
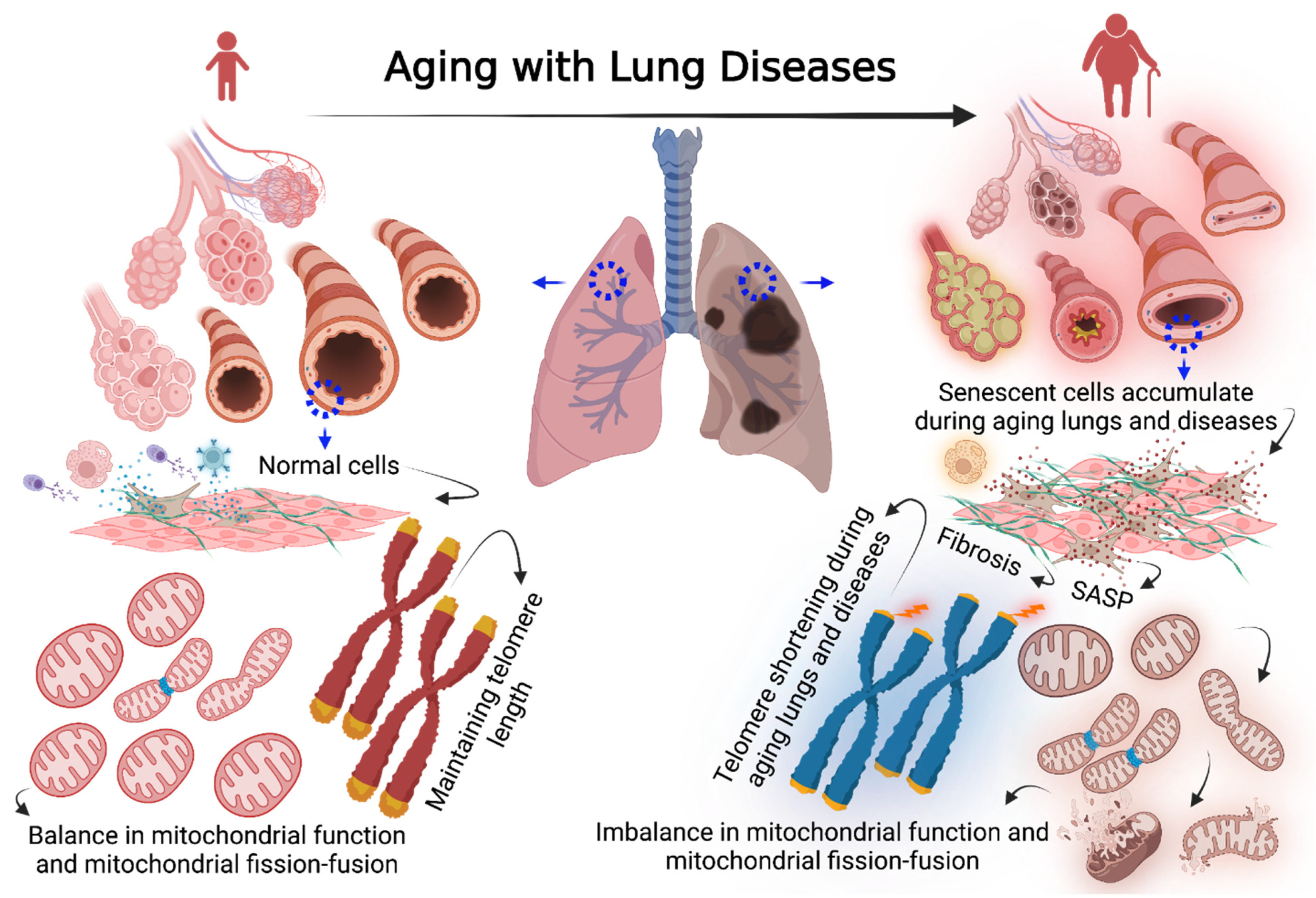
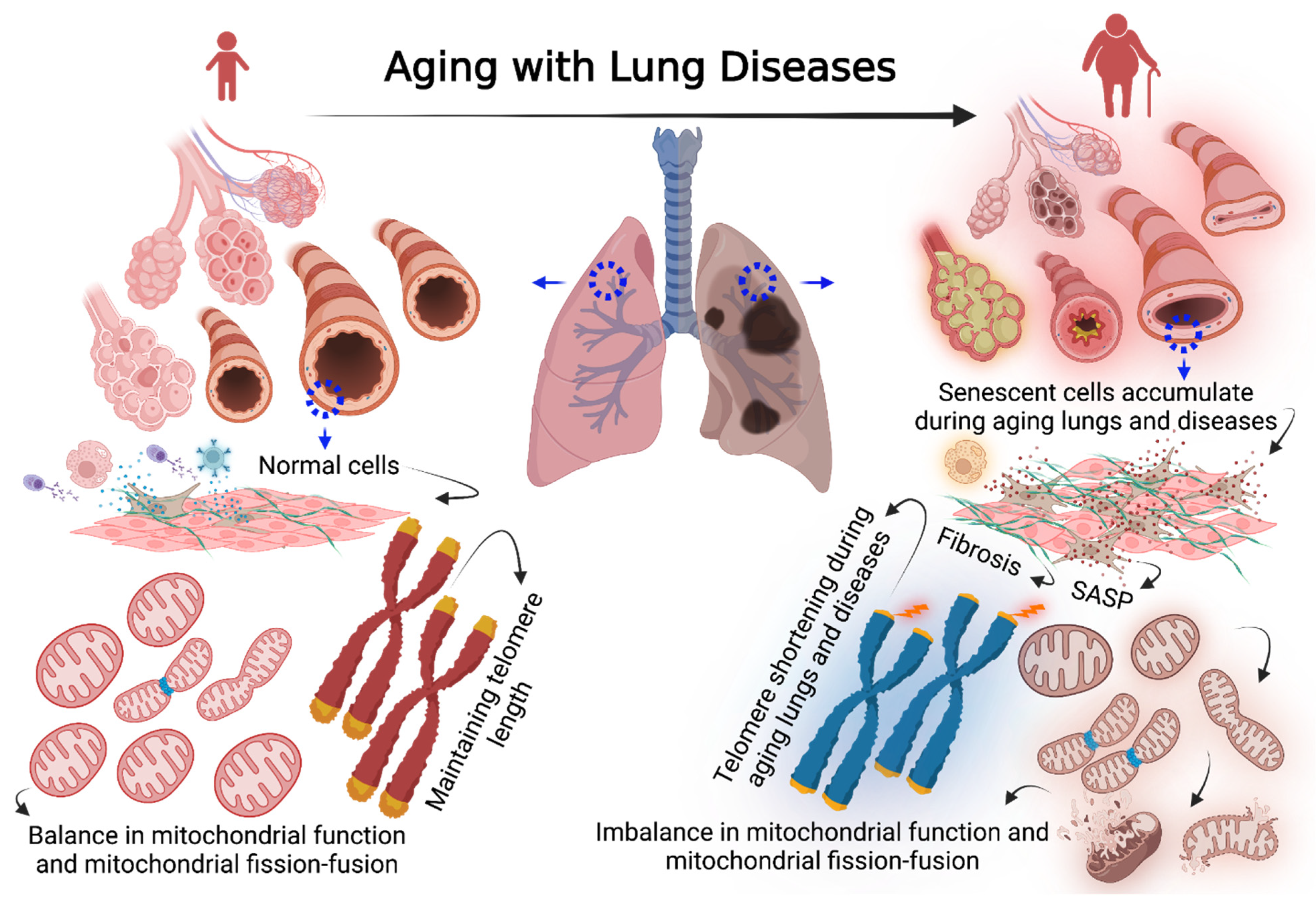
Cellular Senescence in Aging Lungs and Diseases

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Overview of Cellular Senescence
3. Cellular Senescence Signaling Pathways
4. Biomarkers of Cellular Senescence
5. Senescence Signaling in Lung Diseases
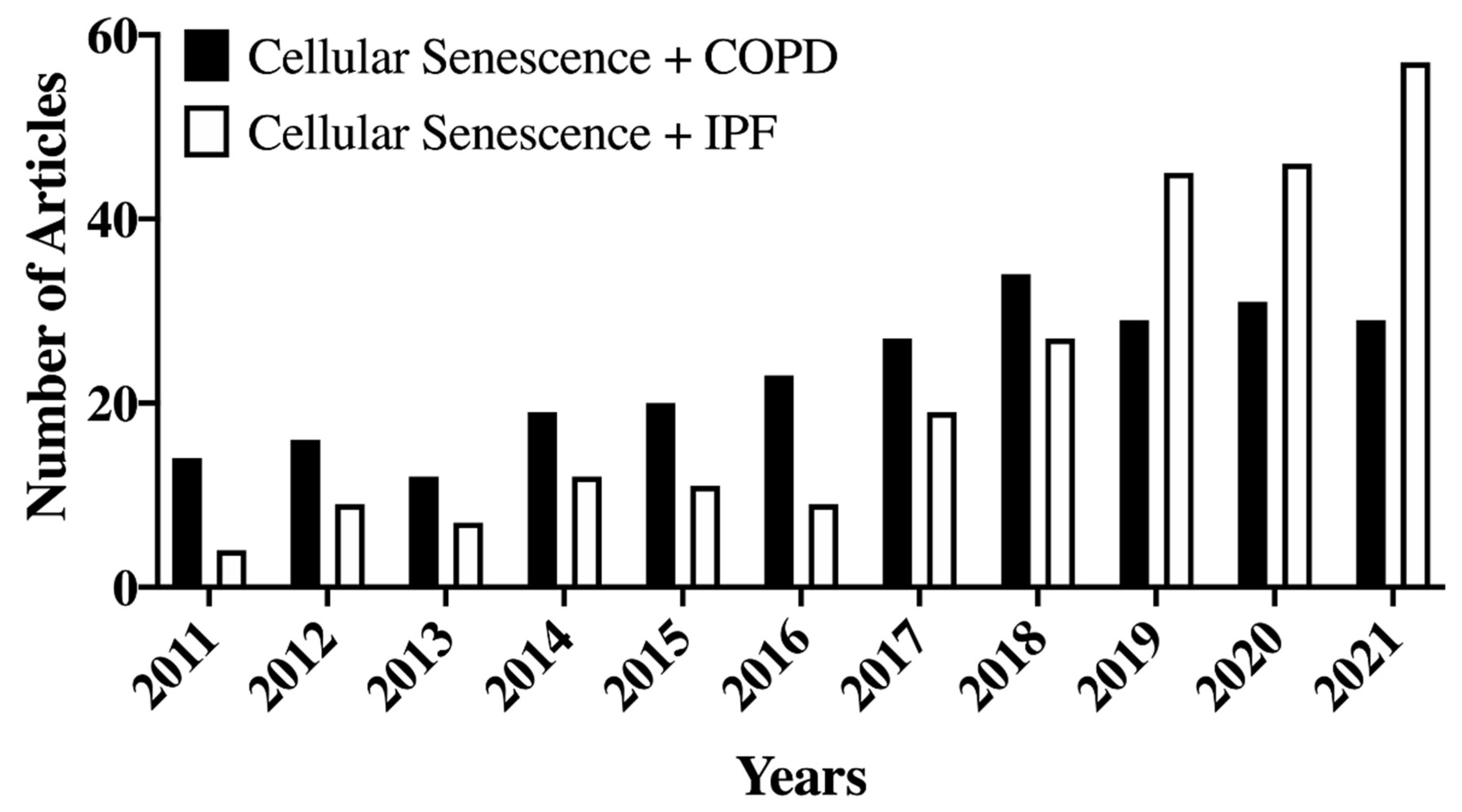
6. Cellular Senescence in COPD
7. Cellular Senescence in IPF
8. Mitochondria in Senescence and Aging
9. Mitochondrial DNA Mutation in Aging Lungs and Diseases
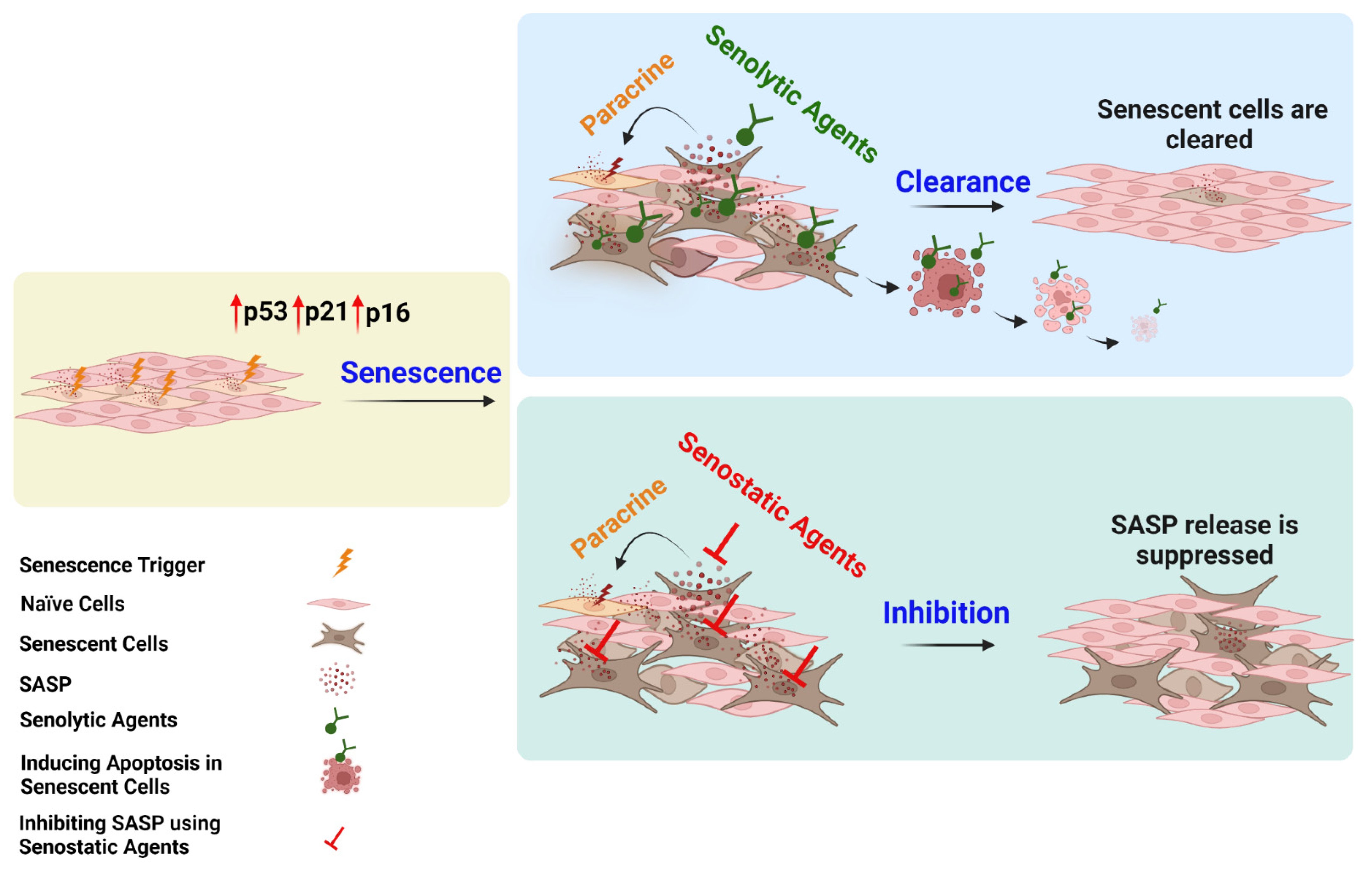
10. Cellular Senescence as a Therapeutic Target
11. Conclusions and Future Insights
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data availability Statement
Acknowledgments
Conflicts of Interest
References
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuilman, T.; Peeper, D.S. Senescence-messaging secretome: SMS-ing cellular stress. Nat. Cancer 2009, 9, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Parikh, P.; Wicher, S.; Khandalavala, K.; Pabelick, C.M.; Britt, R.D., Jr.; Prakash, Y.S. Cellular senescence in the lung across the age spectrum. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2019, 316, L826–L842. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, T.B.L. Understanding the Odd Science of Aging. Cell 2005, 120, 437–447. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Barnes, P.J. COPD as a Disease of Accelerated Lung Aging. Chest 2009, 135, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Mora, A.L.; Bueno, M.; Rojas, M. Mitochondria in the spotlight of aging and idiopathic pulmonary fibrosis. J. Clin. Investig. 2017, 127, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Yue, L.; Yao, H. Mitochondrial dysfunction in inflammatory responses and cellular senescence: Pathogenesis and pharmacological targets for chronic lung diseases. J. Cereb. Blood Flow Metab. 2016, 173, 2305–2318. [Google Scholar] [CrossRef] [Green Version]
- Prakash, Y.; Pabelick, C.M.; Sieck, G. Mitochondrial Dysfunction in Airway Disease. Chest 2017, 152, 618–626. [Google Scholar] [CrossRef]
- Zhou, W.-C.; Qu, J.; Xie, S.-Y.; Sun, Y.; Yao, H.-W. Mitochondrial Dysfunction in Chronic Respiratory Diseases: Implications for the Pathogenesis and Potential Therapeutics. Oxidative Med. Cell. Longev. 2021, 2021, 5188306. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Shelton, D.N.; Chang, E.; Whittier, P.S.; Choi, D.; Funk, W.D. Microarray analysis of replicative senescence. Curr. Biol. 1999, 9, 939–945. [Google Scholar] [CrossRef] [Green Version]
- Denchi, E.L.; Attwooll, C.; Pasini, D.; Helin, K. Deregulated E2F Activity Induces Hyperplasia and Senescence-Like Features in the Mouse Pituitary Gland. Mol. Cell. Biol. 2005, 25, 2660–2672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campisi, J. The biology of replicative senescence. Eur. J. Cancer 1997, 33, 703–709. [Google Scholar] [CrossRef]
- Storer, M.; Mas, A.; Robert-Moreno, À.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence Is a Developmental Mechanism that Contributes to Embryonic Growth and Patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed Cell Senescence during Mammalian Embryonic Development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence: Figure 1. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef] [Green Version]
- Parikh, P.; Britt, R.D., Jr.; Manlove, L.J.; Wicher, S.; Roesler, A.; Ravix, J.; Teske, J.; Thompson, M.A.; Sieck, G.C.; Kirkland, J.L.; et al. Hyperoxia-induced Cellular Senescence in Fetal Airway Smooth Muscle Cells. Am. J. Respir. Cell Mol. Biol. 2019, 61, 51–60. [Google Scholar] [CrossRef]
- Kawamura, T.; Suzuki, J.; Wang, Y.V.; Menendez, S.; Morera, L.B.; Raya, A.; Wahl, G.M.; Belmonte, J.C.I. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature 2009, 460, 1140–1144. [Google Scholar] [CrossRef] [Green Version]
- Barnes, P.J. Senescence in COPD and Its Comorbidities. Annu. Rev. Physiol. 2017, 79, 517–539. [Google Scholar] [CrossRef]
- Greider, C.W. Telomeres Do D-Loop–T-Loop. Cell 1999, 97, 419–422. [Google Scholar] [CrossRef] [Green Version]
- Birch, J.; Victorelli, S.; Rahmatika, D.; Anderson, R.K.; Jiwa, K.; Moisey, E.; Ward, C.; Fisher, A.J.; De Soyza, A.; Passos, J.F. Telomere Dysfunction and Senescence-associated Pathways in Bronchiectasis. Am. J. Respir. Crit. Care Med. 2016, 193, 929–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- d’Adda di Fagagna, F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA Damage Checkpoint Response in Telomere-Initiated Senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Birch, J.; Anderson, R.K.; Correia-Melo, C.; Jurk, D.; Hewitt, G.; Marques, F.M.; Green, N.J.; Moisey, E.; Birrell, M.A.; Belvisi, M.G.; et al. DNA damage response at telomeres contributes to lung aging and chronic obstructive pulmonary disease. Am. J. Physiol. Cell. Mol. Physiol. 2015, 309, L1124–L1137. [Google Scholar] [CrossRef] [Green Version]
- Córdoba-Lanús, E.; Cazorla-Rivero, S.; Espinoza-Jiménez, A.; De-Torres, J.P.; Pajares, M.J.; Aguirre-Jaime, A.; Celli, B.; Casanova, C. Telomere shortening and accelerated aging in COPD: Findings from the BODE cohort. Respir. Res. 2017, 18, 59. [Google Scholar] [CrossRef] [Green Version]
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A.; et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010, 6, 347. [Google Scholar] [CrossRef]
- Korolchuk, V.I.; Miwa, S.; Carroll, B.; von Zglinicki, T. Mitochondria in Cell Senescence: Is Mitophagy the Weakest Link? EBioMedicine 2017, 21, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Passos, J.F.; Saretzki, G.; Ahmed, S.; Nelson, G.; Richter, T.; Peters, H.; Wappler, I.; Birket, M.J.; Harold, G.; Schaeuble, K.; et al. Mitochondrial Dysfunction Accounts for the Stochastic Heterogeneity in Telomere-Dependent Senescence. PLoS Biol. 2007, 5, e110. [Google Scholar] [CrossRef]
- van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Ovadya, Y.; Landsberger, T.; Leins, H.; Vadai, E.; Gal, H.; Biran, A.; Yosef, R.; Sagiv, A.; Agrawal, A.; Shapira, A.; et al. Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat. Commun. 2018, 9, 5435. [Google Scholar] [CrossRef] [Green Version]
- Childs, B.G.; Durik, M.; Baker, D.J.; Van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herranz, N.; Gil, J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Lasry, A.; Ben-Neriah, Y. Senescence-associated inflammatory responses: Aging and cancer perspectives. Trends Immunol. 2015, 36, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Zamudio, R.I.; Robinson, L.; Roux, P.-F.; Bischof, O. SnapShot: Cellular Senescence Pathways. Cell 2017, 170, 816. [Google Scholar] [CrossRef] [PubMed]
- Birch, J.; Barnes, P.J.; Passos, J.F. Mitochondria, telomeres and cell senescence: Implications for lung ageing and disease. Pharmacol. Ther. 2017, 183, 34–49. [Google Scholar] [CrossRef] [Green Version]
- Herbig, U.; Jobling, W.A.; Chen, B.P.; Chen, D.J.; Sedivy, J.M. Telomere Shortening Triggers Senescence of Human Cells through a Pathway Involving ATM, p53, and p21CIP1, but Not p16INK4a. Mol. Cell 2004, 14, 501–513. [Google Scholar] [CrossRef]
- Di Fagagna, F.D. Living on a break: Cellular senescence as a DNA-damage response. Nat. Cancer 2008, 8, 512–522. [Google Scholar] [CrossRef]
- Panier, S.; Boulton, S.J. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2013, 15, 7–18. [Google Scholar] [CrossRef]
- DiTullio, R.A., Jr.; Mochan, T.A.; Venere, M.; Bartkova, J.; Sehested, M.; Bartek, J.; Halazonetis, T.D. 53BP1 functions in an ATM-dependent checkpoint pathway that is constitutively activated in human cancer. Nat. Cell Biol. 2002, 4, 998–1002. [Google Scholar] [CrossRef]
- Mochan, T.A.; Venere, M.; DiTullio, R.A.; Halazonetis, T.D. 53BP1, an activator of ATM in response to DNA damage. DNA Repair 2004, 3, 945–952. [Google Scholar] [CrossRef]
- Childs, B.G.; Baker, D.J.; Kirkland, J.L.; Campisi, J.; Van Deursen, J.M. Senescence and apoptosis: Dueling or complementary cell fates? EMBO Rep. 2014, 15, 1139–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverman, J.; Takai, H.; Buonomo, S.B.; Eisenhaber, F.; de Lange, T. Human Rif1, ortholog of a yeast telomeric protein, is regulated by ATM and 53BP1 and functions in the S-phase checkpoint. Genes Dev. 2004, 18, 2108–2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Capetillo, O.; Chen, H.-T.; Celeste, A.; Ward, I.; Romanienko, P.J.; Morales, J.; Naka, K.; Xia, Z.; Camerini-Otero, R.D.; Motoyama, N.; et al. DNA damage-induced G2–M checkpoint activation by histone H2AX and 53BP1. Nat. Curell Biol. 2002, 4, 993–997. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Matsuoka, S.; Carpenter, P.B.; Elledge, S.J. 53BP1, a Mediator of the DNA Damage Checkpoint. Science 2002, 298, 1435–1438. [Google Scholar] [CrossRef]
- Ward, I.M.; Minn, K.; van Deursen, J.; Chen, J. p53 Binding Protein 53BP1 Is Required for DNA Damage Responses and Tumor Suppression in Mice. Mol. Cell. Biol. 2003, 23, 2556–2563. [Google Scholar] [CrossRef] [Green Version]
- Romagosa, C.; Simonetti, S.; López-Vicente, L.; Mazo, A.; Lleonart, M.E.; Castellvi, J.; Cajal, S.R.Y. p16Ink4a overexpression in cancer: A tumor suppressor gene associated with senescence and high-grade tumors. Oncogene 2011, 30, 2087–2097. [Google Scholar] [CrossRef] [Green Version]
- Sturmlechner, I.; Zhang, C.; Sine, C.C.; van Deursen, E.-J.; Jeganathan, K.B.; Hamada, N.; Grasic, J.; Friedman, D.; Stutchman, J.T.; Can, I.; et al. p21 produces a bioactive secretome that places stressed cells under immunosurveillance. Science 2021, 374. [Google Scholar] [CrossRef]
- Gil, J.; Peters, G. Regulation of the INK4b–ARF–INK4a tumour suppressor locus: All for one or one for all. Nat. Rev. Mol. Cell Biol. 2006, 7, 667–677. [Google Scholar] [CrossRef]
- Sharpless, N.E. INK4a/ARF: A multifunctional tumor suppressor locus. Mutat. Res. Mol. Mech. Mutagen. 2005, 576, 22–38. [Google Scholar] [CrossRef]
- Debacq-Chainiaux, F.; Erusalimsky, J.D.; Campisi, J.; Toussaint, O. Protocols to detect senescence-associated beta-galactosidase (SA-βgal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 2009, 4, 1798–1806. [Google Scholar] [CrossRef]
- Tilstra, J.S.; Robinson, A.R.; Wang, J.; Gregg, S.Q.; Clauson, C.L.; Reay, D.P.; Nasto, L.A.; Croix, C.M.S.; Usas, A.; Vo, N.; et al. NF-κB inhibition delays DNA damage–induced senescence and aging in mice. J. Clin. Investig. 2012, 122, 2601–2612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Segura, A.; de Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; DeMaria, M. Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr. Biol. 2017, 27, 2652–2660.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casella, G.; Munk, R.; Kim, K.M.; Piao, Y.; De, S.; Abdelmohsen, K.; Gorospe, M. Transcriptome signature of cellular senescence. Nucleic Acids Res. 2019, 47, 7294–7305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurz, D.; Decary, S.; Hong, Y.; Erusalimsky, J. Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J. Cell Sci. 2000, 113, 3613–3622. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, G.; von Zglinicki, T.; Passos, J.F. Cell Sorting of Young and Senescent Cells. Methods Mol. Biol. 2013, 1048, 31–47. [Google Scholar] [CrossRef]
- Nicaise, A.M.; Wagstaff, L.J.; Willis, C.M.; Paisie, C.; Chandok, H.; Robson, P.; Fossati, V.; Williams, A.; Crocker, S.J. Cellular senescence in progenitor cells contributes to diminished remyelination potential in progressive multiple sclerosis. Proc. Natl. Acad. Sci. USA 2019, 116, 9030–9039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagano, T.; Nakano, M.; Nakashima, A.; Onishi, K.; Yamao, S.; Enari, M.; Kikkawa, U.; Kamada, S. Identification of cellular senescence-specific genes by comparative transcriptomics. Sci. Rep. 2016, 6, 31758. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Dong, X.; Lee, M.; Maslov, A.Y.; Wang, T.; Vijg, J. Single-cell whole-genome sequencing reveals the functional landscape of somatic mutations in B lymphocytes across the human lifespan. Proc. Natl. Acad. Sci. USA 2019, 116, 9014–9019. [Google Scholar] [CrossRef] [Green Version]
- Frescas, D.; Roux, C.M.; Aygun-Sunar, S.; Gleiberman, A.S.; Krasnov, P.; Kurnasov, O.V.; Strom, E.; Virtuoso, L.P.; Wrobel, M.; Osterman, A.L.; et al. Senescent cells expose and secrete an oxidized form of membrane-bound vimentin as revealed by a natural polyreactive antibody. Proc. Natl. Acad. Sci. USA 2017, 114, E1668–E1677. [Google Scholar] [CrossRef] [Green Version]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef]
- Yao, C.; Guan, X.; Carraro, G.; Parimon, T.; Liu, X.; Huang, G.; Mulay, A.; Soukiasian, H.J.; David, G.; Weigt, S.S.; et al. Senescence of Alveolar Type 2 Cells Drives Progressive Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2021, 203, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; Lebrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. eBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cottage, C.T.; Peterson, N.; Kearley, J.; Berlin, A.; Xiong, X.; Huntley, A.; Zhao, W.; Brown, C.; Migneault, A.; Zerrouki, K.; et al. Targeting p16-induced senescence prevents cigarette smoke-induced emphysema by promoting IGF1/Akt1 signaling in mice. Commun. Biol. 2019, 2, 307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashid, K.; Sundar, I.K.; Gerloff, J.; Li, D.; Rahman, I. Lung cellular senescence is independent of aging in a mouse model of COPD/emphysema. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuji, T.; Aoshiba, K.; Nagai, A. Alveolar Cell Senescence in Patients with Pulmonary Emphysema. Am. J. Respir. Crit. Care Med. 2006, 174, 886–893. [Google Scholar] [CrossRef]
- Amsellem, V.; Gary-Bobo, G.; Marcos, E.; Maitre, B.; Chaar, V.; Validire, P.; Stern, J.-B.; Noureddine, H.; Sapin, E.; Rideau, D.; et al. Telomere Dysfunction Causes Sustained Inflammation in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2011, 184, 1358–1366. [Google Scholar] [CrossRef]
- Rutten, E.P.; Gopal, P.; Wouters, E.F.; Franssen, F.M.; Hageman, G.J.; Vanfleteren, L.E.; Spruit, M.A.; Reynaert, N. Various Mechanistic Pathways Representing the Aging Process Are Altered in COPD. Chest 2016, 149, 53–61. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T. Senolytic drugs: From discovery to translation. J. Intern. Med. 2020, 288, 518–536. [Google Scholar] [CrossRef]
- Pignolo, R.J.; Passos, J.F.; Khosla, S.; Tchkonia, T.; Kirkland, J.L. Reducing Senescent Cell Burden in Aging and Disease. Trends Mol. Med. 2020, 26, 630–638. [Google Scholar] [CrossRef]
- John-Schuster, G.; Günter, S.; Hager, K.; Conlon, T.M.; Eickelberg, O.; Yildirim, A. Inflammaging increases susceptibility to cigarette smoke-induced COPD. Oncotarget 2015, 7, 30068–30083. [Google Scholar] [CrossRef] [Green Version]
- Kujoth, G.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Mitochondrial DNA Mutations, Oxidative Stress, and Apoptosis in Mammalian Aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Mizumura, K.; Cloonan, S.; Nakahira, K.; Bhashyam, A.R.; Cervo, M.; Kitada, T.; Glass, K.; Owen, C.A.; Mahmood, A.; Washko, G.R.; et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J. Clin. Investig. 2014, 124, 3987–4003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paschalaki, K.; Rossios, C.; Pericleous, C.; MacLeod, M.; Rothery, S.; Donaldson, G.C.; Wedzicha, J.A.; Gorgoulis, V.; Randi, A.M.; Barnes, P.J. Inhaled corticosteroids reduce senescence in endothelial progenitor cells from patients with COPD. Thorax 2022, 77, 616–620. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, Y.; Roth, M.; Zhang, L.; Shi, R.; Yang, X.; Li, Y.; Zhang, J. Sirtuin 3 Inhibits Airway Epithelial Mitochondrial Oxidative Stress in Cigarette Smoke-Induced COPD. Oxidative Med. Cell. Longev. 2020, 2020, 7582980. [Google Scholar] [CrossRef]
- Okuda, R.; Aoshiba, K.; Matsushima, H.; Ogura, T.; Okudela, K.; Ohashi, K. Cellular senescence and senescence-associated secretory phenotype: Comparison of idiopathic pulmonary fibrosis, connective tissue disease-associated interstitial lung disease, and chronic obstructive pulmonary disease. J. Thorac. Dis. 2019, 11, 857–864. [Google Scholar] [CrossRef]
- Woldhuis, R.R.; Heijink, I.H.; van den Berge, M.; Timens, W.; Oliver, B.G.G.; de Vries, M.; Brandsma, C.-A. COPD-derived fibroblasts secrete higher levels of senescence-associated secretory phenotype proteins. Thorax 2021, 76, 508–511. [Google Scholar] [CrossRef]
- Shammas, M.A. Telomeres, lifestyle, cancer, and aging. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 28–34. [Google Scholar] [CrossRef] [Green Version]
- Finkel, T.; Hwang, P.M. The Krebs cycle meets the cell cycle: Mitochondria and the G 1 –S transition. Proc. Natl. Acad. Sci. USA 2009, 106, 11825–11826. [Google Scholar] [CrossRef] [Green Version]
- Weir, H.J.; Yao, P.; Huynh, F.K.; Escoubas, C.C.; Goncalves, R.L.; Burkewitz, K.; Laboy, R.; Hirschey, M.D.; Mair, W.B. Dietary Restriction and AMPK Increase Lifespan via Mitochondrial Network and Peroxisome Remodeling. Cell Metab. 2017, 26, 884–896.e5. [Google Scholar] [CrossRef] [Green Version]
- Conti, V.; Corbi, G.; Manzo, V.; Pelaia, G.; Filippelli, A.; Vatrella, A. Sirtuin 1 and Aging Theory for Chronic Obstructive Pulmonary Disease. Anal. Cell. Pathol. 2015, 2015, 897327. [Google Scholar] [CrossRef] [Green Version]
- Johnson, S.; Rabinovitch, P.S.; Kaeberlein, M. mTOR is a key modulator of ageing and age-related disease. Nature 2013, 493, 338–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitani, A.; Ito, K.; Vuppusetty, C.; Barnes, P.J.; Mercado, N. Restoration of Corticosteroid Sensitivity in Chronic Obstructive Pulmonary Disease by Inhibition of Mammalian Target of Rapamycin. Am. J. Respir. Crit. Care Med. 2016, 193, 143–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freund, A.; Patil, C.K.; Campisi, J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011, 30, 1536–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, H.M.; Maarbjerg, S.J.; Crane, J.D.; Jeppesen, J.; Jørgensen, S.B.; Schertzer, J.D.; Shyroka, O.; Kiens, B.; van Denderen, B.J.; Tarnopolsky, M.A.; et al. AMP-activated protein kinase (AMPK) β1β2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc. Natl. Acad. Sci. USA 2011, 108, 16092–16097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyama, E.Q.; Herzig, S.; Courchet, J.; Lewis, T.L., Jr.; Losón, O.C.; Hellberg, K.; Young, N.P.; Chen, H.; Polleux, F.; Chan, D.C.; et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 2016, 351, 275–281. [Google Scholar] [CrossRef] [Green Version]
- Laker, R.C.; Drake, J.C.; Wilson, R.J.; Lira, V.A.; Lewellen, B.M.; Ryall, K.A.; Fisher, C.C.; Zhang, M.; Saucerman, J.J.; Goodyear, L.J.; et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat. Commun. 2017, 8, 548. [Google Scholar] [CrossRef]
- Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J. miR-34a repression of SIRT1 regulates apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13421–13426. [Google Scholar] [CrossRef] [Green Version]
- Ito, Y.; Inoue, A.; Seers, T.; Hato, Y.; Igarashi, A.; Toyama, T.; Taganov, K.D.; Boldin, M.P.; Asahara, H. Identification of targets of tumor suppressor microRNA-34a using a reporter library system. Proc. Natl. Acad. Sci. USA 2017, 114, 3927–3932. [Google Scholar] [CrossRef] [Green Version]
- Kauppinen, A.; Suuronen, T.; Ojala, J.; Kaarniranta, K.; Salminen, A. Antagonistic crosstalk between NF-κB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell. Signal. 2013, 25, 1939–1948. [Google Scholar] [CrossRef] [PubMed]
- Renda, T.; Baraldo, S.; Pelaia, G.; Bazzan, E.; Turato, G.; Papi, A.; Maestrelli, P.; Maselli, R.; Vatrella, A.; Fabbri, L.M.; et al. Increased activation of p38 MAPK in COPD. Eur. Respir. J. 2008, 31, 62–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaffey, K.; Reynolds, S.; Plumb, J.; Kaur, M.; Singh, D. Increased phosphorylated p38 mitogen-activated protein kinase in COPD lungs. Eur. Respir. J. 2012, 42, 28–41. [Google Scholar] [CrossRef] [Green Version]
- Vallese, D.; Ricciardolo, F.L.; Gnemmi, I.; Casolari, P.; Brun, P.; Sorbello, V.; Capelli, A.; Cappello, F.; Cavallesco, G.N.; Papi, A.; et al. Phospho-p38 MAPK Expression in COPD Patients and Asthmatics and in Challenged Bronchial Epithelium. Respiration 2015, 89, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Lea, S.; Li, J.; Plumb, J.; Gaffey, K.; Mason, S.; Gaskell, R.; Harbron, C.; Singh, D. P38 MAPK and glucocorticoid receptor crosstalk in bronchial epithelial cells. Klin. Wochenschr. 2020, 98, 361–374. [Google Scholar] [CrossRef] [Green Version]
- Sgalla, G.; Biffi, A.; Richeldi, L. Idiopathic pulmonary fibrosis: Diagnosis, epidemiology and natural history. Respirology 2015, 21, 427–437. [Google Scholar] [CrossRef] [Green Version]
- Martinez, F.J.; Collard, H.R.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.J.; Taniguchi, H.; Wells, A.U. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Prim. 2017, 3, 17074. [Google Scholar] [CrossRef]
- Sosulski, M.L.; Gongora, R.; Feghali-Bostwick, C.; Lasky, J.A.; Sanchez, C.G. Sirtuin 3 Deregulation Promotes Pulmonary Fibrosis. Journals Gerontol. Ser. A 2016, 72, 595–602. [Google Scholar] [CrossRef]
- Pearce, S.F.; Rebelo-Guiomar, P.; D’Souza, A.R.; Powell, C.A.; Van Haute, L.; Minczuk, M. Regulation of Mammalian Mitochondrial Gene Expression: Recent Advances. Trends Biochem. Sci. 2017, 42, 625–639. [Google Scholar] [CrossRef] [Green Version]
- Fernie, A.R.; Carrari, F.; Sweetlove, L.J. Respiratory metabolism: Glycolysis, the TCA cycle and mitochondrial electron transport. Curr. Opin. Plant Biol. 2004, 7, 254–261. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bueno, M.; Lai, Y.-C.; Romero, Y.; Brands, J.; Croix, C.M.S.; Kamga, C.; Corey, C.; Herazo-Maya, J.D.; Sembrat, J.; Lee, J.; et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Investig. 2014, 125, 521–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwong, F.N.K.; Nicholson, A.G.; Harrison, C.L.; Hansbro, P.M.; Adcock, I.M.; Chung, K.F. Is mitochondrial dysfunction a driving mechanism linking COPD to nonsmall cell lung carcinoma? Eur. Respir. Rev. 2017, 26, 170040. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.; Wang, C.; Qian, G.; Wu, G.; Guo, R.; Li, Q.; Chen, Y.; Li, J.; Li, H.; He, B.; et al. Role of mtDNA haplogroups in COPD susceptibility in a southwestern Han Chinese population. Free Radic. Biol. Med. 2012, 53, 473–481. [Google Scholar] [CrossRef]
- Calvo, S.E.; Clauser, K.; Mootha, V.K. MitoCarta2.0: An updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2015, 44, D1251–D1257. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Chen, R.; Hu, B.; Zein, J.G.; Liu, C.; Comhair, S.A.A.; Aldred, M.A.; Hawkins, G.A.; Meyers, D.A.; Bleecker, E.R.; et al. Mitochondrial DNA Variation and Severe Asthma. American Thoracic Society International Conference Abstracts B33. ASTHMA: MECHANISMS OF DISEASE II 2019. Am. J. Respir. Crit. Care Med. 2019, 199, 1–2. [Google Scholar]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2015, 23, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Birch, J.; Passos, J.F. Targeting the SASP to combat ageing: Mitochondria as possible intracellular allies? BioEssays 2017, 39, 1600235. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, S. Nuclear initiated NF-κB signaling: NEMO and ATM take center stage. Cell Res. 2010, 21, 116–130. [Google Scholar] [CrossRef] [Green Version]
- Holt, J.I.; Harding, A.E.; Morgan-Hughes, J.A. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 1988, 331, 717–719. [Google Scholar] [CrossRef]
- E Giles, R.; Blanc, H.; Cann, H.M.; Wallace, D.C. Maternal inheritance of human mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1980, 77, 6715–6719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, S.-I.; Guarente, L. It takes two to tango: NAD+ and sirtuins in aging/longevity control. NPJ Aging Mech. Dis. 2016, 2, 16017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, R.F.; Zarrintan, S.; Brandenburg, S.M.; Kol, A.; De Bruin, H.G.; Jafari, S.; Dijk, F.; Kalicharan, D.; Kelders, M.; Gosker, H.R.; et al. Prolonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cells. Respir. Res. 2013, 14, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schriner, S.E.; Linford, N.J.; Martin, G.M.; Treuting, P.; Ogburn, C.E.; Emond, M.; Coskun, P.E.; Ladiges, W.; Wolf, N.; Van Remmen, H.; et al. Extension of Murine Life Span by Overexpression of Catalase Targeted to Mitochondria. Science 2005, 308, 1909–1911. [Google Scholar] [CrossRef] [Green Version]
- Soulitzis, N.; Neofytou, E.; Psarrou, M.; Anagnostis, A.; Tavernarakis, N.; Siafakas, N.; Tzortzaki, E.G. Downregulation of lung mitochondrial prohibitin in COPD. Respir. Med. 2012, 106, 954–961. [Google Scholar] [CrossRef] [Green Version]
- Sureshbabu, A.; Ebhandari, V. Targeting mitochondrial dysfunction in lung diseases: Emphasis on mitophagy. Front. Physiol. 2013, 4, 384. [Google Scholar] [CrossRef] [Green Version]
- Stewart, R.; Weir, E.; Montgomery, M.; Niewoehner, D. Hydrogen peroxide contracts airway smooth muscle: A possible endogenous mechanism. Respir. Physiol. 1981, 45, 333–342. [Google Scholar] [CrossRef]
- Ballinger, S.W.; Patterson, C.; Yan, C.-N.; Doan, R.; Burow, D.L.; Young, C.G.; Yakes, F.M.; Van Houten, B.; Ballinger, C.A.; Freeman, B.A.; et al. Hydrogen Peroxide– and Peroxynitrite-Induced Mitochondrial DNA Damage and Dysfunction in Vascular Endothelial and Smooth Muscle Cells. Circ. Res. 2000, 86, 960–966. [Google Scholar] [CrossRef]
- Harman, D. Aging: A Theory Based on Free Radical and Radiation Chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [Green Version]
- Aravamudan, B.; Kiel, A.; Freeman, M.; Delmotte, P.; Thompson, M.; Vassallo, R.; Sieck, G.C.; Pabelick, C.M.; Prakash, Y.S. Cigarette smoke-induced mitochondrial fragmentation and dysfunction in human airway smooth muscle. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2014, 306, L840–L854. [Google Scholar] [CrossRef] [Green Version]
- Mishra, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, O.M.; Gorman, G.S.; Lightowlers, R.N.; Turnbull, D.M. Mitochondrial Diseases: Hope for the Future. Cell 2020, 181, 168–188. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Pinton, P. Mitochondrial DNA keeps you young. Cell Death Dis. 2018, 9, 992. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, Y.; Dong, X.; Zhou, J.; Sun, F.; Han, T.; Lei, P.; Mao, R.; Guo, X.; Wang, Q.; et al. Mitochondrial DNA and genomic DNA ratio in embryo culture medium is not a reliable predictor for in vitro fertilization outcome. Sci. Rep. 2019, 9, 5378. [Google Scholar] [CrossRef]
- Loeb, L.A.; Wallace, D.C.; Martin, G.M. The mitochondrial theory of aging and its relationship to reactive oxygen species damage and somatic mtDNA mutations. Proc. Natl. Acad. Sci. USA 2005, 102, 18769–18770. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, J.; Torrice, C.; Ramsey, M.R.; Kovalev, G.I.; Al-Regaiey, K.; Su, L.; Sharpless, N.E. Ink4a/Arf expression is a biomarker of aging. J. Clin. Investig. 2004, 114, 1299–1307. [Google Scholar] [CrossRef]
- Short, S.; Fielder, E.; Miwa, S.; von Zglinicki, T. Senolytics and senostatics as adjuvant tumour therapy. eBioMedicine 2019, 41, 683–692. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C. Mitochondrial genetic medicine. Nat. Genet. 2018, 50, 1642–1649. [Google Scholar] [CrossRef]
- Singh, B.; Schoeb, T.R.; Bajpai, P.; Slominski, A.; Singh, K.K. Reversing wrinkled skin and hair loss in mice by restoring mitochondrial function. Cell Death Dis. 2018, 9, 735. [Google Scholar] [CrossRef]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.; Rovio, A.T.; Bruder, C.E.; Bohlooly-Y, M.; Gidlöf, S.; Oldfors, A.; Wibom, R.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef]
- Trifunovic, A.; Hansson, A.; Wredenberg, A.; Rovio, A.T.; Dufour, E.; Khvorostov, I.; Spelbrink, J.N.; Wibom, R.; Jacobs, H.T.; Larsson, N.-G. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc. Natl. Acad. Sci. USA 2005, 102, 17993–17998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.I.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Campa, C.C.; Silva, R.; Margaria, J.P.; Pirali, T.; Mattos, M.; Kraemer, L.R.; Reis, D.C.; Grosa, G.; Copperi, F.; Dalmarco, E.M.; et al. Inhalation of the prodrug PI3K inhibitor CL27c improves lung function in asthma and fibrosis. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef]
- Nelson, G.; Wordsworth, J.; Wang, C.; Jurk, D.; Lawless, C.; Martin-Ruiz, C.; von Zglinicki, T. A senescent cell bystander effect: Senescence-induced senescence. Aging Cell 2012, 11, 345–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correia-Melo, C.; Marques, F.D.M.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016, 35, 724–742. [Google Scholar] [CrossRef]
- Coskun, P.E.; Beal, M.F.; Wallace, D.C. Alzheimer’s brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc. Natl. Acad. Sci. USA 2004, 101, 10726–10731. [Google Scholar] [CrossRef] [Green Version]
- Coskun, P.; Wyrembak, J.; Schriner, S.E.; Chen, H.-W.; Marciniack, C.; LaFerla, F.; Wallace, D.C. A mitochondrial etiology of Alzheimer and Parkinson disease. Biochim. Biophys. Acta (BBA) Gen. Subj. 2012, 1820, 553–564. [Google Scholar] [CrossRef] [Green Version]
- Corral-Debrinski, M.; Stepien, G.; Shoffner, J.M.; Lott, M.T.; Kanter, K.; Wallace, D.C. Hypoxemia Is Associated with Mitochondrial DNA Damage and Gene Induction. JAMA 1991, 266, 1812–1816. [Google Scholar] [CrossRef]
- Zifa, E.; Daniil, Z.; Skoumi, E.; Stavrou, M.; Papadimitriou, K.; Terzenidou, M.; Kostikas, K.; Bagiatis, V.; Gourgoulianis, K.I.; Mamuris, Z. Mitochondrial genetic background plays a role in increasing risk to asthma. Mol. Biol. Rep. 2011, 39, 4697–4708. [Google Scholar] [CrossRef]
- Velarde, M.C.; Flynn, J.; Day, N.U.; Melov, S.; Campisi, J. Mitochondrial oxidative stress caused by Sod2 deficiency promotes cellular senescence and aging phenotypes in the skin. Aging 2012, 4, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Carpagnano, G.E.; Lacedonia, D.; Malerba, M.; Palmiotti, G.A.; Cotugno, G.; Carone, M.; Foschino-Barbaro, M.P. Analysis of mitochondrial DNA alteration in new phenotype ACOS. BMC Pulm. Med. 2016, 16, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpagnano, G.E.; Lacedonia, D.; Carone, M.; Soccio, P.; Cotugno, G.; Palmiotti, G.A.; Scioscia, G.; Barbaro, M.P.F. Study of mitochondrial DNA alteration in the exhaled breath condensate of patients affected by obstructive lung diseases. J. Breath Res. 2016, 10, 26005. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Camell, C.D.; Yousefzadeh, M.J.; Zhu, Y.; Prata, L.G.P.L.; Huggins, M.A.; Pierson, M.; Zhang, L.; O’Kelly, R.D.; Pirtskhalava, T.; Xun, P.; et al. Senolytics reduce coronavirus-related mortality in old mice. Science 2021, 373, eabe4832. [Google Scholar] [CrossRef] [PubMed]
- Hubackova, S.; Davidova, E.; Rohlenova, K.; Stursa, J.; Werner, L.; Andera, L.; Dong, L.; Terp, M.; Hodny, Z.; Ditzel, H.J.; et al. Selective elimination of senescent cells by mitochondrial targeting is regulated by ANT2. Cell Death Differ. 2018, 26, 276–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Li, R.; Zhong, R. Extracellular matrix promotes proliferation, migration and adhesion of airway smooth muscle cells in a rat model of chronic obstructive pulmonary disease via upregulation of the PI3K/AKT signaling pathway. Mol. Med. Rep. 2018, 18, 3143–3152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, G.; Kucheryavenko, O.; Wordsworth, J.; Von Zglinicki, T. The senescent bystander effect is caused by ROS-activated NF-κB signalling. Mech. Ageing Dev. 2018, 170, 30–36. [Google Scholar] [CrossRef]
- Pezze, P.D.; Nelson, G.; Otten, E.G.; Korolchuk, V.I.; Kirkwood, T.B.L.; Von Zglinicki, T.; Shanley, D.P. Dynamic Modelling of Pathways to Cellular Senescence Reveals Strategies for Targeted Interventions. PLoS Comput. Biol. 2014, 10, e1003728. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Yang, G.; Kim, Y.; Kim, J.; Ha, J. AMPK activators: Mechanisms of action and physiological activities. Exp. Mol. Med. 2016, 48, e224. [Google Scholar] [CrossRef] [Green Version]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348 Pt 3, 607–614. [Google Scholar] [CrossRef]
- Mimaki, M.; Wang, X.; McKenzie, M.; Thorburn, D.R.; Ryan, M.T. Understanding mitochondrial complex I assembly in health and disease. Biochim. Biophys. Acta (BBA)-Bioenerg. 2012, 1817, 851–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chenggui, W.; Xiaolei, Z.; Pan, Z.; Xu, D.; Zhou, Y.; Wu, Y.; Cai, N.; Tang, Q.; Wang, C.; Yan, M.; et al. Metformin protects against apoptosis and senescence in nucleus pulposus cells and ameliorates disc degeneration in vivo. Cell Death Dis. 2016, 7, e2441. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aghali, A.; Koloko Ngassie, M.L.; Pabelick, C.M.; Prakash, Y.S. Cellular Senescence in Aging Lungs and Diseases. Cells 2022, 11, 1781. https://doi.org/10.3390/cells11111781
Aghali A, Koloko Ngassie ML, Pabelick CM, Prakash YS. Cellular Senescence in Aging Lungs and Diseases. Cells. 2022; 11(11):1781. https://doi.org/10.3390/cells11111781
Chicago/Turabian StyleAghali, Arbi, Maunick Lefin Koloko Ngassie, Christina M. Pabelick, and Y. S. Prakash. 2022. "Cellular Senescence in Aging Lungs and Diseases" Cells 11, no. 11: 1781. https://doi.org/10.3390/cells11111781
APA StyleAghali, A., Koloko Ngassie, M. L., Pabelick, C. M., & Prakash, Y. S. (2022). Cellular Senescence in Aging Lungs and Diseases. Cells, 11(11), 1781. https://doi.org/10.3390/cells11111781