
Immune Cell Plasticity in Inflammation: Insights into Description and Regulation of Immune Cell Phenotypes


Abstract
:1. Introduction
2. Techniques for Immune Cell Phenotype Assessment: A Matter of Perspective
3. Mechanisms of Immune Cell Phenotype Switching: Outside In and Inside Out
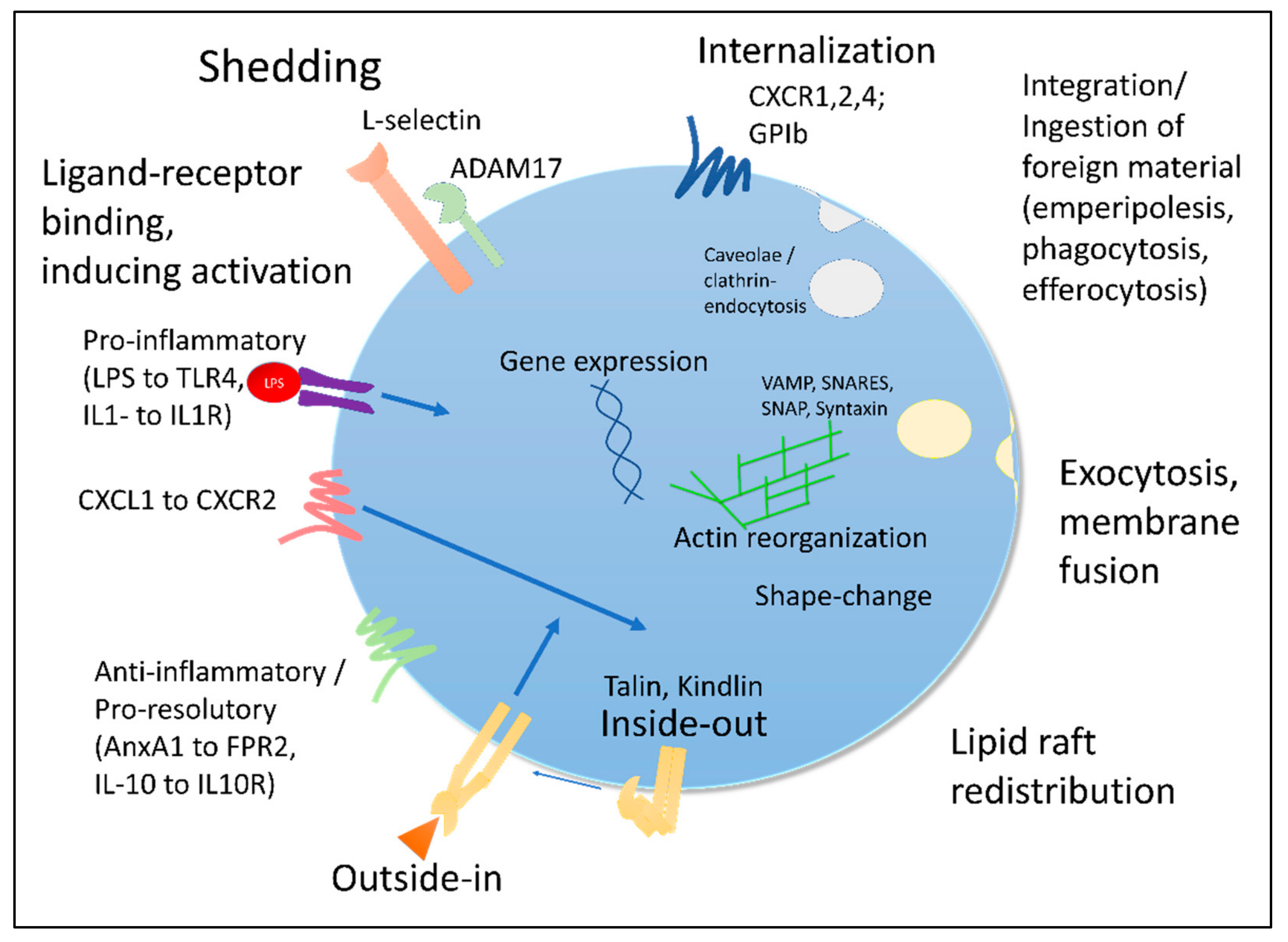
3.1. Surface: Receptor Shedding, Internalization and Desensitization
3.2. Regulation of Gene Expression
3.3. Activation-Dependent Vesicle Release and Membrane Fusion
3.4. Re-Production and Re-Distribution
3.5. Changed Physical Properties
3.6. Integrin-Mediated Signaling
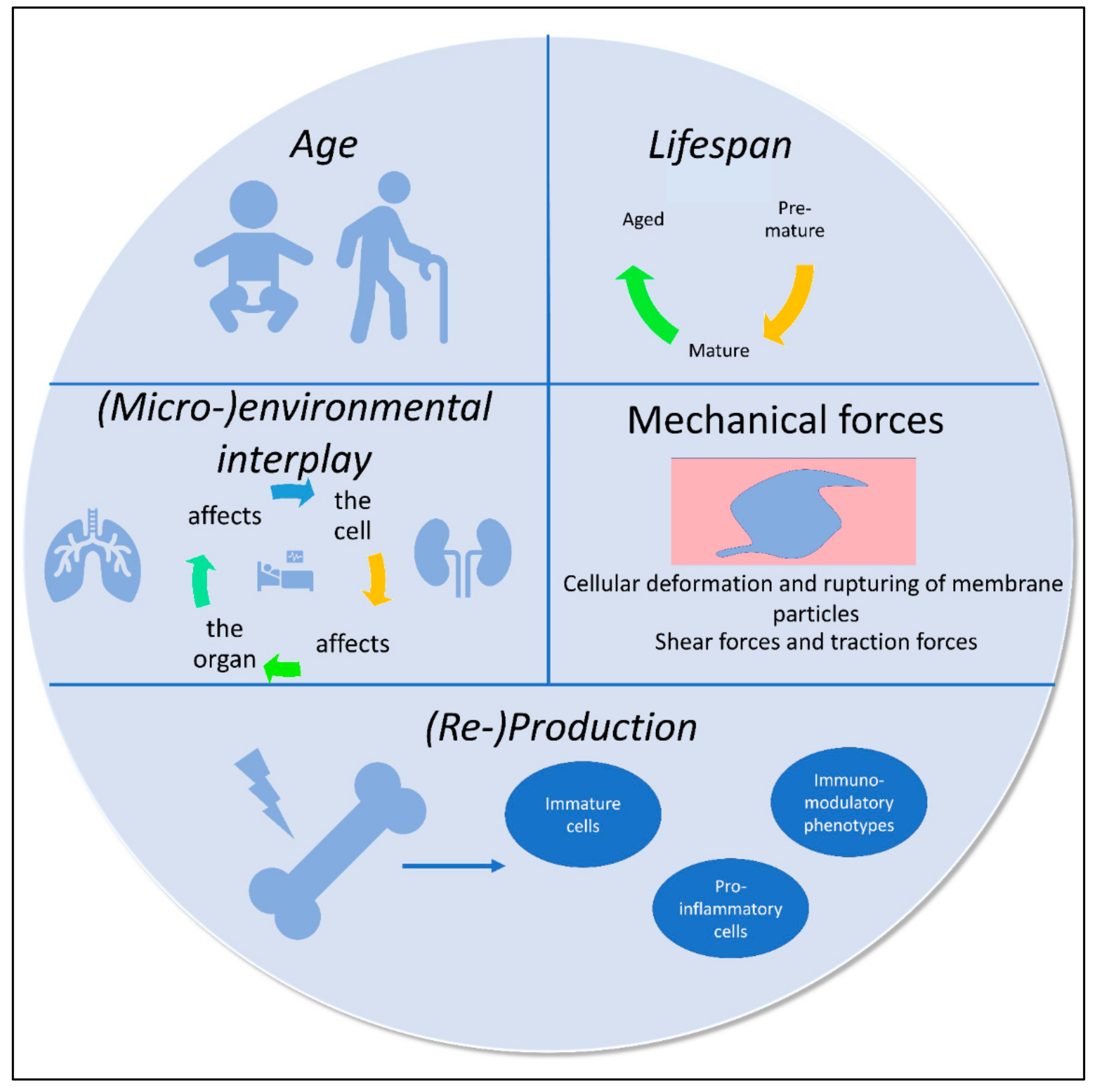
4. Principles and Regulatory Factors Affecting Immune Phenotypes: The Environment Shapes the Cell, and the Cell Impacts Its Environment
4.1. Circadian Rhythms
4.2. Aging
4.3. Stress
4.4. Environment/Compartment (Organ)
4.5. Mechanical Forces
4.6. Inflammation and Injury—A Combination of Extremes
5. Macrophage Phenotypes and Plasticity
5.1. M1, M2 and Beyond
5.2. Tissue-Specific Macrophages
5.3. Overarching Macrophage Phenotypes
6. Neutrophil Phenotypes—The Many Faces of a Single Cell
7. Platelets as Immune Cells and Platelet Phenotypes during Inflammation

{kind=link}
{kind=link}
| Cell Type | Phenotype | Mechanism | Characteristics |
|---|---|---|---|
| Platelets | Immature platelets | Emergency thrombopoiesis |
|
| Sepsis-induced splenic platelets | IL-3-mediated splenic megakaryocyte maturation |
| |
| Lung-derived platelets | S1P-gradient-dependent platelet shedding in pulmonary microvasculature |
| |
| Young vs. old | Life-cycle dependent aging |
| |
| COATed platelets | Dual stimulation with collagen and thrombin |
| |
| Post-emperipolesis platelets | Megakaryocyte emperipolesis followed by thrombopoiesis |
| |
| Plat 1 vs. Plat 2 | Differing stimuli and microenvironments |
| |
| Neutrophils | NDN | Density gradient purification |
|
| LDN | Density gradient purification |
| |
| N1 | Increased following TGF-beta blockade |
| |
| N2 | PPARy-dependent |
| |
| Reverse-migrated neutrophils | LTB4–Neutrophil Elastase-dependent |
| |
| Aged neutrophils | Life-cycle-dependent aging, microbiota-TLR-MyD88-dependent |
| |
| Macrophages | M1 | LPS-, IFN-dependent |
|
| M2 (a, b, c, d) | IL-10, IL-4-dependent |
| |
| Alveolar | Localization |
| |
| Interstitial | Localization |
| |
| Resident | Organ-inherent resident immune cells |
| |
| Perivascular | Localization vs. organ-specific expression patterns |
| |
| Profibrotic | Increased in pulmonary bleomycin model |
| |
| Monocyte derived | Recruitment of monocytes and maturation to macrophages |
| |
| Splenic macrophages | Anatomic localization |
|
8. Regulatory Aspects of DAMPS, PAMPs, Microenvironments and Cellular Interplay
9. Translational Findings and Immune Phenomics of COVID-19
10. Outlook and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Leeuwenhoek, A.V. Arcana Naturae Detecta; Delphis Batavorum, Apud Henricum a Krooneveld: Delft, The Netherlands, 1695. [Google Scholar]
- Andral, G. Essai D’hématologie Pathologique; Fortin, Masson et Cie: Paris, France, 1843. [Google Scholar]
- Margraf, A.; Germena, G.; Drexler, H.C.A.; Rossaint, J.; Ludwig, N.; Prystaj, B.; Mersmann, S.; Thomas, K.; Block, H.; Gottschlich, W.; et al. The integrin-linked kinase is required for chemokine-triggered high-affinity conformation of the neutrophil beta2-integrin LFA-1. Blood 2020, 136, 2200–2205. [Google Scholar] [CrossRef] [PubMed]
- Tracz, J.; Handschuh, L.; Lalowski, M.; Marczak, L.; Kostka-Jeziorny, K.; Perek, B.; Wanic-Kossowska, M.; Podkowinska, A.; Tykarski, A.; Formanowicz, D.; et al. Proteomic Profiling of Leukocytes Reveals Dysregulation of Adhesion and Integrin Proteins in Chronic Kidney Disease-Related Atherosclerosis. J. Proteome Res. 2021, 20, 3053–3067. [Google Scholar] [CrossRef] [PubMed]
- Alarcon-Barrera, J.C.; von Hegedus, J.H.; Brouwers, H.; Steenvoorden, E.; Ioan-Facsinay, A.; Mayboroda, O.A.; Ondo-Mendez, A.; Giera, M. Lipid metabolism of leukocytes in the unstimulated and activated states. Anal. Bioanal. Chem. 2020, 412, 2353–2363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schafflick, D.; Xu, C.A.; Hartlehnert, M.; Cole, M.; Schulte-Mecklenbeck, A.; Lautwein, T.; Wolbert, J.; Heming, M.; Meuth, S.G.; Kuhlmann, T.; et al. Integrated single cell analysis of blood and cerebrospinal fluid leukocytes in multiple sclerosis. Nat. Commun. 2020, 11, 247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leite, G.G.F.; Ferreira, B.L.; Tashima, A.K.; Nishiduka, E.S.; Cunha-Neto, E.; Brunialti, M.K.C.; Assuncao, M.; Azevedo, L.C.P.; Freitas, F.; van der Poll, T.; et al. Combined Transcriptome and Proteome Leukocyte’s Profiling Reveals Up-Regulated Module of Genes/Proteins Related to Low Density Neutrophils and Impaired Transcription and Translation Processes in Clinical Sepsis. Front. Immunol. 2021, 12, 744799. [Google Scholar] [CrossRef] [PubMed]
- Fay, M.E.; Myers, D.R.; Kumar, A.; Turbyfield, C.T.; Byler, R.; Crawford, K.; Mannino, R.G.; Laohapant, A.; Tyburski, E.A.; Sakurai, Y.; et al. Cellular softening mediates leukocyte demargination and trafficking, thereby increasing clinical blood counts. Proc. Natl. Acad. Sci. USA 2016, 113, 1987–1992. [Google Scholar] [CrossRef] [Green Version]
- Tse, H.T.; Gossett, D.R.; Moon, Y.S.; Masaeli, M.; Sohsman, M.; Ying, Y.; Mislick, K.; Adams, R.P.; Rao, J.; Di Carlo, D. Quantitative diagnosis of malignant pleural effusions by single-cell mechanophenotyping. Sci. Transl. Med. 2013, 5, 212ra163. [Google Scholar] [CrossRef] [Green Version]
- Margraf, A.; Cappenberg, A.; Vadillo, E.; Ludwig, N.; Thomas, K.; Korner, K.; Zondler, L.; Rossaint, J.; Germena, G.; Hirsch, E.; et al. ArhGAP15, a RacGAP, Acts as a Temporal Signaling Regulator of Mac-1 Affinity in Sterile Inflammation. J. Immunol. 2020, 205, 1365–1375. [Google Scholar] [CrossRef]
- Crainiciuc, G.; Palomino-Segura, M.; Molina-Moreno, M.; Sicilia, J.; Aragones, D.G.; Li, J.L.Y.; Madurga, R.; Adrover, J.M.; Aroca-Crevillen, A.; Martin-Salamanca, S.; et al. Behavioural immune landscapes of inflammation. Nature 2022, 601, 415–421. [Google Scholar] [CrossRef]
- Nicolai, L.; Leunig, A.; Brambs, S.; Kaiser, R.; Weinberger, T.; Weigand, M.; Muenchhoff, M.; Hellmuth, J.C.; Ledderose, S.; Schulz, H.; et al. Immunothrombotic Dysregulation in COVID-19 Pneumonia Is Associated With Respiratory Failure and Coagulopathy. Circulation 2020, 142, 1176–1189. [Google Scholar] [CrossRef]
- Ballesteros, I.; Rubio-Ponce, A.; Genua, M.; Lusito, E.; Kwok, I.; Fernandez-Calvo, G.; Khoyratty, T.E.; van Grinsven, E.; Gonzalez-Hernandez, S.; Nicolas-Avila, J.A.; et al. Co-option of Neutrophil Fates by Tissue Environments. Cell 2020, 183, 1282–1297. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Geue, S.; Coman, C.; Munzer, P.; Kopczynski, D.; Has, C.; Hoffmann, N.; Manke, M.C.; Lang, F.; Sickmann, A.; et al. Identification of key lipids critical for platelet activation by comprehensive analysis of the platelet lipidome. Blood 2018, 132, e1–e12. [Google Scholar] [CrossRef] [PubMed]
- Anders, C.B.; Lawton, T.M.W.; Smith, H.L.; Garret, J.; Doucette, M.M.; Ammons, M.C.B. Use of integrated metabolomics, transcriptomics, and signal protein profile to characterize the effector function and associated metabotype of polarized macrophage phenotypes. J. Leukoc. Biol. 2022, 111, 667–693. [Google Scholar] [CrossRef] [PubMed]
- Zeming, K.K.; Lu, R.; Woo, K.L.; Sun, G.; Quek, K.Y.; Cheow, L.F.; Chen, C.H.; Han, J.; Lim, S.L. Multiplexed Single-Cell Leukocyte Enzymatic Secretion Profiling from Whole Blood Reveals Patient-Specific Immune Signature. Anal. Chem. 2021, 93, 4374–4382. [Google Scholar] [CrossRef] [PubMed]
- Rossaint, J.; Berger, C.; Van Aken, H.; Scheld, H.H.; Zahn, P.K.; Rukosujew, A.; Zarbock, A. Cardiopulmonary bypass during cardiac surgery modulates systemic inflammation by affecting different steps of the leukocyte recruitment cascade. PLoS ONE 2012, 7, e45738. [Google Scholar] [CrossRef] [Green Version]
- Edwards, D.R.; Handsley, M.M.; Pennington, C.J. The ADAM metalloproteinases. Mol. Asp. Med. 2008, 29, 258–289. [Google Scholar] [CrossRef]
- Cappenberg, A.; Margraf, A.; Thomas, K.; Bardel, B.; McCreedy, D.A.; Van Marck, V.; Mellmann, A.; Lowell, C.A.; Zarbock, A. L-selectin shedding affects bacterial clearance in the lung: A new regulatory pathway for integrin outside-in signaling. Blood 2019, 134, 1445–1457. [Google Scholar] [CrossRef]
- Zhu, X.D.; Zhuang, Y.; Ben, J.J.; Qian, L.L.; Huang, H.P.; Bai, H.; Sha, J.H.; He, Z.G.; Chen, Q. Caveolae-dependent endocytosis is required for class A macrophage scavenger receptor-mediated apoptosis in macrophages. J. Biol. Chem. 2011, 286, 8231–8239. [Google Scholar] [CrossRef] [Green Version]
- Kumari, S.; Mg, S.; Mayor, S. Endocytosis unplugged: Multiple ways to enter the cell. Cell Res. 2010, 20, 256–275. [Google Scholar] [CrossRef] [Green Version]
- Rose, J.J.; Foley, J.F.; Murphy, P.M.; Venkatesan, S. On the mechanism and significance of ligand-induced internalization of human neutrophil chemokine receptors CXCR1 and CXCR2. J. Biol. Chem. 2004, 279, 24372–24386. [Google Scholar] [CrossRef] [Green Version]
- Forster, R.; Kremmer, E.; Schubel, A.; Breitfeld, D.; Kleinschmidt, A.; Nerl, C.; Bernhardt, G.; Lipp, M. Intracellular and surface expression of the HIV-1 coreceptor CXCR4/fusin on various leukocyte subsets: Rapid internalization and recycling upon activation. J. Immunol. 1998, 160, 1522–1531. [Google Scholar] [PubMed]
- Cramer, E.M.; Lu, H.; Caen, J.P.; Soria, C.; Berndt, M.C.; Tenza, D. Differential redistribution of platelet glycoproteins Ib and IIb-IIIa after plasmin stimulation. Blood 1991, 77, 694–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabl, M.; Holdfeldt, A.; Sundqvist, M.; Lomei, J.; Dahlgren, C.; Forsman, H. FPR2 signaling without beta-arrestin recruitment alters the functional repertoire of neutrophils. Biochem. Pharmacol. 2017, 145, 114–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forsman, H.; Onnheim, K.; Andreasson, E.; Christenson, K.; Karlsson, A.; Bylund, J.; Dahlgren, C. Reactivation of desensitized formyl peptide receptors by platelet activating factor: A novel receptor cross talk mechanism regulating neutrophil superoxide anion production. PLoS ONE 2013, 8, e60169. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Kluger, Y.; Nakayama, Y.; Poddar, R.; Whitney, C.; DeTora, A.; Weissman, S.M.; Newburger, P.E. Gene expression in mature neutrophils: Early responses to inflammatory stimuli. J. Leukoc. Biol. 2004, 75, 358–372. [Google Scholar] [CrossRef] [Green Version]
- Andres-Benito, P.; Moreno, J.; Dominguez, R.; Aso, E.; Povedano, M.; Ferrer, I. Inflammatory Gene Expression in Whole Peripheral Blood at Early Stages of Sporadic Amyotrophic Lateral Sclerosis. Front. Neurol. 2017, 8, 546. [Google Scholar] [CrossRef] [Green Version]
- Ramilo, O.; Allman, W.; Chung, W.; Mejias, A.; Ardura, M.; Glaser, C.; Wittkowski, K.M.; Piqueras, B.; Banchereau, J.; Palucka, A.K.; et al. Gene expression patterns in blood leukocytes discriminate patients with acute infections. Blood 2007, 109, 2066–2077. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.M.; Xavier, R.; Good, K.L.; Chtanova, T.; Newton, R.; Sisavanh, M.; Zimmer, S.; Deng, C.; Silva, D.G.; Frost, M.J.; et al. Immune cell transcriptome datasets reveal novel leukocyte subset-specific genes and genes associated with allergic processes. J. Allergy Clin. Immunol. 2006, 118, 496–503. [Google Scholar] [CrossRef]
- Lequerre, T.; Gauthier-Jauneau, A.C.; Bansard, C.; Derambure, C.; Hiron, M.; Vittecoq, O.; Daveau, M.; Mejjad, O.; Daragon, A.; Tron, F.; et al. Gene profiling in white blood cells predicts infliximab responsiveness in rheumatoid arthritis. Arthritis Res. Ther. 2006, 8, R105. [Google Scholar] [CrossRef] [Green Version]
- Godini, R.; Fallahi, H.; Ebrahimie, E. Network analysis of inflammatory responses to sepsis by neutrophils and peripheral blood mononuclear cells. PLoS ONE 2018, 13, e0201674. [Google Scholar] [CrossRef] [Green Version]
- Scicluna, B.P.; Uhel, F.; van Vught, L.A.; Wiewel, M.A.; Hoogendijk, A.J.; Baessman, I.; Franitza, M.; Nurnberg, P.; Horn, J.; Cremer, O.L.; et al. The leukocyte non-coding RNA landscape in critically ill patients with sepsis. Elife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Margraf, A.; Nussbaum, C.; Rohwedder, I.; Klapproth, S.; Kurz, A.R.M.; Florian, A.; Wiebking, V.; Pircher, J.; Pruenster, M.; Immler, R.; et al. Maturation of Platelet Function During Murine Fetal Development In Vivo. Arter. Thromb. Vasc. Biol. 2017, 37, 1076–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koseoglu, S.; Peters, C.G.; Fitch-Tewfik, J.L.; Aisiku, O.; Danglot, L.; Galli, T.; Flaumenhaft, R. VAMP-7 links granule exocytosis to actin reorganization during platelet activation. Blood 2015, 126, 651–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wersall, A.; Williams, C.M.; Brown, E.; Iannitti, T.; Williams, N.; Poole, A.W. Mouse Platelet Ral GTPases Control P-Selectin Surface Expression, Regulating Platelet-Leukocyte Interaction. Arter. Thromb. Vasc. Biol. 2018, 38, 787–800. [Google Scholar] [CrossRef] [Green Version]
- Rorvig, S.; Ostergaard, O.; Heegaard, N.H.; Borregaard, N. Proteome profiling of human neutrophil granule subsets, secretory vesicles, and cell membrane: Correlation with transcriptome profiling of neutrophil precursors. J. Leukoc. Biol. 2013, 94, 711–721. [Google Scholar] [CrossRef]
- Sheshachalam, A.; Srivastava, N.; Mitchell, T.; Lacy, P.; Eitzen, G. Granule protein processing and regulated secretion in neutrophils. Front. Immunol. 2014, 5, 448. [Google Scholar] [CrossRef] [Green Version]
- Margraf, A.; Zarbock, A. Platelets in Inflammation and Resolution. J. Immunol. 2019, 203, 2357–2367. [Google Scholar] [CrossRef]
- Margraf, A.; Lowell, C.A.; Zarbock, A. Neutrophils in acute inflammation: Current concepts and translational implications. Blood 2022, 139, 2130–2144. [Google Scholar] [CrossRef]
- Nagai, Y.; Garrett, K.P.; Ohta, S.; Bahrun, U.; Kouro, T.; Akira, S.; Takatsu, K.; Kincade, P.W. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity 2006, 24, 801–812. [Google Scholar] [CrossRef] [Green Version]
- Mitroulis, I.; Kalafati, L.; Bornhauser, M.; Hajishengallis, G.; Chavakis, T. Regulation of the Bone Marrow Niche by Inflammation. Front. Immunol. 2020, 11, 1540. [Google Scholar] [CrossRef]
- Frodermann, V.; Rohde, D.; Courties, G.; Severe, N.; Schloss, M.J.; Amatullah, H.; McAlpine, C.S.; Cremer, S.; Hoyer, F.F.; Ji, F.; et al. Exercise reduces inflammatory cell production and cardiovascular inflammation via instruction of hematopoietic progenitor cells. Nat. Med. 2019, 25, 1761–1771. [Google Scholar] [CrossRef] [PubMed]
- Weber, G.F.; Chousterman, B.G.; He, S.; Fenn, A.M.; Nairz, M.; Anzai, A.; Brenner, T.; Uhle, F.; Iwamoto, Y.; Robbins, C.S.; et al. Interleukin-3 amplifies acute inflammation and is a potential therapeutic target in sepsis. Science 2015, 347, 1260–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulte-Schrepping, J.; Reusch, N.; Paclik, D.; Bassler, K.; Schlickeiser, S.; Zhang, B.; Kramer, B.; Krammer, T.; Brumhard, S.; Bonaguro, L.; et al. Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell 2020, 182, 1419–1440.e1423. [Google Scholar] [CrossRef]
- Kwok, I.; Becht, E.; Xia, Y.; Ng, M.; Teh, Y.C.; Tan, L.; Evrard, M.; Li, J.L.Y.; Tran, H.T.N.; Tan, Y.; et al. Combinatorial Single-Cell Analyses of Granulocyte-Monocyte Progenitor Heterogeneity Reveals an Early Uni-potent Neutrophil Progenitor. Immunity 2020, 53, 303–318.e305. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Podstawka, J.; Lou, Y.; Li, L.; Lee, E.K.S.; Divangahi, M.; Petri, B.; Jirik, F.R.; Kelly, M.M.; Yipp, B.G. Aged polymorphonuclear leukocytes cause fibrotic interstitial lung disease in the absence of regulation by B cells. Nat. Immunol. 2018, 19, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Yipp, B.G.; Kim, J.H.; Lima, R.; Zbytnuik, L.D.; Petri, B.; Swanlund, N.; Ho, M.; Szeto, V.G.; Tak, T.; Koenderman, L.; et al. The Lung is a Host Defense Niche for Immediate Neutrophil-Mediated Vascular Protection. Sci. Immunol. 2017, 2, eaam8929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margraf, A.; Ley, K.; Zarbock, A. Neutrophil Recruitment: From Model Systems to Tissue-Specific Patterns. Trends Immunol. 2019, 40, 613–634. [Google Scholar] [CrossRef] [PubMed]
- Valet, C.; Magnen, M.; Qiu, L.; Cleary, S.J.; Wang, K.M.; Ranucci, S.; Grockowiak, E.; Boudra, R.; Conrad, C.; Seo, Y.; et al. Sepsis promotes splenic production of a protective platelet pool with high CD40 ligand expression. J. Clin. Investig. 2022, 132. [Google Scholar] [CrossRef]
- Rossaint, J.; Thomas, K.; Mersmann, S.; Skupski, J.; Margraf, A.; Tekath, T.; Jouvene, C.C.; Dalli, J.; Hidalgo, A.; Meuth, S.G.; et al. Platelets orchestrate the resolution of pulmonary inflammation in mice by T reg cell repositioning and macrophage education. J. Exp. Med. 2021, 218, e20201353. [Google Scholar] [CrossRef]
- Kamnev, A.; Lacouture, C.; Fusaro, M.; Dupre, L. Molecular Tuning of Actin Dynamics in Leukocyte Migration as Revealed by Immune-Related Actinopathies. Front. Immunol. 2021, 12, 750537. [Google Scholar] [CrossRef]
- Sprenkeler, E.G.G.; Webbers, S.D.S.; Kuijpers, T.W. When Actin is Not Actin’ Like It Should: A New Category of Distinct Primary Immunodeficiency Disorders. J. Innate Immun. 2021, 13, 3–25. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.; Yago, T.; Setiadi, H.; Wang, Y.; Mehta-D’souza, P.; Fu, J.; Crocker, P.R.; Rodgers, W.; Xia, L.; McEver, R.P. O-glycans direct selectin ligands to lipid rafts on leukocytes. Proc. Natl. Acad. Sci. USA 2015, 112, 8661–8666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabbri, M.; Di Meglio, S.; Gagliani, M.C.; Consonni, E.; Molteni, R.; Bender, J.R.; Tacchetti, C.; Pardi, R. Dynamic partitioning into lipid rafts controls the endo-exocytic cycle of the alphaL/beta2 integrin, LFA-1, during leukocyte chemotaxis. Mol. Biol. Cell 2005, 16, 5793–5803. [Google Scholar] [CrossRef] [Green Version]
- Poggi, P.; Mirabella, R.; Neri, S.; Assirelli, E.; Dolzani, P.; Mariani, E.; Calder, P.C.; Chatgilialoglu, A. Membrane fatty acid heterogeneity of leukocyte classes is altered during in vitro cultivation but can be restored with ad-hoc lipid supplementation. Lipids Health Dis. 2015, 14, 165. [Google Scholar] [CrossRef] [Green Version]
- Morgan, P.K.; Huynh, K.; Pernes, G.; Miotto, P.M.; Mellett, N.A.; Giles, C.; Meikle, P.J.; Murphy, A.J.; Lancaster, G.I. Macrophage polarization state affects lipid composition and the channeling of exogenous fatty acids into endogenous lipid pools. J. Biol. Chem. 2021, 297, 101341. [Google Scholar] [CrossRef] [PubMed]
- Benlachgar, N.; Doghmi, K.; Masrar, A.; Mahtat, E.M.; Harmouche, H.; Tazi Mezalek, Z. Immature platelets: A review of the available evidence. Thromb. Res. 2020, 195, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; McArdle, S.; Marki, A.; Mikulski, Z.; Gutierrez, E.; Engelhardt, B.; Deutsch, U.; Ginsberg, M.; Groisman, A.; Ley, K. Neutrophil recruitment limited by high-affinity bent beta2 integrin binding ligand in cis. Nat. Commun. 2016, 7, 12658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Z.; Kiosses, W.B.; Sun, H.; Orecchioni, M.; Ghosheh, Y.; Zajonc, D.M.; Arnaout, M.A.; Gutierrez, E.; Groisman, A.; Ginsberg, M.H.; et al. High-Affinity Bent beta2-Integrin Molecules in Arresting Neutrophils Face Each Other through Binding to ICAMs In cis. Cell Rep. 2019, 26, 119–130.e115. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Zhu, L.; Dudiki, T.; Gabanic, B.; Good, L.; Podrez, E.A.; Cherepanova, O.A.; Qin, J.; Byzova, T.V. Macrophage Migration and Phagocytosis Are Controlled by Kindlin-3’s Link to the Cytoskeleton. J. Immunol. 2020, 204, 1954–1967. [Google Scholar] [CrossRef]
- Xue, Z.H.; Feng, C.; Liu, W.L.; Tan, S.M. A role of kindlin-3 in integrin alphaMbeta2 outside-in signaling and the Syk-Vav1-Rac1/Cdc42 signaling axis. PLoS ONE 2013, 8, e56911. [Google Scholar] [CrossRef] [Green Version]
- Feng, C.; Li, Y.F.; Yau, Y.H.; Lee, H.S.; Tang, X.Y.; Xue, Z.H.; Zhou, Y.C.; Lim, W.M.; Cornvik, T.C.; Ruedl, C.; et al. Kindlin-3 mediates integrin alphaLbeta2 outside-in signaling, and it interacts with scaffold protein receptor for activated-C kinase 1 (RACK1). J. Biol. Chem. 2012, 287, 10714–10726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mocsai, A.; Walzog, B.; Lowell, C.A. Intracellular signalling during neutrophil recruitment. Cardiovasc. Res. 2015, 107, 373–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Vijaykumar, A.; Raghavan, J.V.; Rananaware, S.R.; Alakesh, A.; Bodele, J.; Rehman, J.U.; Shukla, S.; Wagde, V.; Nadig, S.; et al. Particle uptake driven phagocytosis in macrophages and neutrophils enhances bacterial clearance. J. Control. Release 2022, 343, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Welin, A.; Winberg, M.E.; Abdalla, H.; Sarndahl, E.; Rasmusson, B.; Stendahl, O.; Lerm, M. Incorporation of Mycobacterium tuberculosis lipoarabinomannan into macrophage membrane rafts is a prerequisite for the phagosomal maturation block. Infect. Immun. 2008, 76, 2882–2887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunin, P.; Nigrovic, P.A. Megakaryocytes as immune cells. J. Leukoc. Biol. 2019, 105, 1111–1121. [Google Scholar] [CrossRef]
- Cunin, P.; Bouslama, R.; Machlus, K.R.; Martinez-Bonet, M.; Lee, P.Y.; Wactor, A.; Nelson-Maney, N.; Morris, A.; Guo, L.; Weyrich, A.; et al. Megakaryocyte emperipolesis mediates membrane transfer from intracytoplasmic neutrophils to platelets. Elife 2019, 8, e44031. [Google Scholar] [CrossRef]
- Aroca-Crevillen, A.; Adrover, J.M.; Hidalgo, A. Circadian Features of Neutrophil Biology. Front. Immunol. 2020, 11, 576. [Google Scholar] [CrossRef]
- Nguyen, K.D.; Fentress, S.J.; Qiu, Y.; Yun, K.; Cox, J.S.; Chawla, A. Circadian gene Bmal1 regulates diurnal oscillations of Ly6C(hi) inflammatory monocytes. Science 2013, 341, 1483–1488. [Google Scholar] [CrossRef] [Green Version]
- Scheiermann, C.; Kunisaki, Y.; Lucas, D.; Chow, A.; Jang, J.E.; Zhang, D.; Hashimoto, D.; Merad, M.; Frenette, P.S. Adrenergic nerves govern circadian leukocyte recruitment to tissues. Immunity 2012, 37, 290–301. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Holtkamp, S.; Hergenhan, S.M.; Kraus, K.; de Juan, A.; Weber, J.; Bradfield, P.; Grenier, J.M.P.; Pelletier, J.; Druzd, D.; et al. Circadian Expression of Migratory Factors Establishes Lineage-Specific Signatures that Guide the Homing of Leukocyte Subsets to Tissues. Immunity 2018, 49, 1175–1190.e1177. [Google Scholar] [CrossRef] [Green Version]
- Holtkamp, S.J.; Ince, L.M.; Barnoud, C.; Schmitt, M.T.; Sinturel, F.; Pilorz, V.; Pick, R.; Jemelin, S.; Muhlstadt, M.; Boehncke, W.H.; et al. Circadian clocks guide dendritic cells into skin lymphatics. Nat. Immunol. 2021, 22, 1375–1381. [Google Scholar] [CrossRef]
- Druzd, D.; Matveeva, O.; Ince, L.; Harrison, U.; He, W.; Schmal, C.; Herzel, H.; Tsang, A.H.; Kawakami, N.; Leliavski, A.; et al. Lymphocyte Circadian Clocks Control Lymph Node Trafficking and Adaptive Immune Responses. Immunity 2017, 46, 120–132. [Google Scholar] [CrossRef] [Green Version]
- Pick, R.; He, W.; Chen, C.S.; Scheiermann, C. Time-of-Day-Dependent Trafficking and Function of Leukocyte Subsets. Trends Immunol. 2019, 40, 524–537. [Google Scholar] [CrossRef] [Green Version]
- Colas, R.A.; Souza, P.R.; Walker, M.E.; Burton, M.; Zaslona, Z.; Curtis, A.M.; Marques, R.M.; Dalli, J. Impaired Production and Diurnal Regulation of Vascular RvDn-3 DPA Increase Systemic Inflammation and Cardiovascular Disease. Circ. Res. 2018, 122, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Sperandio, M.; Quackenbush, E.J.; Sushkova, N.; Altstatter, J.; Nussbaum, C.; Schmid, S.; Pruenster, M.; Kurz, A.; Margraf, A.; Steppner, A.; et al. Ontogenetic regulation of leukocyte recruitment in mouse yolk sac vessels. Blood 2013, 121, e118–e128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, A.C.; Goldstein, D.R.; Montgomery, R.R. Age-dependent dysregulation of innate immunity. Nat. Rev. Immunol. 2013, 13, 875–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolich-Zugich, J. Author Correction: The twilight of immunity: Emerging concepts in aging of the immune system. Nat. Immunol. 2018, 19, 1146. [Google Scholar] [CrossRef]
- Barkaway, A.; Rolas, L.; Joulia, R.; Bodkin, J.; Lenn, T.; Owen-Woods, C.; Reglero-Real, N.; Stein, M.; Vazquez-Martinez, L.; Girbl, T.; et al. Age-related changes in the local milieu of inflamed tissues cause aberrant neutrophil trafficking and subsequent remote organ damage. Immunity 2021, 54, 1494–1510.e1497. [Google Scholar] [CrossRef]
- Nathan, P.; Gibbs, J.E.; Rainger, G.E.; Chimen, M. Changes in Circadian Rhythms Dysregulate Inflammation in Ageing: Focus on Leukocyte Trafficking. Front. Immunol. 2021, 12, 673405. [Google Scholar] [CrossRef]
- Kondratov, R.V.; Kondratova, A.A.; Gorbacheva, V.Y.; Vykhovanets, O.V.; Antoch, M.P. Early aging and age-related pathologies in mice deficient in BMAL1, the core componentof the circadian clock. Genes Dev. 2006, 20, 1868–1873. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Chen, G.; Manwani, D.; Mortha, A.; Xu, C.; Faith, J.J.; Burk, R.D.; Kunisaki, Y.; Jang, J.E.; Scheiermann, C.; et al. Neutrophil ageing is regulated by the microbiome. Nature 2015, 525, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Uhl, B.; Vadlau, Y.; Zuchtriegel, G.; Nekolla, K.; Sharaf, K.; Gaertner, F.; Massberg, S.; Krombach, F.; Reichel, C.A. Aged neutrophils contribute to the first line of defense in the acute inflammatory response. Blood 2016, 128, 2327–2337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adrover, J.M.; Del Fresno, C.; Crainiciuc, G.; Cuartero, M.I.; Casanova-Acebes, M.; Weiss, L.A.; Huerga-Encabo, H.; Silvestre-Roig, C.; Rossaint, J.; Cossio, I.; et al. A Neutrophil Timer Coordinates Immune Defense and Vascular Protection. Immunity 2019, 50, 390–402.e310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhabhar, F.S. Enhancing versus suppressive effects of stress on immune function: Implications for immunoprotection and immunopathology. Neuroimmunomodulation 2009, 16, 300–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhabhar, F.S.; McEwen, B.S. Enhancing versus suppressive effects of stress hormones on skin immune function. Proc. Natl. Acad. Sci. USA 1999, 96, 1059–1064. [Google Scholar] [CrossRef] [Green Version]
- McEwen, B.S. Protective and damaging effects of stress mediators. N. Engl. J. Med. 1998, 338, 171–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viswanathan, K.; Dhabhar, F.S. Stress-induced enhancement of leukocyte trafficking into sites of surgery or immune activation. Proc. Natl. Acad. Sci. USA 2005, 102, 5808–5813. [Google Scholar] [CrossRef] [Green Version]
- Maes, M.; Van Bockstaele, D.R.; Gastel, A.; Song, C.; Schotte, C.; Neels, H.; DeMeester, I.; Scharpe, S.; Janca, A. The effects of psychological stress on leukocyte subset distribution in humans: Evidence of immune activation. Neuropsychobiology 1999, 39, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ince, L.M.; Weber, J.; Scheiermann, C. Control of Leukocyte Trafficking by Stress-Associated Hormones. Front. Immunol. 2018, 9, 3143. [Google Scholar] [CrossRef] [Green Version]
- Cosentino, M.; Marino, F.; Maestroni, G.J. Sympathoadrenergic modulation of hematopoiesis: A review of available evidence and of therapeutic perspectives. Front. Cell Neurosci. 2015, 9, 302. [Google Scholar] [CrossRef]
- Buckley, C.D.; Ross, E.A.; McGettrick, H.M.; Osborne, C.E.; Haworth, O.; Schmutz, C.; Stone, P.C.; Salmon, M.; Matharu, N.M.; Vohra, R.K.; et al. Identification of a phenotypically and functionally distinct population of long-lived neutrophils in a model of reverse endothelial migration. J. Leukoc. Biol. 2006, 79, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Grieshaber-Bouyer, R.; Radtke, F.A.; Cunin, P.; Stifano, G.; Levescot, A.; Vijaykumar, B.; Nelson-Maney, N.; Blaustein, R.B.; Monach, P.A.; Nigrovic, P.A.; et al. The neutrotime transcriptional signature defines a single continuum of neutrophils across biological compartments. Nat. Commun. 2021, 12, 2856. [Google Scholar] [CrossRef] [PubMed]
- Damazo, A.S.; Yona, S.; Flower, R.J.; Perretti, M.; Oliani, S.M. Spatial and temporal profiles for anti-inflammatory gene expression in leukocytes during a resolving model of peritonitis. J. Immunol. 2006, 176, 4410–4418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damazo, A.S.; Yona, S.; D’Acquisto, F.; Flower, R.J.; Oliani, S.M.; Perretti, M. Critical protective role for annexin 1 gene expression in the endotoxemic murine microcirculation. Am. J. Pathol. 2005, 166, 1607–1617. [Google Scholar] [CrossRef] [Green Version]
- Babbin, B.A.; Laukoetter, M.G.; Nava, P.; Koch, S.; Lee, W.Y.; Capaldo, C.T.; Peatman, E.; Severson, E.A.; Flower, R.J.; Perretti, M.; et al. Annexin A1 regulates intestinal mucosal injury, inflammation, and repair. J. Immunol. 2008, 181, 5035–5044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nusbaum, P.; Laine, C.; Bouaouina, M.; Seveau, S.; Cramer, E.M.; Masse, J.M.; Lesavre, P.; Halbwachs-Mecarelli, L. Distinct signaling pathways are involved in leukosialin (CD43) down-regulation, membrane blebbing, and phospholipid scrambling during neutrophil apoptosis. J. Biol. Chem. 2005, 280, 5843–5853. [Google Scholar] [CrossRef] [Green Version]
- Graziano, B.R.; Town, J.P.; Sitarska, E.; Nagy, T.L.; Fosnaric, M.; Penic, S.; Iglic, A.; Kralj-Iglic, V.; Gov, N.S.; Diz-Munoz, A.; et al. Cell confinement reveals a branched-actin independent circuit for neutrophil polarity. PLoS Biol. 2019, 17, e3000457. [Google Scholar] [CrossRef] [Green Version]
- Babiychuk, E.B.; Monastyrskaya, K.; Potez, S.; Draeger, A. Blebbing confers resistance against cell lysis. Cell Death Differ. 2011, 18, 80–89. [Google Scholar] [CrossRef] [Green Version]
- Guilliams, M.; Svedberg, F.R. Does tissue imprinting restrict macrophage plasticity? Nat. Immunol. 2021, 22, 118–127. [Google Scholar] [CrossRef]
- Goldsmith, H.L.; Quinn, T.A.; Drury, G.; Spanos, C.; McIntosh, F.A.; Simon, S.I. Dynamics of neutrophil aggregation in couette flow revealed by videomicroscopy: Effect of shear rate on two-body collision efficiency and doublet lifetime. Biophys. J. 2001, 81, 2020–2034. [Google Scholar] [CrossRef] [Green Version]
- Moazzam, F.; DeLano, F.A.; Zweifach, B.W.; Schmid-Schonbein, G.W. The leukocyte response to fluid stress. Proc. Natl. Acad. Sci. USA 1997, 94, 5338–5343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cougoule, C.; Van Goethem, E.; Le Cabec, V.; Lafouresse, F.; Dupre, L.; Mehraj, V.; Mege, J.L.; Lastrucci, C.; Maridonneau-Parini, I. Blood leukocytes and macrophages of various phenotypes have distinct abilities to form podosomes and to migrate in 3D environments. Eur. J. Cell Biol. 2012, 91, 938–949. [Google Scholar] [CrossRef] [PubMed]
- Renkawitz, J.; Sixt, M. Mechanisms of force generation and force transmission during interstitial leukocyte migration. EMBO Rep. 2010, 11, 744–750. [Google Scholar] [CrossRef] [Green Version]
- Renkawitz, J.; Kopf, A.; Stopp, J.; de Vries, I.; Driscoll, M.K.; Merrin, J.; Hauschild, R.; Welf, E.S.; Danuser, G.; Fiolka, R.; et al. Nuclear positioning facilitates amoeboid migration along the path of least resistance. Nature 2019, 568, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Lupu, F.; Kinasewitz, G.; Dormer, K. The role of endothelial shear stress on haemodynamics, inflammation, coagulation and glycocalyx during sepsis. J. Cell Mol. Med. 2020, 24, 12258–12271. [Google Scholar] [CrossRef]
- Ploppa, A.; Schmidt, V.; Hientz, A.; Reutershan, J.; Haeberle, H.A.; Nohe, B. Mechanisms of leukocyte distribution during sepsis: An experimental study on the interdependence of cell activation, shear stress and endothelial injury. Crit. Care 2010, 14, R201. [Google Scholar] [CrossRef] [Green Version]
- Marki, A.; Buscher, K.; Lorenzini, C.; Meyer, M.; Saigusa, R.; Fan, Z.; Yeh, Y.T.; Hartmann, N.; Dan, J.M.; Kiosses, W.B.; et al. Elongated neutrophil-derived structures are blood-borne microparticles formed by rolling neutrophils during sepsis. J. Exp. Med. 2021, 218, e20200551. [Google Scholar] [CrossRef]
- Radley, G.; Ali, S.; Pieper, I.L.; Thornton, C.A. Mechanical shear stress and leukocyte phenotype and function: Implications for ventricular assist device development and use. Int. J. Artif. Organs 2019, 42, 133–142. [Google Scholar] [CrossRef]
- Margraf, A.; Ludwig, N.; Zarbock, A.; Rossaint, J. Systemic Inflammatory Response Syndrome After Surgery: Mechanisms and Protection. Anesth. Analg. 2020, 131, 1693–1707. [Google Scholar] [CrossRef]
- Schyns, J.; Bai, Q.; Ruscitti, C.; Radermecker, C.; De Schepper, S.; Chakarov, S.; Farnir, F.; Pirottin, D.; Ginhoux, F.; Boeckxstaens, G.; et al. Non-classical tissue monocytes and two functionally distinct populations of interstitial macrophages populate the mouse lung. Nat. Commun. 2019, 10, 3964. [Google Scholar] [CrossRef] [Green Version]
- Ley, K. M1 Means Kill; M2 Means Heal. J. Immunol. 2017, 199, 2191–2193. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Chavez-Galan, L.; Olleros, M.L.; Vesin, D.; Garcia, I. Much More than M1 and M2 Macrophages, There are also CD169(+) and TCR(+) Macrophages. Front. Immunol. 2015, 6, 263. [Google Scholar] [CrossRef]
- Rostam, H.M.; Reynolds, P.M.; Alexander, M.R.; Gadegaard, N.; Ghaemmaghami, A.M. Image based Machine Learning for identification of macrophage subsets. Sci. Rep. 2017, 7, 3521. [Google Scholar] [CrossRef] [PubMed]
- Krausgruber, T.; Blazek, K.; Smallie, T.; Alzabin, S.; Lockstone, H.; Sahgal, N.; Hussell, T.; Feldmann, M.; Udalova, I.A. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat. Immunol. 2011, 12, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Liang, H.; Zen, K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front. Immunol. 2014, 5, 614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, M.S.; Jung, D.Y.; Morel, C.; Lakhani, S.A.; Kim, J.K.; Flavell, R.A.; Davis, R.J. JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science 2013, 339, 218–222. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Xu, X.H.; Jin, L. Macrophage Polarization in Physiological and Pathological Pregnancy. Front. Immunol. 2019, 10, 792. [Google Scholar] [CrossRef]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Corrigendum: Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front. Immunol. 2020, 11, 234. [Google Scholar] [CrossRef]
- Wang, L.X.; Zhang, S.X.; Wu, H.J.; Rong, X.L.; Guo, J. M2b macrophage polarization and its roles in diseases. J. Leukoc. Biol. 2019, 106, 345–358. [Google Scholar] [CrossRef] [Green Version]
- Mehta, A.; Brewington, R.; Chatterji, M.; Zoubine, M.; Kinasewitz, G.T.; Peer, G.T.; Chang, A.C.; Taylor, F.B., Jr.; Shnyra, A. Infection-induced modulation of m1 and m2 phenotypes in circulating monocytes: Role in immune monitoring and early prognosis of sepsis. Shock 2004, 22, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Benoit, M.; Desnues, B.; Mege, J.L. Macrophage polarization in bacterial infections. J. Immunol. 2008, 181, 3733–3739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, X.; Sharma, N.; Kapadia, F.; Zhou, G.; Lu, Y.; Hong, H.; Paruchuri, K.; Mahabeleshwar, G.H.; Dalmas, E.; Venteclef, N.; et al. Kruppel-like factor 4 regulates macrophage polarization. J. Clin. Investig. 2011, 121, 2736–2749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Tang, J.; Shuai, W.; Meng, J.; Feng, J.; Han, Z. Macrophage polarization and its role in the pathogenesis of acute lung injury/acute respiratory distress syndrome. Inflamm. Res. 2020, 69, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Zhongqian, L.; Chunmei, S.; Pingfa, C.; Lei, H.; Qin, J.; Genhua, M.; Yijun, D. Activation of M1 macrophages in sepsis-induced acute kidney injury in response to heparin-binding protein. PLoS ONE 2018, 13, e0196423. [Google Scholar] [CrossRef] [Green Version]
- Lv, L.L.; Tang, P.M.; Li, C.J.; You, Y.K.; Li, J.; Huang, X.R.; Ni, J.; Feng, M.; Liu, B.C.; Lan, H.Y. The pattern recognition receptor, Mincle, is essential for maintaining the M1 macrophage phenotype in acute renal inflammation. Kidney Int. 2017, 91, 587–602. [Google Scholar] [CrossRef]
- Xu, F.; Ma, Y.; Huang, W.; Gao, J.; Guo, M.; Li, J.; Kong, L.; Liang, G.; Du, R.; Xu, Q.; et al. Typically inhibiting USP14 promotes autophagy in M1-like macrophages and alleviates CLP-induced sepsis. Cell Death Dis. 2020, 11, 666. [Google Scholar] [CrossRef]
- Rackov, G.; Hernandez-Jimenez, E.; Shokri, R.; Carmona-Rodriguez, L.; Manes, S.; Alvarez-Mon, M.; Lopez-Collazo, E.; Martinez, A.C.; Balomenos, D. p21 mediates macrophage reprogramming through regulation of p50-p50 NF-kappaB and IFN-beta. J. Clin. Investig. 2016, 126, 3089–3103. [Google Scholar] [CrossRef]
- Watanabe, N.; Suzuki, Y.; Inokuchi, S.; Inoue, S. Sepsis induces incomplete M2 phenotype polarization in peritoneal exudate cells in mice. J. Intensive Care 2016, 4, 6. [Google Scholar] [CrossRef] [Green Version]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Spite, M.; Norling, L.V.; Summers, L.; Yang, R.; Cooper, D.; Petasis, N.A.; Flower, R.J.; Perretti, M.; Serhan, C.N. Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature 2009, 461, 1287–1291. [Google Scholar] [CrossRef] [Green Version]
- Chiang, N.; Fredman, G.; Backhed, F.; Oh, S.F.; Vickery, T.; Schmidt, B.A.; Serhan, C.N. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature 2012, 484, 524–528. [Google Scholar] [CrossRef] [Green Version]
- Morita, M.; Kuba, K.; Ichikawa, A.; Nakayama, M.; Katahira, J.; Iwamoto, R.; Watanebe, T.; Sakabe, S.; Daidoji, T.; Nakamura, S.; et al. The lipid mediator protectin D1 inhibits influenza virus replication and improves severe influenza. Cell 2013, 153, 112–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bang, S.; Donnelly, C.R.; Luo, X.; Toro-Moreno, M.; Tao, X.; Wang, Z.; Chandra, S.; Bortsov, A.V.; Derbyshire, E.R.; Ji, R.R. Activation of GPR37 in macrophages confers protection against infection-induced sepsis and pain-like behaviour in mice. Nat. Commun. 2021, 12, 1704. [Google Scholar] [CrossRef] [PubMed]
- Bang, S.; Xie, Y.K.; Zhang, Z.J.; Wang, Z.; Xu, Z.Z.; Ji, R.R. GPR37 regulates macrophage phagocytosis and resolution of inflammatory pain. J. Clin. Investig. 2018, 128, 3568–3582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruckerl, D.; Campbell, S.M.; Duncan, S.; Sutherland, T.E.; Jenkins, S.J.; Hewitson, J.P.; Barr, T.A.; Jackson-Jones, L.H.; Maizels, R.M.; Allen, J.E. Macrophage origin limits functional plasticity in helminth-bacterial co-infection. PLoS Pathog. 2017, 13, e1006233. [Google Scholar] [CrossRef] [PubMed]
- Schyns, J.; Bureau, F.; Marichal, T. Lung Interstitial Macrophages: Past, Present, and Future. J. Immunol. Res. 2018, 2018, 5160794. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Looney, A.P.; Liu, L.; Wu, E.; Fong, V.; Hsu, A.; Chak, S.; Naikawadi, R.P.; Wolters, P.J.; Abate, A.R.; et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019, 20, 163–172. [Google Scholar] [CrossRef]
- Borges da Silva, H.; Fonseca, R.; Pereira, R.M.; Cassado Ados, A.; Alvarez, J.M.; D’Imperio Lima, M.R. Splenic Macrophage Subsets and Their Function during Blood-Borne Infections. Front. Immunol. 2015, 6, 480. [Google Scholar] [CrossRef] [Green Version]
- Ludewig, B.; Cervantes-Barragan, L. CD169(+) macrophages take the bullet. Nat. Immunol. 2011, 13, 13–14. [Google Scholar] [CrossRef]
- Honke, N.; Shaabani, N.; Cadeddu, G.; Sorg, U.R.; Zhang, D.E.; Trilling, M.; Klingel, K.; Sauter, M.; Kandolf, R.; Gailus, N.; et al. Enforced viral replication activates adaptive immunity and is essential for the control of a cytopathic virus. Nat. Immunol. 2011, 13, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Van der Heide, D.; Weiskirchen, R.; Bansal, R. Therapeutic Targeting of Hepatic Macrophages for the Treatment of Liver Diseases. Front. Immunol. 2019, 10, 2852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triantafyllou, E.; Pop, O.T.; Possamai, L.A.; Wilhelm, A.; Liaskou, E.; Singanayagam, A.; Bernsmeier, C.; Khamri, W.; Petts, G.; Dargue, R.; et al. MerTK expressing hepatic macrophages promote the resolution of inflammation in acute liver failure. Gut 2018, 67, 333–347. [Google Scholar] [CrossRef] [PubMed]
- MacParland, S.A.; Liu, J.C.; Ma, X.Z.; Innes, B.T.; Bartczak, A.M.; Gage, B.K.; Manuel, J.; Khuu, N.; Echeverri, J.; Linares, I.; et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun. 2018, 9, 4383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulsmans, M.; Sager, H.B.; Roh, J.D.; Valero-Munoz, M.; Houstis, N.E.; Iwamoto, Y.; Sun, Y.; Wilson, R.M.; Wojtkiewicz, G.; Tricot, B.; et al. Cardiac macrophages promote diastolic dysfunction. J. Exp. Med. 2018, 215, 423–440. [Google Scholar] [CrossRef]
- Hulsmans, M.; Clauss, S.; Xiao, L.; Aguirre, A.D.; King, K.R.; Hanley, A.; Hucker, W.J.; Wulfers, E.M.; Seemann, G.; Courties, G.; et al. Macrophages Facilitate Electrical Conduction in the Heart. Cell 2017, 169, 510–522.e520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakarov, S.; Lim, H.Y.; Tan, L.; Lim, S.Y.; See, P.; Lum, J.; Zhang, X.M.; Foo, S.; Nakamizo, S.; Duan, K.; et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science 2019, 363, eaau0964. [Google Scholar] [CrossRef]
- Dick, S.A.; Wong, A.; Hamidzada, H.; Nejat, S.; Nechanitzky, R.; Vohra, S.; Mueller, B.; Zaman, R.; Kantores, C.; Aronoff, L.; et al. Three tissue resident macrophage subsets coexist across organs with conserved origins and life cycles. Sci. Immunol. 2022, 7, eabf7777. [Google Scholar] [CrossRef]
- Hoyer, F.F.; Naxerova, K.; Schloss, M.J.; Hulsmans, M.; Nair, A.V.; Dutta, P.; Calcagno, D.M.; Herisson, F.; Anzai, A.; Sun, Y.; et al. Tissue-Specific Macrophage Responses to Remote Injury Impact the Outcome of Subsequent Local Immune Challenge. Immunity 2019, 51, 899–914.e897. [Google Scholar] [CrossRef]
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; de Bruijn, M.F.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015, 518, 547–551. [Google Scholar] [CrossRef]
- Stremmel, C.; Schuchert, R.; Wagner, F.; Thaler, R.; Weinberger, T.; Pick, R.; Mass, E.; Ishikawa-Ankerhold, H.C.; Margraf, A.; Hutter, S.; et al. Author Correction: Yolk sac macrophage progenitors traffic to the embryo during defined stages of development. Nat. Commun. 2018, 9, 3699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honold, L.; Nahrendorf, M. Resident and Monocyte-Derived Macrophages in Cardiovascular Disease. Circ. Res. 2018, 122, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Amann, L.; Monaco, G.; Sankowski, R.; Staszewski, O.; Krueger, M.; Del Gaudio, F.; He, L.; Paterson, N.; Nent, E.; et al. Specification of CNS macrophage subsets occurs postnatally in defined niches. Nature 2022, 604, 740–748. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.W. Current Understanding in Neutrophil Differentiation and Heterogeneity. Immune Netw. 2017, 17, 298–306. [Google Scholar] [CrossRef] [Green Version]
- Silvestre-Roig, C.; Hidalgo, A.; Soehnlein, O. Neutrophil heterogeneity: Implications for homeostasis and pathogenesis. Blood 2016, 127, 2173–2181. [Google Scholar] [CrossRef] [Green Version]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Ohms, M.; Moller, S.; Laskay, T. An Attempt to Polarize Human Neutrophils Toward N1 and N2 Phenotypes in vitro. Front. Immunol. 2020, 11, 532. [Google Scholar] [CrossRef]
- Cuartero, M.I.; Ballesteros, I.; Moraga, A.; Nombela, F.; Vivancos, J.; Hamilton, J.A.; Corbi, A.L.; Lizasoain, I.; Moro, M.A. N2 neutrophils, novel players in brain inflammation after stroke: Modulation by the PPARgamma agonist rosiglitazone. Stroke 2013, 44, 3498–3508. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Yabluchanskiy, A.; Iyer, R.P.; Cannon, P.L.; Flynn, E.R.; Jung, M.; Henry, J.; Cates, C.A.; Deleon-Pennell, K.Y.; Lindsey, M.L. Temporal neutrophil polarization following myocardial infarction. Cardiovasc. Res. 2016, 110, 51–61. [Google Scholar] [CrossRef] [Green Version]
- Headland, S.E.; Jones, H.R.; Norling, L.V.; Kim, A.; Souza, P.R.; Corsiero, E.; Gil, C.D.; Nerviani, A.; Dell’Accio, F.; Pitzalis, C.; et al. Neutrophil-derived microvesicles enter cartilage and protect the joint in inflammatory arthritis. Sci. Transl. Med. 2015, 7, 315ra190. [Google Scholar] [CrossRef]
- Scapini, P.; Marini, O.; Tecchio, C.; Cassatella, M.A. Human neutrophils in the saga of cellular heterogeneity: Insights and open questions. Immunol. Rev. 2016, 273, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Hardisty, G.R.; Llanwarne, F.; Minns, D.; Gillan, J.L.; Davidson, D.J.; Gwyer Findlay, E.; Gray, R.D. High Purity Isolation of Low Density Neutrophils Casts Doubt on Their Exceptionality in Health and Disease. Front. Immunol. 2021, 12, 625922. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, S.M.; Geller, A.E.; Hu, X.; Tieri, D.; Ding, C.; Klaes, C.K.; Cooke, E.A.; Woeste, M.R.; Martin, Z.C.; Chen, O.; et al. A specific low-density neutrophil population correlates with hypercoagulation and disease severity in hospitalized COVID-19 patients. JCI Insight 2021, 6, e148435. [Google Scholar] [CrossRef] [PubMed]
- Evrard, M.; Kwok, I.W.H.; Chong, S.Z.; Teng, K.W.W.; Becht, E.; Chen, J.; Sieow, J.L.; Penny, H.L.; Ching, G.C.; Devi, S.; et al. Developmental Analysis of Bone Marrow Neutrophils Reveals Populations Specialized in Expansion, Trafficking, and Effector Functions. Immunity 2018, 48, 364–379.e368. [Google Scholar] [CrossRef] [Green Version]
- Guerin, E.; Orabona, M.; Raquil, M.A.; Giraudeau, B.; Bellier, R.; Gibot, S.; Bene, M.C.; Lacombe, F.; Droin, N.; Solary, E.; et al. Circulating immature granulocytes with T-cell killing functions predict sepsis deterioration*. Crit. Care Med. 2014, 42, 2007–2018. [Google Scholar] [CrossRef]
- Pillay, J.; Kamp, V.M.; van Hoffen, E.; Visser, T.; Tak, T.; Lammers, J.W.; Ulfman, L.H.; Leenen, L.P.; Pickkers, P.; Koenderman, L. A subset of neutrophils in human systemic inflammation inhibits T cell responses through Mac-1. J. Clin. Investig. 2012, 122, 327–336. [Google Scholar] [CrossRef]
- Kamp, V.M.; Pillay, J.; Lammers, J.W.; Pickkers, P.; Ulfman, L.H.; Koenderman, L. Human suppressive neutrophils CD16bright/CD62Ldim exhibit decreased adhesion. J. Leukoc. Biol. 2012, 92, 1011–1020. [Google Scholar] [CrossRef]
- Millrud, C.R.; Kagedal, A.; Kumlien Georen, S.; Winqvist, O.; Uddman, R.; Razavi, R.; Munck-Wikland, E.; Cardell, L.O. NET-producing CD16(high) CD62L(dim) neutrophils migrate to tumor sites and predict improved survival in patients with HNSCC. Int. J. Cancer 2017, 140, 2557–2567. [Google Scholar] [CrossRef] [Green Version]
- Pillay, J.; Ramakers, B.P.; Kamp, V.M.; Loi, A.L.; Lam, S.W.; Hietbrink, F.; Leenen, L.P.; Tool, A.T.; Pickkers, P.; Koenderman, L. Functional heterogeneity and differential priming of circulating neutrophils in human experimental endotoxemia. J. Leukoc. Biol. 2010, 88, 211–220. [Google Scholar] [CrossRef]
- Grieshaber-Bouyer, R.; Nigrovic, P.A. Neutrophil Heterogeneity as Therapeutic Opportunity in Immune-Mediated Disease. Front. Immunol. 2019, 10, 346. [Google Scholar] [CrossRef] [Green Version]
- Bai, M.; Grieshaber-Bouyer, R.; Wang, J.; Schmider, A.B.; Wilson, Z.S.; Zeng, L.; Halyabar, O.; Godin, M.D.; Nguyen, H.N.; Levescot, A.; et al. CD177 modulates human neutrophil migration through activation-mediated integrin and chemoreceptor regulation. Blood 2017, 130, 2092–2100. [Google Scholar] [CrossRef] [PubMed]
- Levy, Y.; Wiedemann, A.; Hejblum, B.P.; Durand, M.; Lefebvre, C.; Surenaud, M.; Lacabaratz, C.; Perreau, M.; Foucat, E.; Dechenaud, M.; et al. CD177, a specific marker of neutrophil activation, is associated with coronavirus disease 2019 severity and death. iScience 2021, 24, 102711. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Yu, L.; Fang, L.; Yang, W.; Yu, T.; Miao, Y.; Chen, M.; Wu, K.; Chen, F.; Cong, Y.; et al. CD177(+) neutrophils as functionally activated neutrophils negatively regulate IBD. Gut 2018, 67, 1052–1063. [Google Scholar] [CrossRef] [PubMed]
- Nourshargh, S.; Renshaw, S.A.; Imhof, B.A. Reverse Migration of Neutrophils: Where, When, How, and Why? Trends Immunol. 2016, 37, 273–286. [Google Scholar] [CrossRef]
- Ji, J.; Fan, J. Neutrophil in Reverse Migration: Role in Sepsis. Front. Immunol. 2021, 12, 656039. [Google Scholar] [CrossRef]
- Fischer, J.; Walter, C.; Tonges, A.; Aleth, H.; Jordao, M.J.C.; Leddin, M.; Groning, V.; Erdmann, T.; Lenz, G.; Roth, J.; et al. Safeguard function of PU.1 shapes the inflammatory epigenome of neutrophils. Nat. Immunol. 2019, 20, 546–558. [Google Scholar] [CrossRef]
- Kienle, K.; Glaser, K.M.; Eickhoff, S.; Mihlan, M.; Knopper, K.; Reategui, E.; Epple, M.W.; Gunzer, M.; Baumeister, R.; Tarrant, T.K.; et al. Neutrophils self-limit swarming to contain bacterial growth in vivo. Science 2021, 372, eabe7729. [Google Scholar] [CrossRef]
- Lammermann, T.; Afonso, P.V.; Angermann, B.R.; Wang, J.M.; Kastenmuller, W.; Parent, C.A.; Germain, R.N. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature 2013, 498, 371–375. [Google Scholar] [CrossRef]
- Adrover, J.M.; Aroca-Crevillen, A.; Crainiciuc, G.; Ostos, F.; Rojas-Vega, Y.; Rubio-Ponce, A.; Cilloniz, C.; Bonzon-Kulichenko, E.; Calvo, E.; Rico, D.; et al. Programmed ‘disarming’ of the neutrophil proteome reduces the magnitude of inflammation. Nat. Immunol. 2020, 21, 135–144. [Google Scholar] [CrossRef]
- Xie, X.; Shi, Q.; Wu, P.; Zhang, X.; Kambara, H.; Su, J.; Yu, H.; Park, S.Y.; Guo, R.; Ren, Q.; et al. Single-cell transcriptome profiling reveals neutrophil heterogeneity in homeostasis and infection. Nat. Immunol. 2020, 21, 1119–1133. [Google Scholar] [CrossRef]
- Leslie, M. Cell biology. Beyond clotting: The powers of platelets. Science 2010, 328, 562–564. [Google Scholar] [CrossRef] [PubMed]
- Rossaint, J.; Margraf, A.; Zarbock, A. Role of Platelets in Leukocyte Recruitment and Resolution of Inflammation. Front. Immunol. 2018, 9, 2712. [Google Scholar] [CrossRef] [PubMed]
- Nicolai, L.; Massberg, S. Platelets as key players in inflammation and infection. Curr. Opin. Hematol. 2020, 27, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Karpatkin, S. Heterogeneity of human platelets. II. Functional evidence suggestive of young and old platelets. J. Clin. Investig. 1969, 48, 1083–1087. [Google Scholar] [CrossRef] [PubMed]
- Lesyk, G.; Jurasz, P. Advances in Platelet Subpopulation Research. Front. Cardiovasc. Med. 2019, 6, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moraes, L.A.; Paul-Clark, M.J.; Rickman, A.; Flower, R.J.; Goulding, N.J.; Perretti, M. Ligand-specific glucocorticoid receptor activation in human platelets. Blood 2005, 106, 4167–4175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liverani, E.; Banerjee, S.; Roberts, W.; Naseem, K.M.; Perretti, M. Prednisolone exerts exquisite inhibitory properties on platelet functions. Biochem. Pharmacol. 2012, 83, 1364–1373. [Google Scholar] [CrossRef] [Green Version]
- Lindemann, S.; Tolley, N.D.; Dixon, D.A.; McIntyre, T.M.; Prescott, S.M.; Zimmerman, G.A.; Weyrich, A.S. Activated platelets mediate inflammatory signaling by regulated interleukin 1beta synthesis. J. Cell Biol. 2001, 154, 485–490. [Google Scholar] [CrossRef] [Green Version]
- Manne, B.K.; Xiang, S.C.; Rondina, M.T. Platelet secretion in inflammatory and infectious diseases. Platelets 2017, 28, 155–164. [Google Scholar] [CrossRef]
- Oggero, S.; de Gaetano, M.; Marcone, S.; Fitzsimons, S.; Pinto, A.L.; Ikramova, D.; Barry, M.; Burke, D.; Montero-Melendez, T.; Cooper, D.; et al. Extracellular vesicles from monocyte/platelet aggregates modulate human atherosclerotic plaque reactivity. J. Extracell. Vesicles 2021, 10, 12084. [Google Scholar] [CrossRef]
- Boilard, E.; Nigrovic, P.A.; Larabee, K.; Watts, G.F.; Coblyn, J.S.; Weinblatt, M.E.; Massarotti, E.M.; Remold-O’Donnell, E.; Farndale, R.W.; Ware, J.; et al. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science 2010, 327, 580–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, A.P.; Nikols, E.; Freire, D.; Machlus, K.R. The pathobiology of platelet and megakaryocyte extracellular vesicles: A (c)lot has changed. J. Thromb. Haemost. 2022. [Google Scholar] [CrossRef] [PubMed]
- Puhm, F.; Boilard, E.; Machlus, K.R. Platelet Extracellular Vesicles: Beyond the Blood. Arter. Thromb. Vasc. Biol. 2021, 41, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Pick, M.; Perry, C.; Lapidot, T.; Guimaraes-Sternberg, C.; Naparstek, E.; Deutsch, V.; Soreq, H. Stress-induced cholinergic signaling promotes inflammation-associated thrombopoiesis. Blood 2006, 107, 3397–3406. [Google Scholar] [CrossRef] [Green Version]
- Kaser, A.; Brandacher, G.; Steurer, W.; Kaser, S.; Offner, F.A.; Zoller, H.; Theurl, I.; Widder, W.; Molnar, C.; Ludwiczek, O.; et al. Interleukin-6 stimulates thrombopoiesis through thrombopoietin: Role in inflammatory thrombocytosis. Blood 2001, 98, 2720–2725. [Google Scholar] [CrossRef] [Green Version]
- Koyama, K.; Katayama, S.; Muronoi, T.; Tonai, K.; Goto, Y.; Koinuma, T.; Shima, J.; Nunomiya, S. Time course of immature platelet count and its relation to thrombocytopenia and mortality in patients with sepsis. PLoS ONE 2018, 13, e0192064. [Google Scholar] [CrossRef] [Green Version]
- Park, S.H.; Ha, S.O.; Cho, Y.U.; Park, C.J.; Jang, S.; Hong, S.B. Immature platelet fraction in septic patients: Clinical relevance of immature platelet fraction is limited to the sensitive and accurate discrimination of septic patients from non-septic patients, not to the discrimination of sepsis severity. Ann. Lab. Med. 2016, 36, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Enz Hubert, R.M.; Rodrigues, M.V.; Andreguetto, B.D.; Santos, T.M.; de Fatima Pereira Gilberti, M.; de Castro, V.; Annichino-Bizzacchi, J.M.; Dragosavac, D.; Carvalho-Filho, M.A.; De Paula, E.V. Association of the immature platelet fraction with sepsis diagnosis and severity. Sci. Rep. 2015, 5, 8019. [Google Scholar] [CrossRef] [Green Version]
- Imperiali, C.E.; Lopez-Delgado, J.C.; Dastis-Arias, M.; Sanchez-Navarro, L. Evaluation of the delta of immature platelet fraction as a predictive biomarker of inflammatory response after cardiac surgery. J. Clin. Pathol. 2020, 73, 335–340. [Google Scholar] [CrossRef]
- Nishimura, S.; Nagasaki, M.; Kunishima, S.; Sawaguchi, A.; Sakata, A.; Sakaguchi, H.; Ohmori, T.; Manabe, I.; Italiano, J.E., Jr.; Ryu, T.; et al. IL-1alpha induces thrombopoiesis through megakaryocyte rupture in response to acute platelet needs. J. Cell Biol. 2015, 209, 453–466. [Google Scholar] [CrossRef] [Green Version]
- Lefrancais, E.; Ortiz-Munoz, G.; Caudrillier, A.; Mallavia, B.; Liu, F.C.; Sayah, D.M.; Thornton, E.E.; Headley, M.B.; David, T.; Coughlin, S.R.; et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature 2017, 544, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Friese, P.; Heilmann, E.; George, J.N.; Burstein, S.A.; Dale, G.L. Aged platelets have an impaired response to thrombin as quantitated by P-selectin expression. Blood 1994, 83, 161–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allan, H.E.; Hayman, M.A.; Marcone, S.; Chan, M.V.; Edin, M.L.; Maffucci, T.; Joshi, A.; Menke, L.; Crescente, M.; Mayr, M.; et al. Proteome and functional decline as platelets age in the circulation. J. Thromb. Haemost. 2021, 19, 3095–3112. [Google Scholar] [CrossRef] [PubMed]
- Grozovsky, R.; Begonja, A.J.; Liu, K.; Visner, G.; Hartwig, J.H.; Falet, H.; Hoffmeister, K.M. The Ashwell-Morell receptor regulates hepatic thrombopoietin production via JAK2-STAT3 signaling. Nat. Med. 2015, 21, 47–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kile, B.T. Aging platelets stimulate TPO production. Nat. Med. 2015, 21, 11–12. [Google Scholar] [CrossRef]
- Radziwon-Balicka, A.; Lesyk, G.; Back, V.; Fong, T.; Loredo-Calderon, E.L.; Dong, B.; El-Sikhry, H.; El-Sherbeni, A.A.; El-Kadi, A.; Ogg, S.; et al. Differential eNOS-signalling by platelet subpopulations regulates adhesion and aggregation. Cardiovasc. Res. 2017, 113, 1719–1731. [Google Scholar] [CrossRef]
- Prodan, C.I.; Joseph, P.M.; Vincent, A.S.; Dale, G.L. Coated-platelets in ischemic stroke: Differences between lacunar and cortical stroke. J. Thromb. Haemost. 2008, 6, 609–614. [Google Scholar] [CrossRef]
- Alberio, L.; Safa, O.; Clemetson, K.J.; Esmon, C.T.; Dale, G.L. Surface expression and functional characterization of alpha-granule factor V in human platelets: Effects of ionophore A23187, thrombin, collagen, and convulxin. Blood 2000, 95, 1694–1702. [Google Scholar] [CrossRef]
- Dale, G.L.; Friese, P.; Batar, P.; Hamilton, S.F.; Reed, G.L.; Jackson, K.W.; Clemetson, K.J.; Alberio, L. Stimulated platelets use serotonin to enhance their retention of procoagulant proteins on the cell surface. Nature 2002, 415, 175–179. [Google Scholar] [CrossRef]
- Hindle, M.S.; Spurgeon, B.E.J.; Cheah, L.T.; Webb, B.A.; Naseem, K.M. Multidimensional flow cytometry reveals novel platelet subpopulations in response to prostacyclin. J. Thromb. Haemost. 2021, 19, 1800–1812. [Google Scholar] [CrossRef]
- Dunster, J.L.; Bye, A.P.; Kriek, N.; Sage, T.; Mitchell, J.L.; Kempster, C.; Batista, J.; McKinney, H.; Thomas, P.; Jones, C.I.; et al. Multiparameter phenotyping of platelet reactivity for stratification of human cohorts. Blood Adv. 2021, 5, 4017–4030. [Google Scholar] [CrossRef] [PubMed]
- Aslan, J.E. Platelet Proteomes, Pathways, and Phenotypes as Informants of Vascular Wellness and Disease. Arter. Thromb. Vasc. Biol. 2021, 41, 999–1011. [Google Scholar] [CrossRef] [PubMed]
- Senchenkova, E.Y.; Ansari, J.; Becker, F.; Vital, S.A.; Al-Yafeai, Z.; Sparkenbaugh, E.M.; Pawlinski, R.; Stokes, K.Y.; Carroll, J.L.; Dragoi, A.M.; et al. Novel Role for the AnxA1-Fpr2/ALX Signaling Axis as a Key Regulator of Platelet Function to Promote Resolution of Inflammation. Circulation 2019, 140, 319–335. [Google Scholar] [CrossRef]
- Salamah, M.F.; Ravishankar, D.; Kodji, X.; Moraes, L.A.; Williams, H.F.; Vallance, T.M.; Albadawi, D.A.; Vaiyapuri, R.; Watson, K.; Gibbins, J.M.; et al. The endogenous antimicrobial cathelicidin LL37 induces platelet activation and augments thrombus formation. Blood Adv. 2018, 2, 2973–2985. [Google Scholar] [CrossRef] [PubMed]
- Lefrancais, E.; Looney, M.R. Platelet Biogenesis in the Lung Circulation. Physiology (Bethesda) 2019, 34, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Masucci, M.T.; Minopoli, M.; Carriero, M.V. Tumor Associated Neutrophils. Their Role in Tumorigenesis, Metastasis, Prognosis and Therapy. Front. Oncol. 2019, 9, 1146. [Google Scholar] [CrossRef] [Green Version]
- Lapenna, A.; De Palma, M.; Lewis, C.E. Author Correction: Perivascular macrophages in health and disease. Nat. Rev. Immunol. 2021, 21, 752. [Google Scholar] [CrossRef]
- Hughes, C.E.; Nibbs, R.J.B. A guide to chemokines and their receptors. FEBS J. 2018, 285, 2944–2971. [Google Scholar] [CrossRef]
- Jin, J.; Xiao, Y.; Hu, H.; Zou, Q.; Li, Y.; Gao, Y.; Ge, W.; Cheng, X.; Sun, S.C. Proinflammatory TLR signalling is regulated by a TRAF2-dependent proteolysis mechanism in macrophages. Nat. Commun. 2015, 6, 5930. [Google Scholar] [CrossRef] [Green Version]
- Swanson, L.; Katkar, G.D.; Tam, J.; Pranadinata, R.F.; Chareddy, Y.; Coates, J.; Anandachar, M.S.; Castillo, V.; Olson, J.; Nizet, V.; et al. TLR4 signaling and macrophage inflammatory responses are dampened by GIV/Girdin. Proc. Natl. Acad. Sci. USA 2020, 117, 26895–26906. [Google Scholar] [CrossRef]
- Nadra, I.; Mason, J.C.; Philippidis, P.; Florey, O.; Smythe, C.D.; McCarthy, G.M.; Landis, R.C.; Haskard, D.O. Proinflammatory activation of macrophages by basic calcium phosphate crystals via protein kinase C and MAP kinase pathways: A vicious cycle of inflammation and arterial calcification? Circ. Res. 2005, 96, 1248–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Kubes, P. A Reservoir of Mature Cavity Macrophages that Can Rapidly Invade Visceral Organs to Affect Tissue Repair. Cell 2016, 165, 668–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okabe, Y.; Medzhitov, R. Tissue-specific signals control reversible program of localization and functional polarization of macrophages. Cell 2014, 157, 832–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruffell, D.; Mourkioti, F.; Gambardella, A.; Kirstetter, P.; Lopez, R.G.; Rosenthal, N.; Nerlov, C. A CREB-C/EBPbeta cascade induces M2 macrophage-specific gene expression and promotes muscle injury repair. Proc. Natl. Acad. Sci. USA 2009, 106, 17475–17480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, T.; Natoli, G. Transcriptional regulation of macrophage polarization: Enabling diversity with identity. Nat. Rev. Immunol. 2011, 11, 750–761. [Google Scholar] [CrossRef] [PubMed]
- Cheung, P.; Schaffert, S.; Chang, S.E.; Dvorak, M.; Donato, M.; Macaubas, C.; Foecke, M.H.; Li, T.M.; Zhang, L.; Coan, J.P.; et al. Repression of CTSG, ELANE and PRTN3-mediated histone H3 proteolytic cleavage promotes monocyte-to-macrophage differentiation. Nat. Immunol. 2021, 22, 711–722. [Google Scholar] [CrossRef]
- Gabanyi, I.; Muller, P.A.; Feighery, L.; Oliveira, T.Y.; Costa-Pinto, F.A.; Mucida, D. Neuro-immune Interactions Drive Tissue Programming in Intestinal Macrophages. Cell 2016, 164, 378–391. [Google Scholar] [CrossRef] [Green Version]
- Lavin, Y.; Winter, D.; Blecher-Gonen, R.; David, E.; Keren-Shaul, H.; Merad, M.; Jung, S.; Amit, I. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 2014, 159, 1312–1326. [Google Scholar] [CrossRef] [Green Version]
- Gosselin, D.; Link, V.M.; Romanoski, C.E.; Fonseca, G.J.; Eichenfield, D.Z.; Spann, N.J.; Stender, J.D.; Chun, H.B.; Garner, H.; Geissmann, F.; et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 2014, 159, 1327–1340. [Google Scholar] [CrossRef]
- Orecchioni, M.; Kobiyama, K.; Winkels, H.; Ghosheh, Y.; McArdle, S.; Mikulski, Z.; Kiosses, W.B.; Fan, Z.; Wen, L.; Jung, Y.; et al. Olfactory receptor 2 in vascular macrophages drives atherosclerosis by NLRP3-dependent IL-1 production. Science 2022, 375, 214–221. [Google Scholar] [CrossRef]
- Woytschak, J.; Keller, N.; Krieg, C.; Impellizzieri, D.; Thompson, R.W.; Wynn, T.A.; Zinkernagel, A.S.; Boyman, O. Type 2 Interleukin-4 Receptor Signaling in Neutrophils Antagonizes Their Expansion and Migration during Infection and Inflammation. Immunity 2016, 45, 172–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balasubbramanian, D.; Goodlett, B.L.; Mitchell, B.M. Is IL-12 pro-inflammatory or anti-inflammatory? Depends on the blood pressure. Cardiovasc. Res. 2019, 115, 998–999. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Que, B.; Huang, Y.; Lin, Y.; Chen, J.; Liu, L.; Shi, Y.; Wang, Y.; Wang, M.; Zeng, T.; et al. Interleukin-12p35 knockout promotes macrophage differentiation, aggravates vascular dysfunction, and elevates blood pressure in angiotensin II-infused mice. Cardiovasc. Res. 2019, 115, 1102–1113. [Google Scholar] [CrossRef] [PubMed]
- Kourtzelis, I.; Li, X.; Mitroulis, I.; Grosser, D.; Kajikawa, T.; Wang, B.; Grzybek, M.; von Renesse, J.; Czogalla, A.; Troullinaki, M.; et al. DEL-1 promotes macrophage efferocytosis and clearance of inflammation. Nat. Immunol. 2019, 20, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Jetten, N.; Roumans, N.; Gijbels, M.J.; Romano, A.; Post, M.J.; de Winther, M.P.; van der Hulst, R.R.; Xanthoulea, S. Wound administration of M2-polarized macrophages does not improve murine cutaneous healing responses. PLoS ONE 2014, 9, e102994. [Google Scholar] [CrossRef] [Green Version]
- Neupane, A.S.; Willson, M.; Chojnacki, A.K.; Vargas, E.S.C.F.; Morehouse, C.; Carestia, A.; Keller, A.E.; Peiseler, M.; DiGiandomenico, A.; Kelly, M.M.; et al. Patrolling Alveolar Macrophages Conceal Bacteria from the Immune System to Maintain Homeostasis. Cell 2020, 183, 110–125.e111. [Google Scholar] [CrossRef] [PubMed]
- Uderhardt, S.; Martins, A.J.; Tsang, J.S.; Lammermann, T.; Germain, R.N. Resident Macrophages Cloak Tissue Microlesions to Prevent Neutrophil-Driven Inflammatory Damage. Cell 2019, 177, 541–555.e517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, B.; Pittman, K.; Menezes, G.B.; Hirota, S.A.; Slaba, I.; Waterhouse, C.C.; Beck, P.L.; Muruve, D.A.; Kubes, P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 2010, 330, 362–366. [Google Scholar] [CrossRef]
- Volmering, S.; Block, H.; Boras, M.; Lowell, C.A.; Zarbock, A. The Neutrophil Btk Signalosome Regulates Integrin Activation during Sterile Inflammation. Immunity 2016, 44, 73–87. [Google Scholar] [CrossRef] [Green Version]
- Honda, F.; Kano, H.; Kanegane, H.; Nonoyama, S.; Kim, E.S.; Lee, S.K.; Takagi, M.; Mizutani, S.; Morio, T. The kinase Btk negatively regulates the production of reactive oxygen species and stimulation-induced apoptosis in human neutrophils. Nat. Immunol. 2012, 13, 369–378. [Google Scholar] [CrossRef]
- Bromberger, T.; Klapproth, S.; Rohwedder, I.; Weber, J.; Pick, R.; Mittmann, L.; Min-Weissenhorn, S.J.; Reichel, C.A.; Scheiermann, C.; Sperandio, M.; et al. Binding of Rap1 and Riam to Talin1 Fine-Tune beta2 Integrin Activity During Leukocyte Trafficking. Front. Immunol. 2021, 12, 702345. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shiratori, I.; Uehori, J.; Ikawa, M.; Arase, H. Neutrophil infiltration during inflammation is regulated by PILRalpha via modulation of integrin activation. Nat. Immunol. 2013, 14, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Margraf, A.; Volmering, S.; Skupski, J.; Van Marck, V.; Makrigiannis, A.P.; Block, H.; Zarbock, A. The ITIM Domain-Containing NK Receptor Ly49Q Impacts Pulmonary Infection by Mediating Neutrophil Functions. J. Immunol. 2018, 200, 4085–4093. [Google Scholar] [CrossRef] [PubMed]
- Heit, B.; Robbins, S.M.; Downey, C.M.; Guan, Z.; Colarusso, P.; Miller, B.J.; Jirik, F.R.; Kubes, P. PTEN functions to ‘prioritize’ chemotactic cues and prevent ‘distraction’ in migrating neutrophils. Nat. Immunol. 2008, 9, 743–752. [Google Scholar] [CrossRef]
- Xu, J.; Gao, X.P.; Ramchandran, R.; Zhao, Y.Y.; Vogel, S.M.; Malik, A.B. Nonmuscle myosin light-chain kinase mediates neutrophil transmigration in sepsis-induced lung inflammation by activating beta2 integrins. Nat. Immunol. 2008, 9, 880–886. [Google Scholar] [CrossRef] [Green Version]
- Immler, R.; Nadolni, W.; Bertsch, A.; Morikis, V.; Rohwedder, I.; Masgrau-Alsina, S.; Schroll, T.; Yevtushenko, A.; Soehnlein, O.; Moser, M.; et al. The voltage-gated potassium channel KV1.3 regulates neutrophil recruitment during inflammation. Cardiovasc. Res. 2022, 118, 1289–1302. [Google Scholar] [CrossRef]
- Clemens, R.A.; Chong, J.; Grimes, D.; Hu, Y.; Lowell, C.A. STIM1 and STIM2 cooperatively regulate mouse neutrophil store-operated calcium entry and cytokine production. Blood 2017, 130, 1565–1577. [Google Scholar] [CrossRef]
- Clemens, R.A.; Lowell, C.A. Store-operated calcium signaling in neutrophils. J. Leukoc. Biol. 2015, 98, 497–502. [Google Scholar] [CrossRef] [Green Version]
- Grimes, D.; Johnson, R.; Pashos, M.; Cummings, C.; Kang, C.; Sampedro, G.R.; Tycksen, E.; McBride, H.J.; Sah, R.; Lowell, C.A.; et al. ORAI1 and ORAI2 modulate murine neutrophil calcium signaling, cellular activation, and host defense. Proc. Natl. Acad. Sci. USA 2020, 117, 24403–24414. [Google Scholar] [CrossRef]
- Li, J.; Kumari, T.; Barazia, A.; Jha, V.; Jeong, S.Y.; Olson, A.; Kim, M.; Lee, B.K.; Manickam, V.; Song, Z.; et al. Neutrophil DREAM promotes neutrophil recruitment in vascular inflammation. J. Exp. Med. 2022, 219, e20211083. [Google Scholar] [CrossRef]
- Khoyratty, T.E.; Ai, Z.; Ballesteros, I.; Eames, H.L.; Mathie, S.; Martin-Salamanca, S.; Wang, L.; Hemmings, A.; Willemsen, N.; von Werz, V.; et al. Distinct transcription factor networks control neutrophil-driven inflammation. Nat. Immunol. 2021, 22, 1093–1106. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Wannemacher, J.; Christ, S.; Koopmans, T.; Kadri, S.; Zhao, J.; Gouda, M.; Ye, H.; Muck-Hausl, M.; Krenn, P.W.; et al. Neutrophils direct preexisting matrix to initiate repair in damaged tissues. Nat. Immunol. 2022, 23, 518–531. [Google Scholar] [CrossRef] [PubMed]
- Owen-Woods, C.; Joulia, R.; Barkaway, A.; Rolas, L.; Ma, B.; Nottebaum, A.F.; Arkill, K.P.; Stein, M.; Girbl, T.; Golding, M.; et al. Local microvascular leakage promotes trafficking of activated neutrophils to remote organs. J. Clin. Investig. 2020, 130, 2301–2318. [Google Scholar] [CrossRef] [PubMed]
- Colom, B.; Bodkin, J.V.; Beyrau, M.; Woodfin, A.; Ody, C.; Rourke, C.; Chavakis, T.; Brohi, K.; Imhof, B.A.; Nourshargh, S. Leukotriene B4-Neutrophil Elastase Axis Drives Neutrophil Reverse Transendothelial Cell Migration In Vivo. Immunity 2015, 42, 1075–1086. [Google Scholar] [CrossRef] [Green Version]
- Skopelja-Gardner, S.; Tai, J.; Sun, X.; Tanaka, L.; Kuchenbecker, J.A.; Snyder, J.M.; Kubes, P.; Mustelin, T.; Elkon, K.B. Acute skin exposure to ultraviolet light triggers neutrophil-mediated kidney inflammation. Proc. Natl. Acad. Sci. USA 2021, 118, e2019097118. [Google Scholar] [CrossRef]
- Sreeramkumar, V.; Adrover, J.M.; Ballesteros, I.; Cuartero, M.I.; Rossaint, J.; Bilbao, I.; Nacher, M.; Pitaval, C.; Radovanovic, I.; Fukui, Y.; et al. Neutrophils scan for activated platelets to initiate inflammation. Science 2014, 346, 1234–1238. [Google Scholar] [CrossRef] [Green Version]
- De Giovanni, M.; Tam, H.; Valet, C.; Xu, Y.; Looney, M.R.; Cyster, J.G. GPR35 promotes neutrophil recruitment in response to serotonin metabolite 5-HIAA. Cell 2022, 185, 815–830.e819. [Google Scholar] [CrossRef]
- Mauler, M.; Herr, N.; Schoenichen, C.; Witsch, T.; Marchini, T.; Hardtner, C.; Koentges, C.; Kienle, K.; Ollivier, V.; Schell, M.; et al. Platelet Serotonin Aggravates Myocardial Ischemia/Reperfusion Injury via Neutrophil Degranulation. Circulation 2019, 139, 918–931. [Google Scholar] [CrossRef]
- Estevez, B.; Du, X. New Concepts and Mechanisms of Platelet Activation Signaling. Physiology (Bethesda) 2017, 32, 162–177. [Google Scholar] [CrossRef] [Green Version]
- Gaertner, F.; Ahmad, Z.; Rosenberger, G.; Fan, S.; Nicolai, L.; Busch, B.; Yavuz, G.; Luckner, M.; Ishikawa-Ankerhold, H.; Hennel, R.; et al. Migrating Platelets Are Mechano-scavengers that Collect and Bundle Bacteria. Cell 2017, 171, 1368–1382. [Google Scholar] [CrossRef]
- Nicolai, L.; Schiefelbein, K.; Lipsky, S.; Leunig, A.; Hoffknecht, M.; Pekayvaz, K.; Raude, B.; Marx, C.; Ehrlich, A.; Pircher, J.; et al. Vascular surveillance by haptotactic blood platelets in inflammation and infection. Nat. Commun. 2020, 11, 5778. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, Y.; Jin, S.; Wang, M.; Jiao, Y.; Yang, B.; Lu, X.; Ji, X.; Fei, Y.; Yang, H.; et al. Single-cell sequencing of immune cells from anticitrullinated peptide antibody positive and negative rheumatoid arthritis. Nat. Commun. 2021, 12, 4977. [Google Scholar] [CrossRef] [PubMed]
- Koppejan, H.; Hameetman, M.; Beyrend, G.; van Unen, V.; Kwekkeboom, J.C.; van der Helm-van Mil, A.H.; Toes, R.E.M.; van Gaalen, F.A. Immunoprofiling of early, untreated rheumatoid arthritis using mass cytometry reveals an activated basophil subset inversely linked to ACPA status. Arthritis Res. Ther. 2021, 23, 272. [Google Scholar] [CrossRef]
- Reyes, M.; Filbin, M.R.; Bhattacharyya, R.P.; Billman, K.; Eisenhaure, T.; Hung, D.T.; Levy, B.D.; Baron, R.M.; Blainey, P.C.; Goldberg, M.B.; et al. An immune-cell signature of bacterial sepsis. Nat. Med. 2020, 26, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Conroy, J.; Wang, X.; Situ, M.; Namas, R.A.; Vodovotz, Y.; Chen, W.; Singh, H.; Billiar, T.R. The independent prognostic value of global epigenetic alterations: An analysis of single-cell ATAC-seq of circulating leukocytes from trauma patients followed by validation in whole blood leukocyte transcriptomes across three etiologies of critical illness. EBioMedicine 2022, 76, 103860. [Google Scholar] [CrossRef] [PubMed]
- Mitsialis, V.; Wall, S.; Liu, P.; Ordovas-Montanes, J.; Parmet, T.; Vukovic, M.; Spencer, D.; Field, M.; McCourt, C.; Toothaker, J.; et al. Single-Cell Analyses of Colon and Blood Reveal Distinct Immune Cell Signatures of Ulcerative Colitis and Crohn’s Disease. Gastroenterology 2020, 159, 591–608.e510. [Google Scholar] [CrossRef]
- Wauters, E.; Van Mol, P.; Garg, A.D.; Jansen, S.; Van Herck, Y.; Vanderbeke, L.; Bassez, A.; Boeckx, B.; Malengier-Devlies, B.; Timmerman, A.; et al. Discriminating mild from critical COVID-19 by innate and adaptive immune single-cell profiling of bronchoalveolar lavages. Cell Res. 2021, 31, 272–290. [Google Scholar] [CrossRef]
- Wilk, A.J.; Rustagi, A.; Zhao, N.Q.; Roque, J.; Martinez-Colon, G.J.; McKechnie, J.L.; Ivison, G.T.; Ranganath, T.; Vergara, R.; Hollis, T.; et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 2020, 26, 1070–1076. [Google Scholar] [CrossRef]
- Heming, M.; Li, X.; Rauber, S.; Mausberg, A.K.; Borsch, A.L.; Hartlehnert, M.; Singhal, A.; Lu, I.N.; Fleischer, M.; Szepanowski, F.; et al. Neurological Manifestations of COVID-19 Feature T Cell Exhaustion and Dedifferentiated Monocytes in Cerebrospinal Fluid. Immunity 2021, 54, 164–175.e166. [Google Scholar] [CrossRef]
- Garash, R.; Bajpai, A.; Marcinkiewicz, B.M.; Spiller, K.L. Drug delivery strategies to control macrophages for tissue repair and regeneration. Exp. Biol. Med. (Maywood) 2016, 241, 1054–1063. [Google Scholar] [CrossRef] [Green Version]
- Faas, M.; Ipseiz, N.; Ackermann, J.; Culemann, S.; Gruneboom, A.; Schroder, F.; Rothe, T.; Scholtysek, C.; Eberhardt, M.; Bottcher, M.; et al. IL-33-induced metabolic reprogramming controls the differentiation of alternatively activated macrophages and the resolution of inflammation. Immunity 2021, 54, 2531–2546.e2535. [Google Scholar] [CrossRef] [PubMed]
- Horuluoglu, B.; Bayik, D.; Kayraklioglu, N.; Goguet, E.; Kaplan, M.J.; Klinman, D.M. PAM3 supports the generation of M2-like macrophages from lupus patient monocytes and improves disease outcome in murine lupus. J. Autoimmun. 2019, 99, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Horuluoglu, B.H.; Kayraklioglu, N.; Tross, D.; Klinman, D. PAM3 protects against DSS-induced colitis by altering the M2:M1 ratio. Sci. Rep. 2020, 10, 6078. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Xu, M.; Yu, Y.Y.; Hou, Y.; Mi, X.; Sun, Y.X.; Ma, S.; Zuo, X.Y.; Shao, L.L.; Hou, M.; et al. High-dose dexamethasone or all-trans-retinoic acid restores the balance of macrophages towards M2 in immune thrombocytopenia. J. Thromb. Haemost. 2017, 15, 1845–1858. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.S.; Liu, Y.; Wang, J.B.; Peng, J.; Hou, M.; Liu, H.; Feng, R.; Wang, J.W.; Xu, L.P.; Wang, Y.; et al. All-trans retinoic acid plus high-dose dexamethasone as first-line treatment for patients with newly diagnosed immune thrombocytopenia: A multicentre, open-label, randomised, controlled, phase 2 trial. Lancet Haematol. 2021, 8, e688–e699. [Google Scholar] [CrossRef]
- Trilleaud, C.; Gauttier, V.; Biteau, K.; Girault, I.; Belarif, L.; Mary, C.; Pengam, S.; Teppaz, G.; Thepenier, V.; Danger, R.; et al. Agonist anti-ChemR23 mAb reduces tissue neutrophil accumulation and triggers chronic inflammation resolution. Sci. Adv. 2021, 7, eabd1453. [Google Scholar] [CrossRef] [PubMed]
- Ansari, J.; Senchenkova, E.Y.; Vital, S.A.; Al-Yafeai, Z.; Kaur, G.; Sparkenbaugh, E.M.; Orr, A.W.; Pawlinski, R.; Hebbel, R.P.; Granger, D.N.; et al. Targeting the AnxA1/Fpr2/ALX pathway regulates neutrophil function, promoting thromboinflammation resolution in sickle cell disease. Blood 2021, 137, 1538–1549. [Google Scholar] [CrossRef]
- Jobin, K.; Stumpf, N.E.; Schwab, S.; Eichler, M.; Neubert, P.; Rauh, M.; Adamowski, M.; Babyak, O.; Hinze, D.; Sivalingam, S.; et al. A high-salt diet compromises antibacterial neutrophil responses through hormonal perturbation. Sci. Transl. Med. 2020, 12, eaay3850. [Google Scholar] [CrossRef]
- Canzano, P.; Brambilla, M.; Porro, B.; Cosentino, N.; Tortorici, E.; Vicini, S.; Poggio, P.; Cascella, A.; Pengo, M.F.; Veglia, F.; et al. Platelet and Endothelial Activation as Potential Mechanisms Behind the Thrombotic Complications of COVID-19 Patients. JACC Basic Transl. Sci. 2021, 6, 202–218. [Google Scholar] [CrossRef]
- Turnbull, I.R.; Fuchs, A.; Remy, K.E.; Kelly, M.P.; Frazier, E.P.; Ghosh, S.; Chang, S.W.; Mazer, M.B.; Hess, A.; Leonard, J.M.; et al. Dysregulation of the leukocyte signaling landscape during acute COVID-19. PLoS ONE 2022, 17, e0264979. [Google Scholar] [CrossRef]
- Suvarna, K.; Salkar, A.; Palanivel, V.; Bankar, R.; Banerjee, N.; Gayathri, J.P.M.; Srivastava, A.; Singh, A.; Khatri, H.; Agrawal, S.; et al. A Multi-omics Longitudinal Study Reveals Alteration of the Leukocyte Activation Pathway in COVID-19 Patients. J. Proteome Res. 2021, 20, 4667–4680. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, R.; Leunig, A.; Pekayvaz, K.; Popp, O.; Joppich, M.; Polewka, V.; Escaig, R.; Anjum, A.; Hoffknecht, M.L.; Gold, C.; et al. Self-sustaining IL-8 loops drive a prothrombotic neutrophil phenotype in severe COVID-19. JCI Insight 2021, 6, e150862. [Google Scholar] [CrossRef] [PubMed]
- Veras, F.P.; Pontelli, M.C.; Silva, C.M.; Toller-Kawahisa, J.E.; de Lima, M.; Nascimento, D.C.; Schneider, A.H.; Caetite, D.; Tavares, L.A.; Paiva, I.M.; et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J. Exp. Med. 2020, 217, e150862. [Google Scholar] [CrossRef]
- MacDonald, L.; Alivernini, S.; Tolusso, B.; Elmesmari, A.; Somma, D.; Perniola, S.; Paglionico, A.; Petricca, L.; Bosello, S.L.; Carfi, A.; et al. COVID-19 and RA share an SPP1 myeloid pathway that drives PD-L1+ neutrophils and CD14+ monocytes. JCI Insight 2021, 6, e150862. [Google Scholar] [CrossRef] [PubMed]
- Lourda, M.; Dzidic, M.; Hertwig, L.; Bergsten, H.; Palma Medina, L.M.; Sinha, I.; Kvedaraite, E.; Chen, P.; Muvva, J.R.; Gorin, J.B.; et al. High-dimensional profiling reveals phenotypic heterogeneity and disease-specific alterations of granulocytes in COVID-19. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Panda, R.; Castanheira, F.V.; Schlechte, J.M.; Surewaard, B.G.; Shim, H.B.; Zucoloto, A.Z.; Slavikova, Z.; Yipp, B.G.; Kubes, P.; McDonald, B. A functionally distinct neutrophil landscape in severe COVID-19 reveals opportunities for adjunctive therapies. JCI Insight 2022, 7. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Pitmon, E.; Wen, L.; Miller, J.; Ehinger, E.; Herro, R.; Liu, W.; Chen, J.; Mikulski, Z.; Conrad, D.J.; et al. Bone Marrow Transplantation Rescues Monocyte Recruitment Defect and Improves Cystic Fibrosis in Mice. J. Immunol. 2022, 208, 745–752. [Google Scholar] [CrossRef]
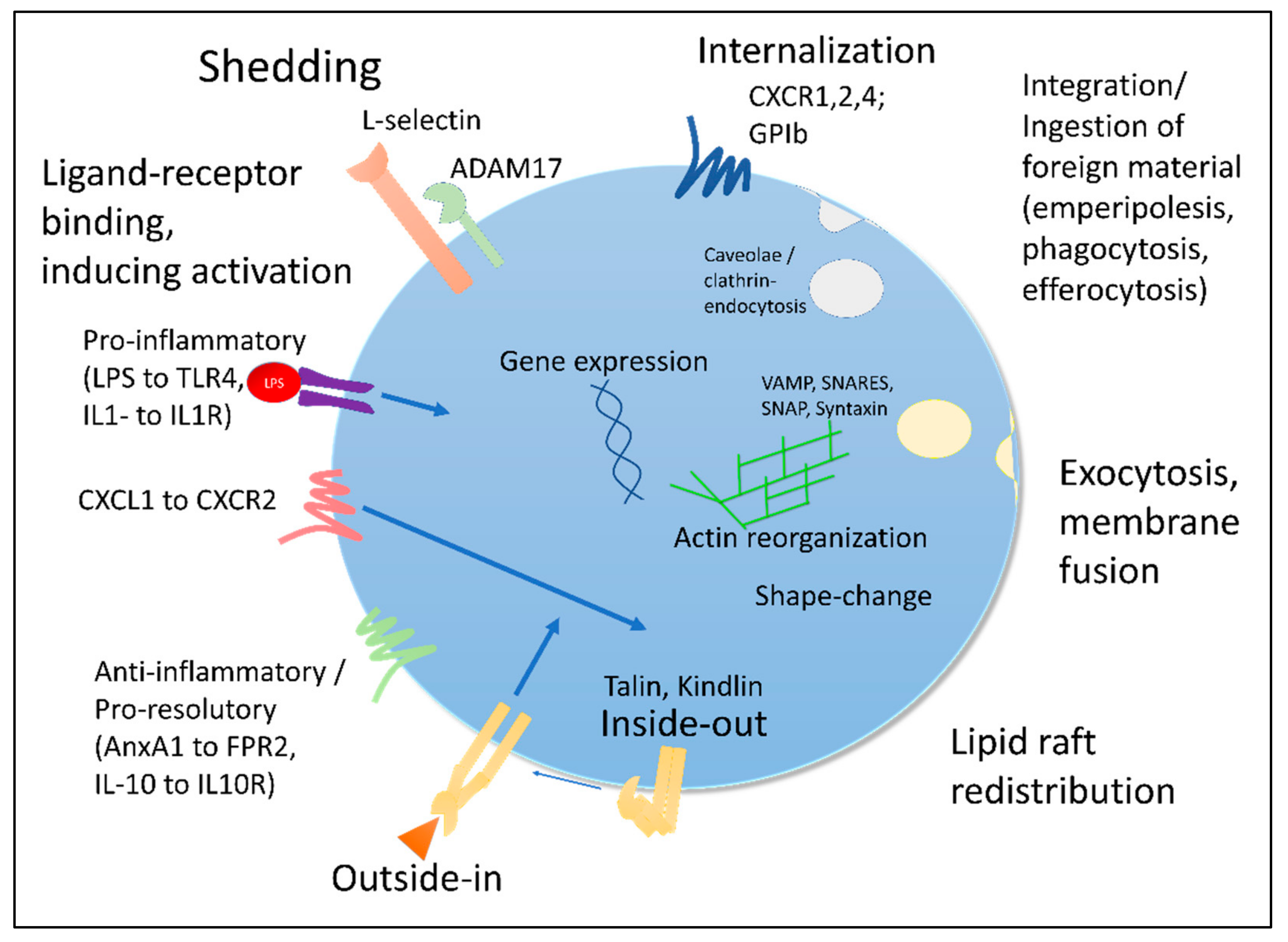

| Type | Technique | Readout | Pro | Con | Example |
|---|---|---|---|---|---|
| Surface receptor analysis | FACS, Microscopy | Detection and expression levels of surface molecules | Quick, relatively cheap | Incomplete, biased (depending on antibody selection), possibility of confounding effects (dilution, shedding, etc.) | Nicolai et al. [12] |
| Proteomics | MassSpec | Protein content | Complete representation of protein composition | Relatively expensive, machinery needed | Leite et al. [7] |
| Transcriptomics | Sequencing | Gene expression analysis | “Wholistic” perspective on regulatory mechanisms and phenotypes | Expensive, time-consuming | Ballesteros et al. [13] |
| Lipidomics | MassSpec | Lipid mediator composition | Detailed lipid analysis | Expensive, equipment intensive | Peng et al. [14] |
| Metabolic profiling | UHPLC/MS/MS “Metabotype” | Leukocyte specific metabolite analysis | More detailed cellular characterization | Confounder, expensive | Anders et al. [15] |
| Single-cell enzyme secretion | Single-cell multiplex profiling | Leukocyte enzyme secretion phenotyping | Patient characteristic immune secretory signatures | Not routinely available | Zeming et al. [16] |
| Functional assays | Flow chambers, ROS production, ligand binding, … | Functionality in activation assays | Direct functional readout | Not routinely available | Rossaint et al. [17] |
| Behavioral landscape | Integrated phenotype assessment | Integrated multiparametric overview of cellular motion | Generalistic perspective on cellular behavior and functionality | Labor-intensive workup, not routinely available | Crainiciuc et al. [11] |
| Disease | Technique | Observation | Publication |
|---|---|---|---|
| Sero-positive/sero-negative rheumatoid arthritis (RA) | Single-cell sequencing | Upregulation of CCL13, 18 and MMP3 in circulating myeloid cells; lack of HLA-DRB5 of ACPA− vs. ACPA+ RA patients. | Wu et al. [264] |
| Mass cytometry | Identification of increased CD62L+ basophil subset in ACPA+ vs. ACPA− patients. | Koppejan et al. [265] | |
| Multiple sclerosis (MS) phenotyping | scRNAseq | Compartmentalized immune cell mechanisms and altered expression profiles | Schafflick et al. [6] |
| Sepsis phenotyping | scRNAseq | PBMC assessment reveals PLAC8 and CLU-dependent CD14+ IL1R2hiHLA-DRlo monocyte discrimination of bacterial sepsis vs. non-sepsis patients with visible expansion in septic patients | Reyes et al. [266] |
| scATAC-seq | PBMCs show prognostic value of overall epigenetic heterogeneity (EG-hi: worse survival) | Chen et al. [267] | |
| Inflammatory bowel disease (IBD) | scRNAseq | Identification of mucosal IL1B+ macrophages and monocytes in IBD vs. control; PBMCs with increased IL-1β+ circulating monocytes in active Crohn’s disease vs. ulcerative colitis. | Mitsialis et al. [268] |
| COVID-19 phenotyping | scRNAseq of BAL | Discrimination of disease severity | Wauters et al. [269] |
| scRNAseq of Blood | Cellular atlas of blood immune responses during COVID-19, including developing neutrophil population and HLAII downregulation within PBMCs | Wilk et al. [270] | |
| scRNAseq of cerebrospinal fluid | Dedifferentiated monocytes in CSF in neuro-COVID-19 patients | Heming et al. [271] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Margraf, A.; Perretti, M. Immune Cell Plasticity in Inflammation: Insights into Description and Regulation of Immune Cell Phenotypes. Cells 2022, 11, 1824. https://doi.org/10.3390/cells11111824
Margraf A, Perretti M. Immune Cell Plasticity in Inflammation: Insights into Description and Regulation of Immune Cell Phenotypes. Cells. 2022; 11(11):1824. https://doi.org/10.3390/cells11111824
Chicago/Turabian StyleMargraf, Andreas, and Mauro Perretti. 2022. "Immune Cell Plasticity in Inflammation: Insights into Description and Regulation of Immune Cell Phenotypes" Cells 11, no. 11: 1824. https://doi.org/10.3390/cells11111824
APA StyleMargraf, A., & Perretti, M. (2022). Immune Cell Plasticity in Inflammation: Insights into Description and Regulation of Immune Cell Phenotypes. Cells, 11(11), 1824. https://doi.org/10.3390/cells11111824