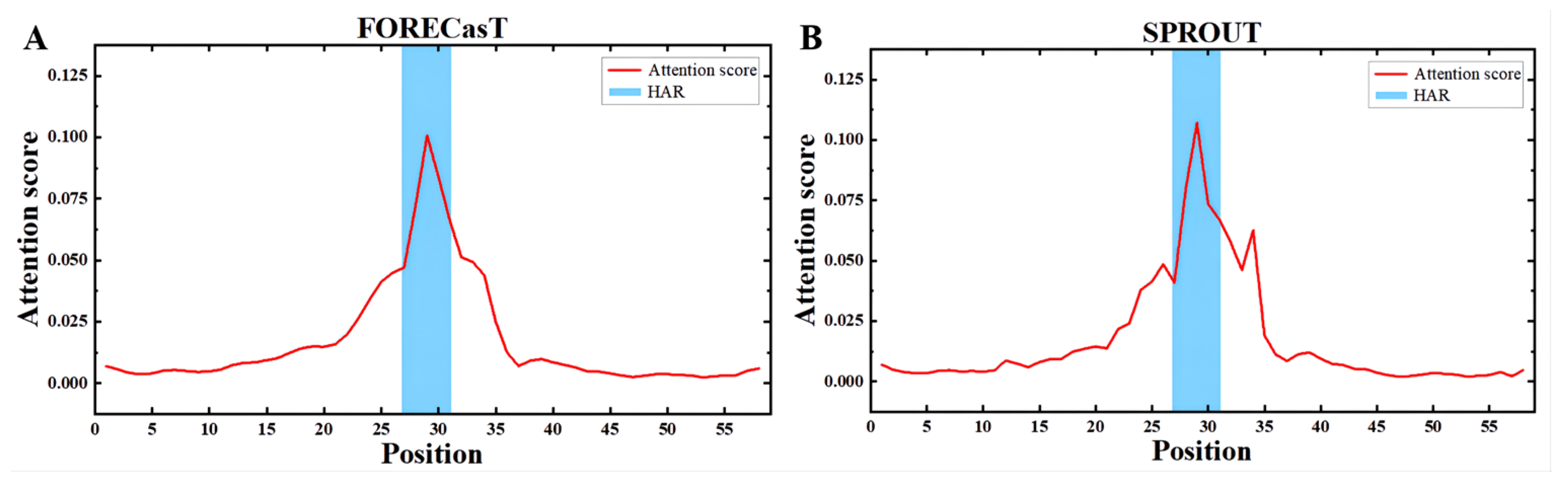
3.4. Apindel Reveals Important Sequence Loci Associated with Repair Outcomes
An important advantage of Apindel over other deep-learning-based frameworks was that Apindel further incorporated an attention mechanism, thus allowing one to capture important sequence loci affecting predicted DNA repair, by detecting the attention vectors of samples. Here, we examined the distribution of the attention scores, by averaging the attention vector
over all samples in the test dataset. In the FORECasT test set (
Figure 5A), the High-attention regions (HAR) [
26] appeared near the cut site (i.e., the 25th Kmer to the 35st Kmer), and a similar finding was shown in the SPROUT test set (
Figure 5B). The above observations suggested that contextual sequences, around the 10 bp window on either side of the cleavage sites, were critical for predicting repair outcomes, and sequences farther away, relative to the cleavage sites (such as the 10th site near the
end), had a weaker effect on repair outcomes, which was consistent with previous findings [
14]. Therefore, our study confirmed that nucleotides at different locations had different degrees of effect on CRISPR/Cas9 editing outcomes, and the nucleotides near the cleavage sites had the greatest influence.
4. Discussion
As a simple and programmable nuclease-based genome editing tool, the CRISPR/Cas9 system has greatly improved the ability to perform precise editing in the human genome [
27]. In recent years, the rapid development of CRISPR-based technology has expanded its application scope, and CRISPR technology has been applied to human diseases, cancer, plant biology [
28,
29,
30], etc. It can be seen that the CRISPR/Cas9 system has great application prospects. Experiments demonstrated that the cellular-repair outcomes generated by CRISPR/Cas9 were determined by local-sequence features. An attention-based deep learning model was proposed, to accurately predict DNA-repair outcomes. Compared with previous models, Apindel considered more comprehensive repair labels. It can predict repair outcomes in more detail, contains a higher amount of information, and was more practical. By fully taking advantage of the superior predictive capacity of deep learning models [
31] and the interpretability of the Attention mechanism [
32], Apindel can, accurately, predict DNA mutational outcomes and capture the important sequence sites that affect the repair outcomes.
According to previous research [
14], we knew that bases at different positions had different effects on repair outcomes. Therefore, we considered adding Positional Encoding, to represent the location information in the process of sequences encoding. In the “Model Selection” section, it can be seen that Positional Encoding had a great influence on the repair outcomes, and the Apindel model with Positional Encoding had better performance than the Apindel_NoPE model, in predicting repair outcomes, indicating the necessity of including Positional Encoding in the model.
In order to study the impact of location information on the prediction model more intuitively, we added an attention mechanism to the model. By averaging the attention vectors, we got a set of weights. Through weighting the output feature matrix, the prediction performance of Apindel was improved. By observation, we found that bases near the cleavage sites were the most important for the prediction of template-free CRISPR/Cas9 editing outcomes. It was consistent with the previous conclusion [
14] and would suggest researchers should pay more attention to the impact of sequences near the cleavage sites on the repair outcomes.
Frameshift mutation refers to the deletion or addition of non-three multiples of bases in the normal DNA molecule, which causes a series of coding errors to occur after this position. Since frameshift mutation can lead to some serious consequences, we introduced the prediction task of frameshift-mutation frequency, when testing the model performance. However, to our surprise, the method based on deep learning is not doing well in predicting frameshift (e.g., Apindel and CROTON), which will become the focus of our later research work.
5. Conclusions
In this paper, a novel DNA-repair-outcomes prediction model, Apindel was constructed. With introduction of the GloVe model and Positional Encoding, Apindel attempted new feature representation methods, to embed sequence information into the deep learning model; the BiLSTM model was used to extract contextual sequence features, and the Attention mechanism was calculated to assign weights to the output matrix, to characterize the different influence of different loci nucleotides on the repair outcomes. The superior performance of Apindel was, further, confirmed by comparison with existing models. Our model used deep learning to comprehend the automatic learning of sequence features between DNA and corresponding repair outcomes, avoiding the unknown influence of the manual-feature-construction process on the model-prediction outcomes. Thus, it is able to predict more comprehensive and detailed repair labels, with higher accuracy and better utility. It is a new attempt of deep learning, in the direction of DNA-repair-outcome prediction.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}