
The Outside-In Journey of Tissue Transglutaminase in Cancer



Abstract
:1. Structure and Functions
2. TG2 in Cancer
3. Intracellular Functions of TG2 in Tumor Cells
4. TG2/FN/β-Integrin Complex
5. TG2 in Cancer Stem Cells
6. TG2 in the Extracellular Matrix in Cancer
7. TG2 in Cancer-Associated Fibroblasts (CAFs)
8. TG2 in Immune Cells
9. TG2 in Endothelial Cells
10. TG2 and Matrix Metalloproteases in Metastatic Progression
11. TG2 and the Stiff Matrix
12. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Huang, X.; Lin, T.; Gu, J.; Zhang, L.; Roth, J.A.; Stephens, L.C.; Yu, Y.; Liu, J.; Fang, B. Combined TRAIL and Bax gene therapy prolonged survival in mice with ovarian cancer xenograft. Gene Ther. 2002, 9, 1379–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephens, P.; Grenard, P.; Aeschlimann, P.; Langley, M.; Blain, E.; Errington, R.; Kipling, D.; Thomas, D.; Aeschlimann, D. Crosslinking and G-protein functions of transglutaminase 2 contribute differentially to fibroblast wound healing responses. J. Cell Sci. 2004, 117, 3389–3403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Begg, G.E.; Carrington, L.; Stokes, P.H.; Matthews, J.M.; Wouters, M.A.; Husain, A.; Lorand, L.; Iismaa, S.E.; Graham, R.M. Mechanism of allosteric regulation of transglutaminase 2 by GTP. Proc. Natl. Acad. Sci. USA 2006, 103, 19683–19688. [Google Scholar] [CrossRef] [Green Version]
- Fesus, L.; Piacentini, M. Transglutaminase 2: An enigmatic enzyme with diverse functions. Trends Biochem. Sci. 2002, 27, 534–539. [Google Scholar] [CrossRef]
- Akimov, S.S.; Belkin, A.M. Cell surface tissue transglutaminase is involved in adhesion and migration of monocytic cells on fibronectin. Blood 2001, 98, 1567–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akimov, S.S.; Krylov, D.; Fleischman, L.F.; Belkin, A.M. Tissue transglutaminase is an integrin-binding adhesion coreceptor for fibronectin. J. Cell Biol. 2000, 148, 825–838. [Google Scholar] [CrossRef] [Green Version]
- Verderio, E.A.; Telci, D.; Okoye, A.; Melino, G.; Griffin, M. A novel RGD-independent cel adhesion pathway mediated by fibronectin-bound tissue transglutaminase rescues cells from anoikis. J. Biol. Chem. 2003, 278, 42604–42614. [Google Scholar] [CrossRef] [Green Version]
- Hang, J.; Zemskov, E.A.; Lorand, L.; Belkin, A.M. Identification of a novel recognition sequence for fibronectin within the NH2-terminal beta-sandwich domain of tissue transglutaminase. J. Biol. Chem. 2005, 280, 23675–23683. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, I.; Osterlund, E.C.; Stamnaes, J.; Iversen, R.; Andersen, J.T.; Jorgensen, T.J.; Sollid, L.M. Dissecting the interaction between transglutaminase 2 and fibronectin. Amino Acids 2017, 49, 489–500. [Google Scholar] [CrossRef] [Green Version]
- Zemskov, E.A.; Loukinova, E.; Mikhailenko, I.; Coleman, R.A.; Strickland, D.K.; Belkin, A.M. Regulation of platelet-derived growth factor receptor function by integrin-associated cell surface transglutaminase. J. Biol. Chem. 2009, 284, 16693–16703. [Google Scholar] [CrossRef] [Green Version]
- Zemskov, E.A.; Mikhailenko, I.; Smith, E.P.; Belkin, A.M. Tissue transglutaminase promotes PDGF/PDGFR-mediated signaling and responses in vascular smooth muscle cells. J. Cell Physiol. 2012, 227, 2089–2096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Condello, S.; Sima, L.E.; Ivan, C.; Cardenas, H.; Schiltz, G.E.; Mishra, R.K.; Matei, D. Tissue transglutaminase regulates interactions between ovarian cancer stem cells and the tumor niche. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piredda, L.; Farrace, M.G.; Lo Bello, M.; Malorni, W.; Melino, G.; Petruzzelli, R.; Piacentini, M. Identification of ‘tissue’ transglutaminase binding proteins in neural cells committed to apoptosis. FASEB J. 1999, 13, 355–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondapaka, S.B.; Singh, S.S.; Dasmahapatra, G.P.; Sausville, E.A.; Roy, K.K. Perifosine, a novel alkylphospholipid, inhibits protein kinase B activation. Mol. Cancer Ther. 2003, 2, 1093–1103. [Google Scholar] [PubMed]
- Facchiano, F.; Facchiano, A.; Facchiano, A.M. The role of transglutaminase-2 and its substrates in human diseases. Front. Biosci. 2006, 11, 1758–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heath, D.J.; Downes, S.; Verderio, E.; Griffin, M. Characterization of tissue transglutaminase in human osteoblast-like cells. J. Bone Miner. Res. 2001, 16, 1477–1485. [Google Scholar] [CrossRef]
- Balklava, Z.; Verderio, E.; Collighan, R.; Gross, S.; Adams, J.; Griffin, M. Analysis of tissue transglutaminase function in the migration of Swiss 3T3 fibroblasts: The active-state conformation of the enzyme does not affect cell motility but is important for its secretion. J. Biol. Chem. 2002, 277, 16567–16575. [Google Scholar] [CrossRef] [Green Version]
- Beninati, S.; Senger, D.R.; Cordella-Miele, E.; Mukherjee, A.B.; Chackalaparampil, I.; Shanmugam, V.; Singh, K.; Mukherjee, B.B. Osteopontin: Its transglutaminase-catalyzed posttranslational modifications and cross-linking to fibronectin. J. Biochem. 1994, 115, 675–682. [Google Scholar] [CrossRef]
- Yuan, Z.Q.; Sun, M.; Feldman, R.I.; Wang, G.; Ma, X.; Jiang, C.; Coppola, D.; Nicosia, S.V.; Cheng, J.Q. Frequent activation of AKT2 and induction of apoptosis by inhibition of phosphoinositide-3-OH kinase/Akt pathway in human ovarian cancer. Oncogene 2000, 19, 2324–2330. [Google Scholar] [CrossRef] [Green Version]
- Akimov, S.S.; Belkin, A.M. Cell-surface transglutaminase promotes fibronectin assembly via interaction with the gelatin-binding domain of fibronectin: A role in TGFbeta-dependent matrix deposition. J. Cell Sci. 2001, 114, 2989–3000. [Google Scholar] [CrossRef]
- Belkin, A.M.; Tsurupa, G.; Zemskov, E.; Veklich, Y.; Weisel, J.W.; Medved, L. Transglutaminase-mediated oligomerization of the fibrin(ogen) {alpha}C-domains promotes integrin-dependent cell adhesion and signaling. Blood 2005. [Google Scholar] [CrossRef] [PubMed]
- Aeschlimann, D.; Paulsson, M. Cross-linking of laminin-nidogen complexes by tissue transglutaminase. A novel mechanism for basement membrane stabilization. J. Biol. Chem. 1991, 266, 15308–15317. [Google Scholar] [CrossRef]
- Jones, R.A.; Kotsakis, P.; Johnson, T.S.; Chau, D.Y.; Ali, S.; Melino, G.; Griffin, M. Matrix changes induced by transglutaminase 2 lead to inhibition of angiogenesis and tumor growth. Cell Death Differ. 2006, 13, 1442–1453. [Google Scholar] [CrossRef]
- Kleman, J.P.; Aeschlimann, D.; Paulsson, M.; van der Rest, M. Transglutaminase-catalyzed cross-linking of fibrils of collagen V/XI in A204 rhabdomyosarcoma cells. Biochemistry 1995, 34, 13768–13775. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Satpathy, M.; Cao, L.; Pincheira, R.; Emerson, R.; Bigsby, R.; Nakshatri, H.; Matei, D. Enhanced peritoneal ovarian tumor dissemination by tissue transglutaminase. Cancer Res. 2007, 67, 7194–7202. [Google Scholar] [CrossRef] [Green Version]
- Shao, M.; Cao, L.; Shen, C.; Satpathy, M.; Chelladurai, B.; Bigsby, R.M.; Nakshatri, H.; Matei, D. Epithelial-to-mesenchymal transition and ovarian tumor progression induced by tissue transglutaminase. Cancer Res. 2009, 69, 9192–9201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacobuzio-Donahue, C.A.; Ashfaq, R.; Maitra, A.; Adsay, N.V.; Shen-Ong, G.L.; Berg, K.; Hollingsworth, M.A.; Cameron, J.L.; Yeo, C.J.; Kern, S.E.; et al. Highly expressed genes in pancreatic ductal adenocarcinomas: A comprehensive characterization and comparison of the transcription profiles obtained from three major technologies. Cancer Res. 2003, 63, 8614–8622. [Google Scholar]
- Martinet, N.; Bonnard, L.; Regnault, V.; Picard, E.; Burke, L.; Siat, J.; Grosdidier, G.; Martinet, Y.; Vignaud, J.M. In vivo transglutaminase type 1 expression in normal lung, preinvasive bronchial lesions, and lung cancer. Am. J. Respir. Cell Mol. Biol. 2003, 28, 428–435. [Google Scholar] [CrossRef]
- Grigoriev, M.Y.; Suspitsin, E.N.; Togo, A.V.; Pozharisski, K.M.; Ivanova, O.A.; Nardacci, R.; Falasca, L.; Piacentini, M.; Imyanitov, E.N.; Hanson, K.P. Tissue transglutaminase expression in breast carcinomas. J. Exp. Clin. Cancer Res. 2001, 20, 265–268. [Google Scholar]
- Verma, A.; Wang, H.; Manavathi, B.; Fok, J.Y.; Mann, A.P.; Kumar, R.; Mehta, K. Increased expression of tissue transglutaminase in pancreatic ductal adenocarcinoma and its implications in drug resistance and metastasis. Cancer Res. 2006, 66, 10525–10533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, J.Y.; Mangala, L.S.; Fok, J.Y.; Lin, Y.G.; Merritt, W.M.; Spannuth, W.A.; Nick, A.M.; Fiterman, D.J.; Vivas-Mejia, P.E.; Deavers, M.T.; et al. Clinical and biological significance of tissue transglutaminase in ovarian carcinoma. Cancer Res. 2008, 68, 5849–5858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, J.H.; Cho, B.C.; Shim, H.S.; Kim, H.R.; Lim, S.M.; Kim, S.K.; Chung, K.Y.; Islam, S.M.; Song, J.J.; Kim, S.Y.; et al. Transglutaminase 2 expression predicts progression free survival in non-small cell lung cancer patients treated with epidermal growth factor receptor tyrosine kinase inhibitor. J. Korean Med. Sci. 2013, 28, 1005–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Condello, S.; Cao, L.; Matei, D. Tissue transglutaminase regulates beta-catenin signaling through a c-Src-dependent mechanism. FASEB J. 2013, 27, 3100–3112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Yakubov, B.; Ivan, C.; Jones, D.R.; Caperell-Grant, A.; Fishel, M.; Cardenas, H.; Matei, D. Tissue Transglutaminase Activates Cancer-Associated Fibroblasts and Contributes to Gemcitabine Resistance in Pancreatic Cancer. Neoplasia 2016, 18, 689–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Gao, H.; Xu, J.; Reuben, J.; Yu, D.; Mehta, K. Evidence that aberrant expression of tissue transglutaminase promotes stem cell characteristics in mammary epithelial cells. PLoS ONE 2011, 6, e20701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Xu, J.; Brady, S.; Gao, H.; Yu, D.; Reuben, J.; Mehta, K. Tissue transglutaminase promotes drug resistance and invasion by inducing mesenchymal transition in mammary epithelial cells. PLoS ONE 2010, 5, e13390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Xu, J.; Sung, B.; Kumar, S.; Yu, D.; Aggarwal, B.B.; Mehta, K. Evidence that GTP-binding domain but not catalytic domain of transglutaminase 2 is essential for epithelial-to-mesenchymal transition in mammary epithelial cells. Breast Cancer Res. 2012, 14, R4. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.; Petrusca, D.N.; Satpathy, M.; Nakshatri, H.; Petrache, I.; Matei, D. Tissue Transglutaminase Protects Epithelial Ovarian Cancer Cells from Cisplatin Induced Apoptosis by Promoting Cell Survival Signaling. Carcinogenesis 2008. [Google Scholar] [CrossRef] [Green Version]
- Mann, A.P.; Verma, A.; Sethi, G.; Manavathi, B.; Wang, H.; Fok, J.Y.; Kunnumakkara, A.B.; Kumar, R.; Aggarwal, B.B.; Mehta, K. Overexpression of Tissue Transglutaminase Leads to Constitutive Activation of Nuclear Factor-{kappa}B in Cancer Cells: Delineation of a Novel Pathway. Cancer Res. 2006, 66, 8788–8795. [Google Scholar] [CrossRef] [Green Version]
- Mehta, K.; Fok, J.; Miller, F.R.; Koul, D.; Sahin, A.A. Prognostic significance of tissue transglutaminase in drug resistant and metastatic breast cancer. Clin. Cancer Res. 2004, 10, 8068–8076. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.; Shao, M.; Schilder, J.; Guise, T.; Mohammad, K.S.; Matei, D. Tissue transglutaminase links TGF-beta, epithelial to mesenchymal transition and a stem cell phenotype in ovarian cancer. Oncogene 2012, 31, 2521–2534. [Google Scholar] [CrossRef] [Green Version]
- Kerr, C.; Szmacinski, H.; Fisher, M.L.; Nance, B.; Lakowicz, J.R.; Akbar, A.; Keillor, J.W.; Lok Wong, T.; Godoy-Ruiz, R.; Toth, E.A.; et al. Transamidase site-targeted agents alter the conformation of the transglutaminase cancer stem cell survival protein to reduce GTP binding activity and cancer stem cell survival. Oncogene 2017, 36, 2981–2990. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, K.E.; Rojas, K.; Cerione, R.A.; Nakano, I.; Wilson, K.F. The stem cell/cancer stem cell marker ALDH1A3 regulates the expression of the survival factor tissue transglutaminase, in mesenchymal glioma stem cells. Oncotarget 2017, 8, 22325–22343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, A.; Guha, S.; Wang, H.; Fok, J.Y.; Koul, D.; Abbruzzese, J.; Mehta, K. Tissue transglutaminase regulates focal adhesion kinase/AKT activation by modulating PTEN expression in pancreatic cancer cells. Clin. Cancer Res. 2008, 14, 1997–2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, A.; Guha, S.; Diagaradjane, P.; Kunnumakkara, A.B.; Sanguino, A.M.; Lopez-Berestein, G.; Sood, A.K.; Aggarwal, B.B.; Krishnan, S.; Gelovani, J.G.; et al. Therapeutic significance of elevated tissue transglutaminase expression in pancreatic cancer. Clin. Cancer Res. 2008, 14, 2476–2483. [Google Scholar] [CrossRef] [Green Version]
- Singh, U.S.; Kunar, M.T.; Kao, Y.L.; Baker, K.M. Role of transglutaminase II in retinoic acid-induced activation of RhoA-associated kinase-2. EMBO J. 2001, 20, 2413–2423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Condello, S.; Yakubov, B.; Emerson, R.; Caperell-Grant, A.; Hitomi, K.; Xie, J.; Matei, D. Tissue Transglutaminase Mediated Tumor-Stroma Interaction Promotes Pancreatic Cancer Progression. Clin. Cancer Res. 2015, 21, 4482–4493. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Mehta, K. Tissue transglutaminase constitutively activates HIF-1alpha promoter and nuclear factor-kappaB via a non-canonical pathway. PLoS ONE 2012, 7, e49321. [Google Scholar] [CrossRef] [Green Version]
- Fisher, M.L.; Kerr, C.; Adhikary, G.; Grun, D.; Xu, W.; Keillor, J.W.; Eckert, R.L. Transglutaminase Interaction with alpha6/beta4-Integrin Stimulates YAP1-Dependent DeltaNp63alpha Stabilization and Leads to Enhanced Cancer Stem Cell Survival and Tumor Formation. Cancer Res. 2016, 76, 7265–7276. [Google Scholar] [CrossRef] [Green Version]
- Fisher, M.L.; Keillor, J.W.; Xu, W.; Eckert, R.L.; Kerr, C. Transglutaminase Is Required for Epidermal Squamous Cell Carcinoma Stem Cell Survival. Mol. Cancer Res. 2015, 13, 1083–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sima, L.E.; Yakubov, B.; Zhang, S.; Condello, S.; Grigorescu, A.A.; Nwani, N.G.; Chen, L.; Schiltz, G.E.; Arvanitis, C.; Zhang, Z.Y.; et al. Small Molecules Target the Interaction between Tissue Transglutaminase and Fibronectin. Mol. Cancer Ther. 2019, 18, 1057–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yakubov, B.; Chen, L.; Belkin, A.M.; Zhang, S.; Chelladurai, B.; Zhang, Z.Y.; Matei, D. Small molecule inhibitors target the tissue transglutaminase and fibronectin interaction. PLoS ONE 2014, 9, e89285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, K.; Moon, H.G.; Lee, D.S.; Yoo, Y.B. Tissue transglutaminase-interleukin-6 axis facilitates peritoneal tumor spreading and metastasis of human ovarian cancer cells. Lab. Anim. Res. 2015, 31, 188–197. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Yang, Q.Y.; Sai, K.; Chen, F.R.; Pang, J.C.; Ng, H.K.; Kwan, A.L.; Chen, Z.P. TGM2 inhibition attenuates ID1 expression in CD44-high glioma-initiating cells. Neuro-Oncol. 2013, 15, 1353–1365. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Oh, Y.T.; Kim, J.Y.; Kim, S.S.; Choi, E.; Kim, T.H.; Hong, J.H.; Chang, N.; Cho, H.J.; Sa, J.K.; et al. Transglutaminase 2 Inhibition Reverses Mesenchymal Transdifferentiation of Glioma Stem Cells by Regulating C/EBPbeta Signaling. Cancer Res. 2017, 77, 4973–4984. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Oh, S.C.; Min, B.W.; Lee, D.H. Transglutaminase 2 Regulates Self-renewal and Stem Cell Marker of Human Colorectal Cancer Stem Cells. Anticancer Res. 2018, 38, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Bagatur, Y.; Ilter Akulke, A.Z.; Bihorac, A.; Erdem, M.; Telci, D. Tissue transglutaminase expression is necessary for adhesion, metastatic potential and cancer stemness of renal cell carcinoma. Cell Adhes. Migr. 2018, 12, 138–151. [Google Scholar] [CrossRef] [Green Version]
- Yakubov, B.; Chelladurai, B.; Schmitt, J.; Emerson, R.; Turchi, J.J.; Matei, D. Extracellular tissue transglutaminase activates noncanonical NF-kappaB signaling and promotes metastasis in ovarian cancer. Neoplasia 2013, 15, 609–619. [Google Scholar] [CrossRef] [Green Version]
- Biri, B.; Kiss, B.; Kiraly, R.; Schlosser, G.; Lang, O.; Kohidai, L.; Fesus, L.; Nyitray, L. Metastasis-associated S100A4 is a specific amine donor and an activity-independent binding partner of transglutaminase-2. Biochem. J. 2016, 473, 31–42. [Google Scholar] [CrossRef]
- Assi, J.; Srivastava, G.; Matta, A.; Chang, M.C.; Walfish, P.G.; Ralhan, R. Transglutaminase 2 overexpression in tumor stroma identifies invasive ductal carcinomas of breast at high risk of recurrence. PLoS ONE 2013, 8, e74437. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; Wang, G.; Wang, T.; Fu, B.; Zhang, Y.; Huang, L.; Deng, Y.; Chen, G.; Wu, X.; Chen, J.; et al. Cancer-associated Fibroblasts induce epithelial-mesenchymal transition via the Transglutaminase 2-dependent IL-6/IL6R/STAT3 axis in Hepatocellular Carcinoma. Int. J. Biol. Sci. 2020, 16, 2542–2558. [Google Scholar] [CrossRef] [PubMed]
- Eom, S.; Kim, Y.; Kim, M.; Park, D.; Lee, H.; Lee, Y.S.; Choe, J.; Kim, Y.M.; Jeoung, D. Transglutaminase II/microRNA-218/-181a loop regulates positive feedback relationship between allergic inflammation and tumor metastasis. J. Biol. Chem. 2014, 289, 29483–29505. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Hong, J.M.; Jeong, E.M.; Lee, W.J.; Kim, H.R.; Kang, J.S.; Kim, I.G.; Hwang, Y.I. Lack of transglutaminase 2 diminished T-cell responses in mice. Immunology 2014, 142, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.Y.; Oh, Y.; Jeong, E.M.; Park, S.; Lee, D.; Wang, X.; Zeng, Q.; Qin, H.; Hu, F.; Gong, H.; et al. Amplification of transglutaminase 2 enhances tumor-promoting inflammation in gastric cancers. Exp. Mol. Med. 2020, 52, 854–864. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Lee, H.J.; Yoon, S.; Ryu, H.M.; Lee, E.; Jo, Y.; Seo, S.; Kim, D.; Lee, C.H.; Kim, W.; et al. Blockade of CCL2 expression overcomes intrinsic PD-1/PD-L1 inhibitor-resistance in transglutaminase 2-induced PD-L1 positive triple negative breast cancer. Am. J. Cancer Res. 2020, 10, 2878–2894. [Google Scholar]
- Sima, L.E.; Chen, S.; Cardenas, H.; Zhao, G.; Wang, Y.; Ivan, C.; Huang, H.; Zhang, B.; Matei, D. Loss of host tissue transglutaminase boosts antitumor T cell immunity by altering STAT1/STAT3 phosphorylation in ovarian cancer. J. Immunother Cancer 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Li, X.; Tan, S.; Zhou, H.J.; Ji, W.; Bellone, S.; Xu, X.; Zhang, H.; Santin, A.D.; Lou, G.; et al. Tumor-associated macrophages drive spheroid formation during early transcoelomic metastasis of ovarian cancer. J. Clin. Invest. 2016, 126, 4157–4173. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Perez, M.; Caja, S.; Melino, G.; Johnson, T.S.; Lindfors, K.; Griffin, M. A novel extracellular role for tissue transglutaminase in matrix-bound VEGF-mediated angiogenesis. Cell Death Dis. 2013, 4, e808. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Perez, M.; Lee, E.S.; Kojima, S.; Griffin, M. The functional relationship between transglutaminase 2 and transforming growth factor beta1 in the regulation of angiogenesis and endothelial-mesenchymal transition. Cell Death Dis. 2017, 8, e3032. [Google Scholar] [CrossRef] [Green Version]
- Nadalutti, C.; Viiri, K.M.; Kaukinen, K.; Maki, M.; Lindfors, K. Extracellular transglutaminase 2 has a role in cell adhesion, whereas intracellular transglutaminase 2 is involved in regulation of endothelial cell proliferation and apoptosis. Cell Prolif. 2011, 44, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.; Chai, N.; Tian, M.; Zhang, Y.; Wang, G.; Liu, J.; Tian, Z.; Yi, X.; Chen, D.; Li, X.; et al. Novel peptide GX1 inhibits angiogenesis by specifically binding to transglutaminase-2 in the tumorous endothelial cells of gastric cancer. Cell Death Dis. 2018, 9, 579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belkin, A.M.; Akimov, S.S.; Zaritskaya, L.S.; Ratnikov, B.I.; Deryugina, E.I.; Strongin, A.Y. Matrix-dependent proteolysis of surface transglutaminase by membrane-type metalloproteinase regulates cancer cell adhesion and locomotion. J. Biol. Chem. 2001, 276, 18415–18422. [Google Scholar] [CrossRef] [Green Version]
- Satpathy, M.; Shao, M.; Emerson, R.; Donner, D.B.; Matei, D. Tissue transglutaminase regulates matrix metalloproteinase-2 in ovarian cancer by modulating cAMP-response element-binding protein activity. J. Biol. Chem. 2009, 284, 15390–15399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.H.; Lin, C.Y.; Lee, L.T.; Chang, G.D.; Lee, P.P.; Hung, C.C.; Kao, W.T.; Tsai, P.H.; Schally, A.V.; Hwang, J.J.; et al. Up-regulation of fibronectin and tissue transglutaminase promotes cell invasion involving increased association with integrin and MMP expression in A431 cells. Anticancer Res. 2010, 30, 4177–4186. [Google Scholar] [PubMed]
- Delaine-Smith, R.; Wright, N.; Hanley, C.; Hanwell, R.; Bhome, R.; Bullock, M.; Drifka, C.; Eliceiri, K.; Thomas, G.; Knight, M.; et al. Transglutaminase-2 Mediates the Biomechanical Properties of the Colorectal Cancer Tissue Microenvironment that Contribute to Disease Progression. Cancers 2019, 11, 701. [Google Scholar] [CrossRef] [Green Version]
- Antonyak, M.A.; Li, B.; Regan, A.D.; Feng, Q.; Dusaban, S.S.; Cerione, R.A. Tissue transglutaminase is an essential participant in the epidermal growth factor-stimulated signaling pathway leading to cancer cell migration and invasion. J. Biol. Chem. 2009, 284, 17914–17925. [Google Scholar] [CrossRef] [Green Version]
- Vincent, T.; Neve, E.P.; Johnson, J.R.; Kukalev, A.; Rojo, F.; Albanell, J.; Pietras, K.; Virtanen, I.; Philipson, L.; Leopold, P.L.; et al. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-beta mediated epithelial-mesenchymal transition. Nat. Cell Biol. 2009, 11, 943–950. [Google Scholar] [CrossRef] [Green Version]
- Ayinde, O.; Wang, Z.; Griffin, M. Tissue transglutaminase induces Epithelial-Mesenchymal-Transition and the acquisition of stem cell like characteristics in colorectal cancer cells. Oncotarget 2017, 8, 20025–20041. [Google Scholar] [CrossRef] [Green Version]
- Lorand, L.; Dailey, J.E.; Turner, P.M. Fibronectin as a carrier for the transglutaminase from human erythrocytes. Proc. Natl. Acad. Sci. USA 1988, 85, 1057–1059. [Google Scholar] [CrossRef] [Green Version]
- Turner, P.M.; Lorand, L. Complexation of fibronectin with tissue transglutaminase. Biochemistry 1989, 28, 628–635. [Google Scholar] [CrossRef] [PubMed]
- LeMosy, E.K.; Erickson, H.P.; Beyer, W.F., Jr.; Radek, J.T.; Jeong, J.M.; Murthy, S.N.; Lorand, L. Visualization of purified fibronectin-transglutaminase complexes. J. Biol. Chem. 1992, 267, 7880–7885. [Google Scholar] [CrossRef]
- Di Niro, R.; Sulic, A.M.; Mignone, F.; D’Angelo, S.; Boordoni, R.; Iacono, M.; Marzari, R.; Gaiotto, T.; Lavric, M.; Bradbury, A.R.; et al. Rapid interactome profiling by massive sequencing. Nucleic Acids Res. 2010, 38, e110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soluri, M.F.; Boccafoschi, F.; Cotella, D.; Moro, L.; Forestieri, G.; Autiero, I.; Cavallo, L.; Oliva, R.; Griffin, M.; Wang, Z.; et al. Mapping the minimum domain of the fibronectin binding site on transglutaminase 2 (TG2) and its importance in mediating signaling, adhesion, and migration in TG2-expressing cells. FASEB J. 2019, 33, 2327–2342. [Google Scholar] [CrossRef] [Green Version]
- Zemskov, E.A.; Janiak, A.; Hang, J.; Waghray, A.; Belkin, A.M. The role of tissue transglutaminase in cell-matrix interactions. Front. Biosci. 2006, 11, 1057–1076. [Google Scholar] [CrossRef] [Green Version]
- Mangala, L.S.; Fok, J.Y.; Zorrilla-Calancha, I.R.; Verma, A.; Mehta, K. Tissue transglutaminase expression promotes cell attachment, invasion and survival in breast cancer cells. Oncogene 2007, 26, 2459–2470. [Google Scholar] [CrossRef] [Green Version]
- Quintana, E.; Shackleton, M.; Sabel, M.S.; Fullen, D.R.; Johnson, T.M.; Morrison, S.J. Efficient tumour formation by single human melanoma cells. Nature 2008, 456, 593–598. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Wulfkuhle, J.; Zhang, H.; Gu, P.; Yang, Y.; Deng, J.; Margolick, J.B.; Liotta, L.A.; Petricoin, E., 3rd; Zhang, Y. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc. Natl. Acad. Sci. USA 2007, 104, 16158–16163. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Tian, Y.; Yuan, X.; Wu, H.; Liu, Q.; Pestell, R.G.; Wu, K. The role of CD44 in epithelial-mesenchymal transition and cancer development. OncoTargets Ther. 2015, 8, 3783–3792. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Balch, C.; Chan, M.W.; Lai, H.C.; Matei, D.; Schilder, J.M.; Yan, P.S.; Huang, T.H.; Nephew, K.P. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008, 68, 4311–4320. [Google Scholar] [CrossRef] [Green Version]
- Suetsugu, A.; Nagaki, M.; Aoki, H.; Motohashi, T.; Kunisada, T.; Moriwaki, H. Characterization of CD133+ hepatocellular carcinoma cells as cancer stem/progenitor cells. Biochem. Biophys. Res. Commun. 2006, 351, 820–824. [Google Scholar] [CrossRef] [PubMed]
- Silva, I.A.; Bai, S.; McLean, K.; Yang, K.; Griffith, K.; Thomas, D.; Ginestier, C.; Johnston, C.; Kueck, A.; Reynolds, R.K.; et al. Aldehyde dehydrogenase in combination with CD133 defines angiogenic ovarian cancer stem cells that portend poor patient survival. Cancer Res. 2011, 71, 3991–4001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanei, T.; Morimoto, K.; Shimazu, K.; Kim, S.J.; Tanji, Y.; Taguchi, T.; Tamaki, Y.; Noguchi, S. Association of breast cancer stem cells identified by aldehyde dehydrogenase 1 expression with resistance to sequential Paclitaxel and epirubicin-based chemotherapy for breast cancers. Clin. Cancer Res. 2009, 15, 4234–4241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, J.; Suzuki, Y.; Kim, S.J.; Wang, P.C.; Matsumura, M.; Kojima, S. Transactivation via RAR/RXR-Sp1 interaction: Characterization of binding between Sp1 and GC box motif. Mol. Endocrinol. 2001, 15, 1677–1692. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Clevers, H. Wnt signalling in stem cells and cancer. Nature 2005, 434, 843–850. [Google Scholar] [CrossRef]
- Tammela, T.; Sanchez-Rivera, F.J.; Cetinbas, N.M.; Wu, K.; Joshi, N.S.; Helenius, K.; Park, Y.; Azimi, R.; Kerper, N.R.; Wesselhoeft, R.A.; et al. A Wnt-producing niche drives proliferative potential and progression in lung adenocarcinoma. Nature 2017, 545, 355–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Yu, B.; Deng, P.; Cheng, Y.; Yu, Y.; Kevork, K.; Ramadoss, S.; Ding, X.; Li, X.; Wang, C.Y. KDM3 epigenetically controls tumorigenic potentials of human colorectal cancer stem cells through Wnt/beta-catenin signalling. Nat. Commun. 2017, 8, 15146. [Google Scholar] [CrossRef]
- Giustacchini, A.; Thongjuea, S.; Barkas, N.; Woll, P.S.; Povinelli, B.J.; Booth, C.A.G.; Sopp, P.; Norfo, R.; Rodriguez-Meira, A.; Ashley, N.; et al. Single-cell transcriptomics uncovers distinct molecular signatures of stem cells in chronic myeloid leukemia. Nat. Med. 2017, 23, 692–702. [Google Scholar] [CrossRef]
- Condello, S.; Morgan, C.A.; Nagdas, S.; Cao, L.; Turek, J.; Hurley, T.D.; Matei, D. beta-Catenin-regulated ALDH1A1 is a target in ovarian cancer spheroids. Oncogene 2015, 34, 2297–2308. [Google Scholar] [CrossRef] [Green Version]
- Odii, B.O.; Coussons, P. Biological functionalities of transglutaminase 2 and the possibility of its compensation by other members of the transglutaminase family. Sci. World J. 2014, 2014, 714561. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Chen, Z.; Ni, X. Tissue transglutaminase-1 promotes stemness and chemoresistance in gastric cancer cells by regulating Wnt/beta-catenin signaling. Exp. Biol. Med. 2017, 242, 194–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tibbitt, M.W.; Anseth, K.S. Hydrogels as extracellular matrix mimics for 3D cell culture. Biotechnol. Bioeng. 2009, 103, 655–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurose, K.; Hoshaw-Woodard, S.; Adeyinka, A.; Lemeshow, S.; Watson, P.H.; Eng, C. Genetic model of multi-step breast carcinogenesis involving the epithelium and stroma: Clues to tumour-microenvironment interactions. Hum. Mol. Genet. 2001, 10, 1907–1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, T.S.; Liu, Y.; Li, W.; Greenberg, C.S. Identification of two GTP-independent alternatively spliced forms of tissue transglutaminase in human leukocytes, vascular smooth muscle, and endothelial cells. FASEB J. 2007, 21, 4131–4143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tempest, R.; Guarnerio, S.; Maani, R.; Cooper, J.; Peake, N. The Biological and Biomechanical Role of Transglutaminase-2 in the Tumour Microenvironment. Cancers 2021, 13, 2788. [Google Scholar] [CrossRef] [PubMed]
- Lorand, L.; Graham, R.M. Transglutaminases: Crosslinking enzymes with pleiotropic functions. Nat. Rev. Mol. Cell Biol. 2003, 4, 140–156. [Google Scholar] [CrossRef]
- Belkin, A.M. Extracellular TG2: Emerging functions and regulation. FEBS J. 2011, 278, 4704–4716. [Google Scholar] [CrossRef] [Green Version]
- Telci, D.; Wang, Z.; Li, X.; Verderio, E.A.; Humphries, M.J.; Baccarini, M.; Basaga, H.; Griffin, M. Fibronectin-tissue transglutaminase matrix rescues RGD-impaired cell adhesion through syndecan-4 and beta1 integrin co-signaling. J. Biol. Chem. 2008, 283, 20937–20947. [Google Scholar] [CrossRef] [Green Version]
- Faverman, L.; Mikhaylova, L.; Malmquist, J.; Nurminskaya, M. Extracellular transglutaminase 2 activates beta-catenin signaling in calcifying vascular smooth muscle cells. FEBS Lett. 2008, 582, 1552–1557. [Google Scholar] [CrossRef] [Green Version]
- Janiak, A.; Zemskov, E.A.; Belkin, A.M. Cell surface transglutaminase promotes RhoA activation via integrin clustering and suppression of the Src-p190RhoGAP signaling pathway. Mol. Biol. Cell 2006, 17, 1606–1619. [Google Scholar] [CrossRef] [Green Version]
- Deasey, S.; Nurminsky, D.; Shanmugasundaram, S.; Lima, F.; Nurminskaya, M. Transglutaminase 2 as a novel activator of LRP6/beta-catenin signaling. Cell Signal. 2013, 25, 2646–2651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, S.; Murphy, L.J. Tissue transglutaminase has intrinsic kinase activity: Identification of transglutaminase 2 as an insulin-like growth factor-binding protein-3 kinase. J. Biol. Chem. 2004, 279, 23863–23868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, K.; Park, H.B.; Byoun, O.J.; Shin, D.M.; Jeong, E.M.; Kim, Y.W.; Kim, Y.S.; Melino, G.; Kim, I.G.; Lee, D.S. Epithelial transglutaminase 2 is needed for T cell interleukin-17 production and subsequent pulmonary inflammation and fibrosis in bleomycin-treated mice. J. Exp. Med. 2011, 208, 1707–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, S.; Garcia-Palmero, I.; Herrera, M.; Bartolome, R.A.; Pena, C.; Fernandez-Acenero, M.J.; Padilla, G.; Pelaez-Garcia, A.; Lopez-Lucendo, M.; Rodriguez-Merlo, R.; et al. LOXL2 Is Highly Expressed in Cancer-Associated Fibroblasts and Associates to Poor Colon Cancer Survival. Clin. Cancer Res. 2015, 21, 4892–4902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef]
- Irvine, A.F.; Waise, S.; Green, E.W.; Stuart, B.; Thomas, G.J. Characterising cancer-associated fibroblast heterogeneity in non-small cell lung cancer: A systematic review and meta-analysis. Sci. Rep. 2021, 11, 3727. [Google Scholar] [CrossRef]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [Green Version]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Mellone, M.; Hanley, C.J.; Thirdborough, S.; Mellows, T.; Garcia, E.; Woo, J.; Tod, J.; Frampton, S.; Jenei, V.; Moutasim, K.A.; et al. Induction of fibroblast senescence generates a non-fibrogenic myofibroblast phenotype that differentially impacts on cancer prognosis. Aging 2016, 9, 114–132. [Google Scholar] [CrossRef] [Green Version]
- Kieffer, Y.; Hocine, H.R.; Gentric, G.; Pelon, F.; Bernard, C.; Bourachot, B.; Lameiras, S.; Albergante, L.; Bonneau, C.; Guyard, A.; et al. Single-Cell Analysis Reveals Fibroblast Clusters Linked to Immunotherapy Resistance in Cancer. Cancer Discov. 2020, 10, 1330–1351. [Google Scholar] [CrossRef]
- Givel, A.M.; Kieffer, Y.; Scholer-Dahirel, A.; Sirven, P.; Cardon, M.; Pelon, F.; Magagna, I.; Gentric, G.; Costa, A.; Bonneau, C.; et al. miR200-regulated CXCL12beta promotes fibroblast heterogeneity and immunosuppression in ovarian cancers. Nat. Commun. 2018, 9, 1056. [Google Scholar] [CrossRef] [PubMed]
- Magagna, I.; Gourdin, N.; Kieffer, Y.; Licaj, M.; Mhaidly, R.; Andre, P.; Morel, A.; Vincent-Salomon, A.; Paturel, C.; Mechta-Grigoriou, F. CD73-Mediated Immunosuppression Is Linked to a Specific Fibroblast Population That Paves the Way for New Therapy in Breast Cancer. Cancers 2021, 13, 5878. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.M.; Bissell, M.J. Modeling dynamic reciprocity: Engineering three-dimensional culture models of breast architecture, function, and neoplastic transformation. Semin. Cancer Biol. 2005, 15, 342–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohn, J.; Kim, T.I.; Yoon, Y.H.; Kim, J.Y.; Kim, S.Y. Novel transglutaminase inhibitors reverse the inflammation of allergic conjunctivitis. J. Clin. Invest. 2003, 111, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Suh, G.Y.; Ham, H.S.; Lee, S.H.; Choi, J.C.; Koh, W.J.; Kim, S.Y.; Lee, J.; Han, J.; Kim, H.P.; Choi, A.M.; et al. A Peptide with anti-transglutaminase activity decreases lipopolysaccharide-induced lung inflammation in mice. Exp. Lung Res. 2006, 32, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Hong, G.U.; Park, B.S.; Park, J.W.; Kim, S.Y.; Ro, J.Y. IgE production in CD40/CD40L cross-talk of B and mast cells and mediator release via TGase 2 in mouse allergic asthma. Cell Signal. 2013, 25, 1514–1525. [Google Scholar] [CrossRef]
- Moreno, J.J. Effects of antiflammins on transglutaminase and phospholipase A2 activation by transglutaminase. Int. Immunopharmacol. 2006, 6, 300–303. [Google Scholar] [CrossRef]
- Curro, M.; Ferlazzo, N.; Risitano, R.; Condello, S.; Vecchio, M.; Caccamo, D.; Ientile, R. Transglutaminase 2 and phospholipase A(2) interactions in the inflammatory response in human Thp-1 monocytes. Amino Acids 2014, 46, 759–766. [Google Scholar] [CrossRef]
- Jeitner, T.M.; Pinto, J.T.; Krasnikov, B.F.; Horswill, M.; Cooper, A.J. Transglutaminases and neurodegeneration. J. Neurochem. 2009, 109 (Suppl. 1), 160–166. [Google Scholar] [CrossRef] [Green Version]
- Cordella-Miele, E.; Miele, L.; Mukherjee, A.B. A novel transglutaminase-mediated post-translational modification of phospholipase A2 dramatically increases its catalytic activity. J. Biol. Chem. 1990, 265, 17180–17188. [Google Scholar] [CrossRef]
- Cordella-Miele, E.; Miele, L.; Beninati, S.; Mukherjee, A.B. Transglutaminase-catalyzed incorporation of polyamines into phospholipase A2. J. Biochem. 1993, 113, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Zou, W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer 2005, 5, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Stewart, T.J.; Abrams, S.I. How tumours escape mass destruction. Oncogene 2008, 27, 5894–5903. [Google Scholar] [CrossRef] [Green Version]
- Fogg, D.K.; Sibon, C.; Miled, C.; Jung, S.; Aucouturier, P.; Littman, D.R.; Cumano, A.; Geissmann, F. A clonogenic bone marrow progenitor specific for macrophages and dendritic cells. Science 2006, 311, 83–87. [Google Scholar] [CrossRef]
- Nurminskaya, M.V.; Belkin, A.M. Cellular functions of tissue transglutaminase. Int. Rev. Cell Mol. Biol. 2012, 294, 1–97. [Google Scholar] [CrossRef] [Green Version]
- Matic, I.; Sacchi, A.; Rinaldi, A.; Melino, G.; Khosla, C.; Falasca, L.; Piacentini, M. Characterization of transglutaminase type II role in dendritic cell differentiation and function. J. Leukoc. Biol. 2010, 88, 181–188. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Jeong, E.M.; Jeong, Y.J.; Lee, W.J.; Kang, J.S.; Kim, I.G.; Hwang, Y.I. Transglutaminase 2 on the surface of dendritic cells is proposed to be involved in dendritic cell-T cell interaction. Cell Immunol. 2014, 289, 55–62. [Google Scholar] [CrossRef]
- Toth, B.; Garabuczi, E.; Sarang, Z.; Vereb, G.; Vamosi, G.; Aeschlimann, D.; Blasko, B.; Becsi, B.; Erdodi, F.; Lacy-Hulbert, A.; et al. Transglutaminase 2 is needed for the formation of an efficient phagocyte portal in macrophages engulfing apoptotic cells. J. Immunol. 2009, 182, 2084–2092. [Google Scholar] [CrossRef]
- Falasca, L.; Iadevaia, V.; Ciccosanti, F.; Melino, G.; Serafino, A.; Piacentini, M. Transglutaminase type II is a key element in the regulation of the anti-inflammatory response elicited by apoptotic cell engulfment. J. Immunol. 2005, 174, 7330–7340. [Google Scholar] [CrossRef] [Green Version]
- Sarang, Z.; Koroskenyi, K.; Pallai, A.; Duro, E.; Melino, G.; Griffin, M.; Fesus, L.; Szondy, Z. Transglutaminase 2 null macrophages respond to lipopolysaccharide stimulation by elevated proinflammatory cytokine production due to an enhanced alphavbeta3 integrin-induced Src tyrosine kinase signaling. Immunol. Lett. 2011, 138, 71–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curro, M.; Ferlazzo, N.; Condello, S.; Caccamo, D.; Ientile, R. Transglutaminase 2 silencing reduced the beta-amyloid-effects on the activation of human THP-1 cells. Amino Acids 2010, 39, 1427–1433. [Google Scholar] [CrossRef] [PubMed]
- Curro, M.; Gangemi, C.; Giunta, M.L.; Ferlazzo, N.; Navarra, M.; Ientile, R.; Caccamo, D. Transglutaminase 2 is involved in amyloid-beta1–42-induced pro-inflammatory activation via AP1/JNK signalling pathways in THP-1 monocytes. Amino Acids 2017, 49, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, Q.; Zhang, X.; Cui, M.; Li, T.; Zhang, Y.; Liao, Q. Immune subtyping for pancreatic cancer with implication in clinical outcomes and improving immunotherapy. Cancer Cell Int. 2021, 21, 137. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Helming, L.; Milde, R.; Varin, A.; Melgert, B.N.; Draijer, C.; Thomas, B.; Fabbri, M.; Crawshaw, A.; Ho, L.P.; et al. Genetic programs expressed in resting and IL-4 alternatively activated mouse and human macrophages: Similarities and differences. Blood 2013, 121, e57–e69. [Google Scholar] [CrossRef] [PubMed]
- Roby, K.F.; Taylor, C.C.; Sweetwood, J.P.; Cheng, Y.; Pace, J.L.; Tawfik, O.; Persons, D.L.; Smith, P.G.; Terranova, P.F. Development of a syngeneic mouse model for events related to ovarian cancer. Carcinogenesis 2000, 21, 585–591. [Google Scholar] [CrossRef]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef] [Green Version]
- Ziyad, S.; Iruela-Arispe, M.L. Molecular mechanisms of tumor angiogenesis. Genes Cancer 2011, 2, 1085–1096. [Google Scholar] [CrossRef] [Green Version]
- Noman, M.Z.; Hasmim, M.; Messai, Y.; Terry, S.; Kieda, C.; Janji, B.; Chouaib, S. Hypoxia: A key player in antitumor immune response. A Review in the Theme: Cellular Responses to Hypoxia. Am. J. Physiol. Cell Physiol. 2015, 309, C569–C579. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, Z.A.; Barsigian, C.; Chalupowicz, G.D.; Bach, T.L.; Garcia-Manero, G.; Martinez, J. Colocalization of tissue transglutaminase and stress fibers in human vascular smooth muscle cells and human umbilical vein endothelial cells. Exp. Cell Res. 1997, 231, 38–49. [Google Scholar] [CrossRef]
- Gaudry, C.A.; Verderio, E.; Jones, R.A.; Smith, C.; Griffin, M. Tissue transglutaminase is an important player at the surface of human endothelial cells: Evidence for its externalization and its colocalization with the beta(1) integrin. Exp. Cell Res. 1999, 252, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Iismaa, S.E.; Mearns, B.M.; Lorand, L.; Graham, R.M. Transglutaminases and disease: Lessons from genetically engineered mouse models and inherited disorders. Physiol. Rev. 2009, 89, 991–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deasey, S.; Shanmugasundaram, S.; Nurminskaya, M. Tissue-specific responses to loss of transglutaminase 2. Amino Acids 2013, 44, 179–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charytan, D.M.; Padera, R.; Helfand, A.M.; Zeisberg, M.; Xu, X.; Liu, X.; Himmelfarb, J.; Cinelli, A.; Kalluri, R.; Zeisberg, E.M. Increased concentration of circulating angiogenesis and nitric oxide inhibitors induces endothelial to mesenchymal transition and myocardial fibrosis in patients with chronic kidney disease. Int. J. Cardiol. 2014, 176, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Verderio, E.; Gaudry, C.; Gross, S.; Smith, C.; Downes, S.; Griffin, M. Regulation of cell surface tissue transglutaminase: Effects on matrix storage of latent transforming growth factor-beta binding protein-1. J. Histochem. Cytochem. 1999, 47, 1417–1432. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.A.; Wang, Z.; Dookie, S.; Griffin, M. The role of TG2 in ECV304-related vasculogenic mimicry. Amino Acids 2013, 44, 89–101. [Google Scholar] [CrossRef]
- Moller, L.B. Structure and function of the urokinase receptor. Blood Coagul. Fibrinolysis 1993, 4, 293–303. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Banda, M.J.; Leppert, D. Matrix metalloproteinases in immunity. J. Immunol. 1996, 156, 1–4. [Google Scholar]
- Stetler-Stevenson, W.G. Dynamics of matrix turnover during pathologic remodeling of the extracellular matrix. Am. J. Pathol. 1996, 148, 1345–1350. [Google Scholar]
- Himelstein, B.P.; Canete-Soler, R.; Bernhard, E.J.; Dilks, D.W.; Muschel, R.J. Metalloproteinases in tumor progression: The contribution of MMP-9. Invasion Metastasis 1994, 14, 246–258. [Google Scholar]
- Hanahan, D.; Lanzavecchia, A.; Mihich, E. Fourteenth Annual Pezcoller Symposium: The novel dichotomy of immune interactions with tumors. Cancer Res. 2003, 63, 3005–3008. [Google Scholar] [PubMed]
- Franke, F.E.; Von Georgi, R.; Zygmunt, M.; Munstedt, K. Association between fibronectin expression and prognosis in ovarian carcinoma. Anticancer Res. 2003, 23, 4261–4267. [Google Scholar] [PubMed]
- Sternlicht, M.D.; Werb, Z. How matrix metalloproteinases regulate cell behavior. Annu. Rev. Cell Dev. Biol. 2001, 17, 463–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belkin, A.M.; Zemskov, E.A.; Hang, J.; Akimov, S.S.; Sikora, S.; Strongin, A.Y. Cell-surface-associated tissue transglutaminase is a target of MMP-2 proteolysis. Biochemistry 2004, 43, 11760–11769. [Google Scholar] [CrossRef]
- Coughlin, M.F.; Bielenberg, D.R.; Lenormand, G.; Marinkovic, M.; Waghorne, C.G.; Zetter, B.R.; Fredberg, J.J. Cytoskeletal stiffness, friction, and fluidity of cancer cell lines with different metastatic potential. Clin. Exp. Metastasis 2013, 30, 237–250. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Mezencev, R.; Kim, B.; Wang, L.; McDonald, J.; Sulchek, T. Cell stiffness is a biomarker of the metastatic potential of ovarian cancer cells. PLoS ONE 2012, 7, e46609. [Google Scholar] [CrossRef] [Green Version]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [Green Version]
- Cox, T.R.; Erler, J.T. Remodeling and homeostasis of the extracellular matrix: Implications for fibrotic diseases and cancer. Dis. Model. Mech. 2011, 4, 165–178. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Griffin, M. TG2, a novel extracellular protein with multiple functions. Amino Acids 2012, 42, 939–949. [Google Scholar] [CrossRef]
- Thomas, D.; Radhakrishnan, P. Tumor-stromal crosstalk in pancreatic cancer and tissue fibrosis. Mol. Cancer 2019, 18, 14. [Google Scholar] [CrossRef]
- Libring, S.; Shinde, A.; Chanda, M.K.; Nuru, M.; George, H.; Saleh, A.M.; Abdullah, A.; Kinzer-Ursem, T.L.; Calve, S.; Wendt, M.K.; et al. The Dynamic Relationship of Breast Cancer Cells and Fibroblasts in Fibronectin Accumulation at Primary and Metastatic Tumor Sites. Cancers 2020, 12, 1270. [Google Scholar] [CrossRef] [PubMed]
- Coulson-Thomas, V.J.; Coulson-Thomas, Y.M.; Gesteira, T.F.; de Paula, C.A.; Mader, A.M.; Waisberg, J.; Pinhal, M.A.; Friedl, A.; Toma, L.; Nader, H.B. Colorectal cancer desmoplastic reaction up-regulates collagen synthesis and restricts cancer cell invasion. Cell Tissue Res. 2011, 346, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, M.; Levine, H. Cluster size distribution of cells disseminating from a primary tumor. PLoS Comput. Biol. 2021, 17, e1009011. [Google Scholar] [CrossRef] [PubMed]
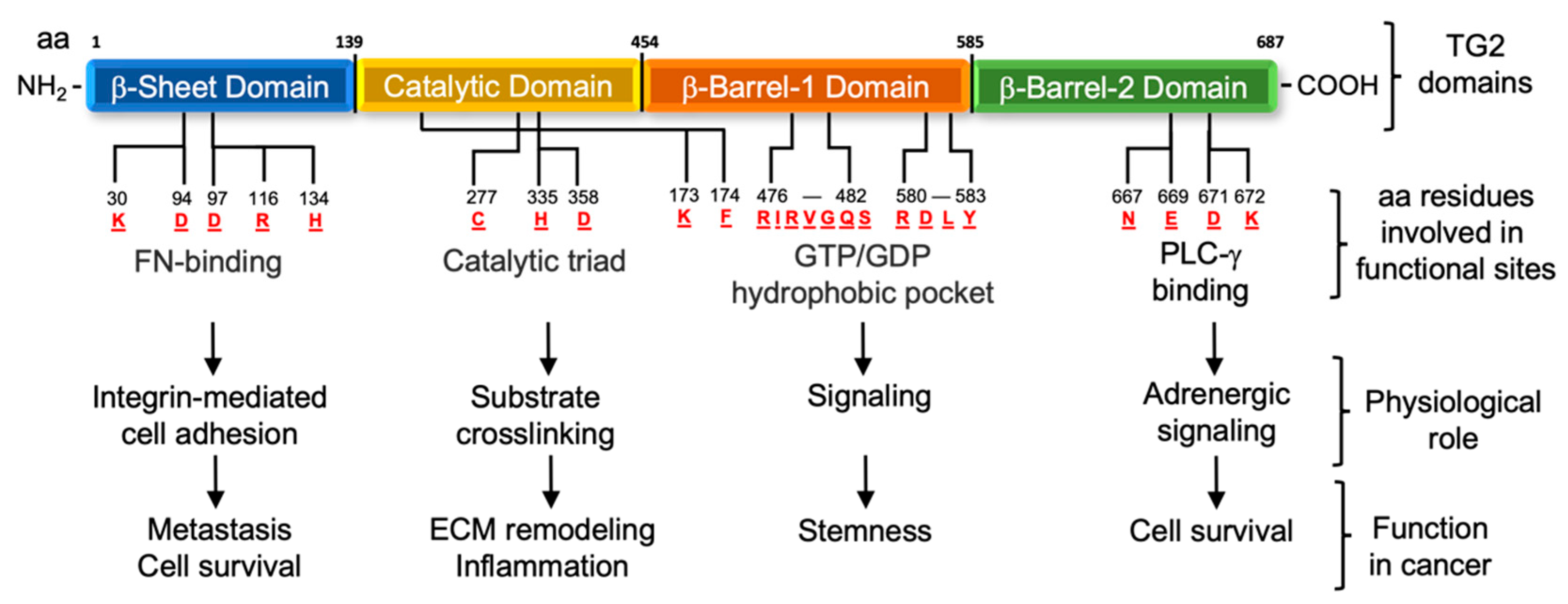
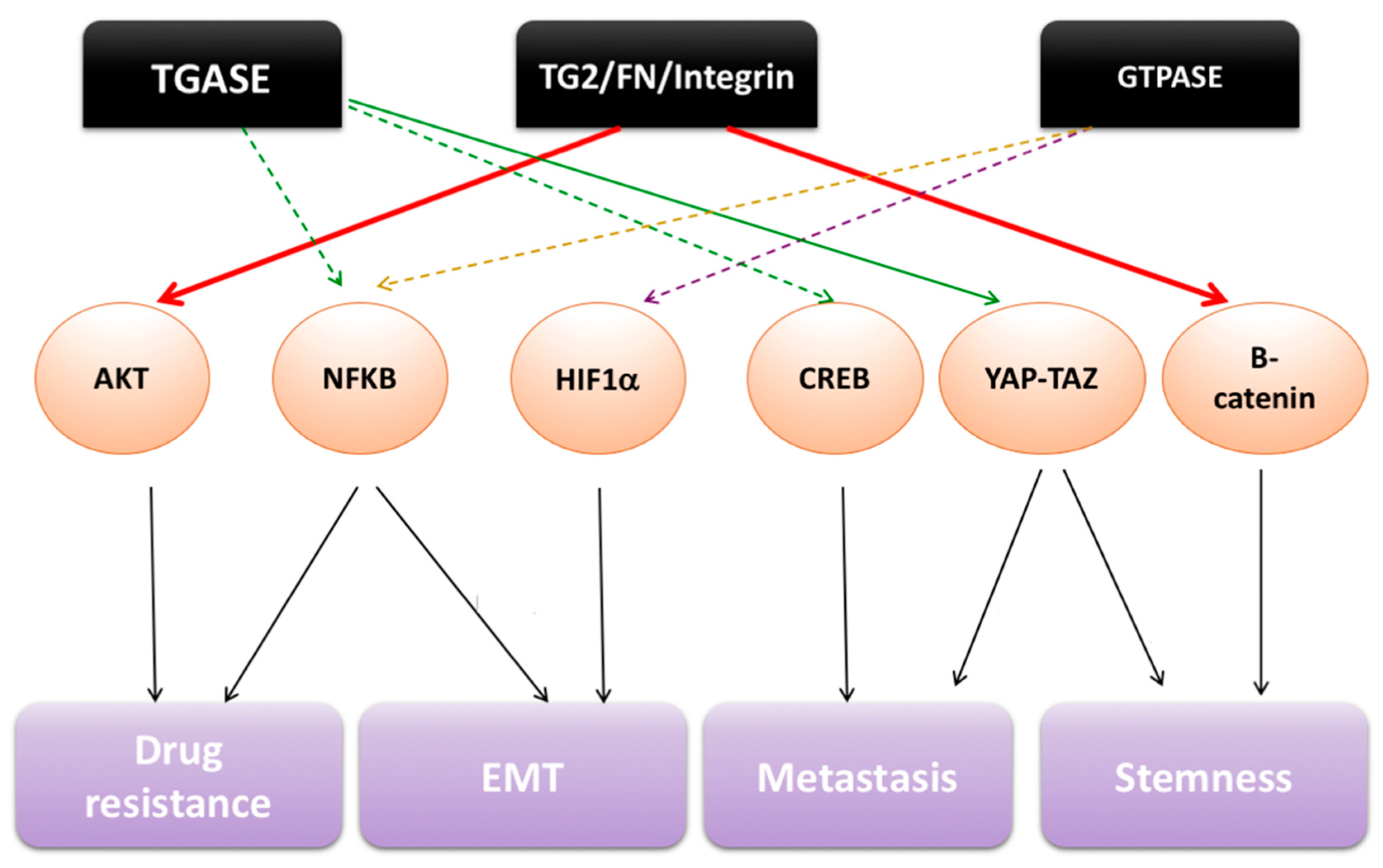
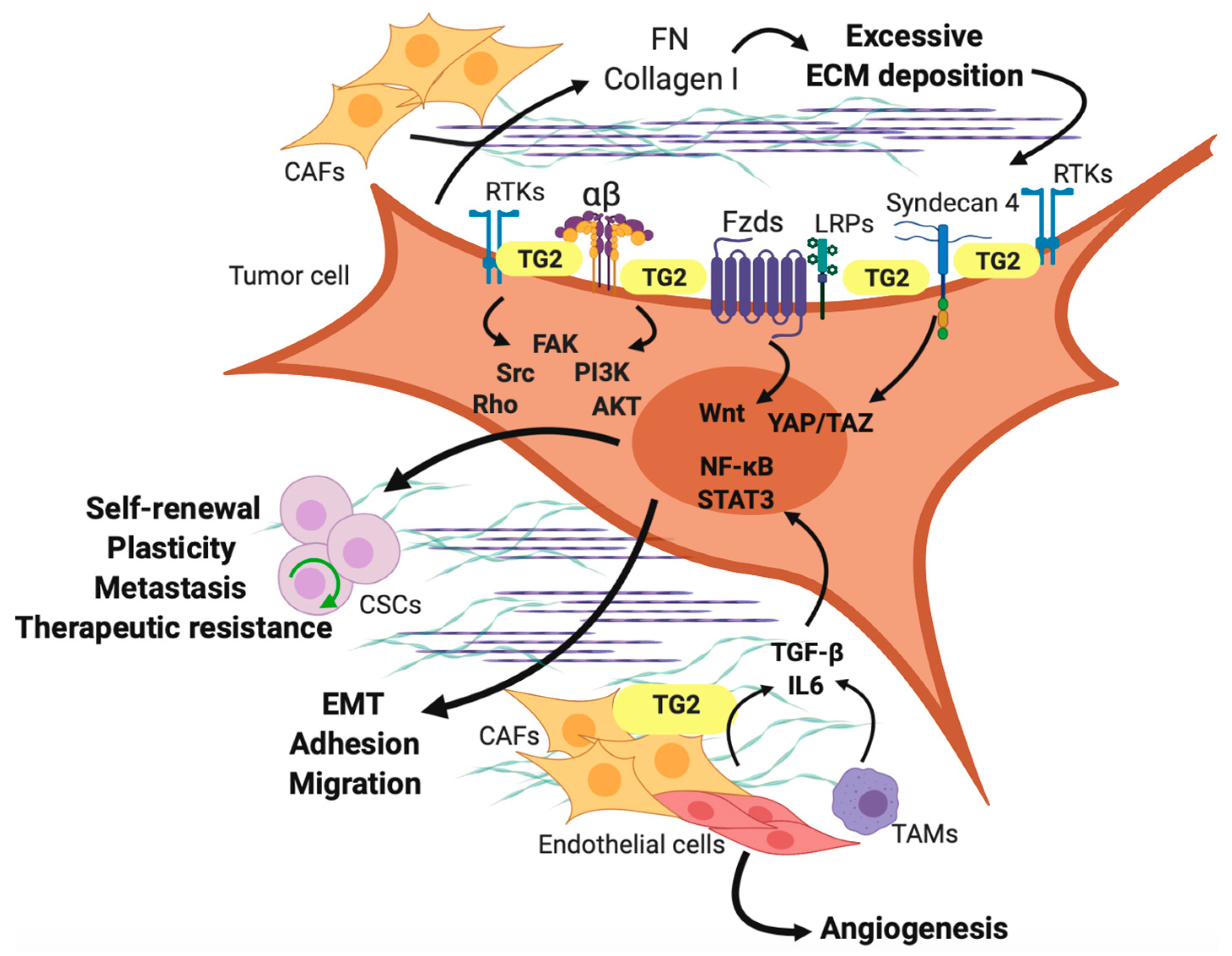

{kind=link}
{kind=link}
{kind=link}
| References | Type of Cancer Model | Oncogenic Signaling | Adhesion and Migration | ECM Remodeling and Invasion | EMT | Metastasis | Angiogenesis | Stemness | Chemotherapy/Radiotherapy Resistance | (Anti-Tumor) Immune Response |
|---|---|---|---|---|---|---|---|---|---|---|
| Condello (2018), [12] | ovarian cancer stem cells and tumors | ● | ||||||||
| Jones (2005), [23] | CT26 colon carcinoma tumors | ● | × | |||||||
| Kleman (1995), [24] | rhabdomyosarcoma cells | ● | ||||||||
| Satpathy (2007), [26] | peritoneal ovarian tumors | ● | ||||||||
| Shao (2009), [27] | ovarian tumors | ● | ||||||||
| Verma (2006), [31] | pancreatic ductal adenocarcinoma (PDA) | ● | ● | |||||||
| Hwang (2008), [32] | ovarian carcinoma cell lines; in vivo chemotherapy-sensitive (HeyA8) and chemotherapy-resistant (HeyA8-MDR and RMG2) models | ● | ● | ● | ||||||
| Jeong (2013), [33] | non-small cell lung cancer patients | ● | ||||||||
| Condello (2013), [34] | ovarian cancer cells and tumors | ● | ||||||||
| Lee (2015, 2016), [35,48] | orthotopic pancreatic xenografts and co-culture of PDA and stromal cells; | ● | ● | ● (TMA secreted TG2 crosstalk with pancreatic cancer-associated fibroblasts; × (PDA cells) | ||||||
| Kumar (2010, 2011, 2012), [36,37,38,49] | human mammary epithelial (MCF10A), breast cancer MCF7, and drug-resistant MCF7-RT cells | ● | ● | ● | ● | ● | ||||
| Cao (2008), [39] | Epithelial ovarian cancer cells | ● | ● | |||||||
| Mann (2006), [40] | pancreatic ductal carcinoma | ● | ||||||||
| Mehta (2004), [41] | metastatic breast cancer cell line MDA-MB-231 and subclones; primary vs. metastatic lymph node breast cancer tumors | ● | ● | ● | ||||||
| Cao (2012), [42] | ovarian cancer cells | ● | ● | ● | ● | |||||
| Kerr (2017), [43] | squamous cell carcinoma—SCC-13 cells | ● | ● | |||||||
| Fisher (2016), [50] | squamous cell carcinoma—SCC-13 cells | ● | ● | |||||||
| Fisher (2015), [51] | squamous cell carcinoma—SCC-13 and A431 cells | ● | ● | |||||||
| Sullivan (2017), [44] | proneural vs. mesenchymal glioma stem cells | ● | ● | ● | ||||||
| Verma (2008), [45,46] | pancreatic cancer cells; athymic nude mouse model; orthotopic PDAC tumors in nude mice; stage II PDAC patient samples | ● | ● | ● | ● | ● | ||||
| Singh (2001), [47] | HeLa endometrial cancer cells | ● | ● | |||||||
| Sima (2019), [52] | ovarian cancer cells; in vivo model measuring intraperitoneal dissemination | ● | ● | |||||||
| Yakubov (2014), [53] | SKOV3 and IGROV1 ovarian cancer cells | ● | ● | |||||||
| Oh (2015), [54] | human ovarian cancer cells | ● | ● | ● | ||||||
| Fu (2013), [55] | glioma-initiating cell lines from fresh surgical glioblastoma samples | ● | ||||||||
| Yin (2017), [56] | xenograft mouse model of glioma | ● | ● | ● | ||||||
| Kang (2018), [57] | human colorectal cancer cells—TU12 cell line derived CSCs subpopulations | ● | ● | ● | ||||||
| Bagatur (2018), [58] | Caki-2 and A-498 primary site and Caki-1 and ACHN metastatic site renal cell carcinoma cell lines | ● | ● | ● | ||||||
| Yakubov (2013), [59] | i.p. and orthotopic ovarian cancer xenografts | ● | ● | ● | ● | |||||
| Biri (2016), [60] | A431 epithelial carcinoma cells | ● | ● | |||||||
| Assi (2013), [61] | stroma of breast invasive ductal carcinomas vs. normal breast tissue | ● | ||||||||
| Jia (2020), [62] | hepatocellular carcinoma cells | ● | ● | |||||||
| Eom (2014), [63] | B16F1 mouse melanoma cells, in vitro and in vivo | ● | ● | ● | ● | |||||
| Kim (2014), [64] | in vivo mouse T cells—contact hypersensitivity reaction; ex vivo restimulation of spleen T cells with tumour lysate-loaded wild-type dendritic cells from immunized mice | × (increased effector and CD8+ memory response) | ||||||||
| Cho (2020), [65] | gastric cancer | ● (tumor-promoting inflammation) | ||||||||
| Choi (2020), [66] | triple negative breast cancer | ● | ● (PD-1/PD-L1 inhibitor-resistance) | |||||||
| Sima (2021), [67] | ovarian cancer syngeneic TG2 null mouse model | ● | ● (decreased CD8+ mediated anti-tumor immune response) | |||||||
| Yin (2016), [68] | tumor-associated macrophages from ovarian cancer | ● (promotion of intraperitoneal spheroid formation) | ||||||||
| Wang (2013), [69] | HUVEC cell culture, aorta ring assay and in vivo angiogenesis models | ● | ● | |||||||
| Wang (2017), [70] | endothelial cells (ECs) and fibroblast co-culture and ECs 3D culture models | ● | ● | ● | ||||||
| Nadalutti (2011), [71] | endothelial cells | ● | ||||||||
| Lei (2018), [72] | Tumor endothelilal cells from gastric cancer | ● | ||||||||
| Belkin (2001), [73] | glioma and fibrosarcoma cells | ● | ● | |||||||
| Satpathy (2009), [74] | ovarian cancer cells | ● | ||||||||
| Chen (2010), [75] | A431 epithelial carcinoma cells | ● | ||||||||
| Delaine-Smith (2019), [76] | organotypic 3D fibroblast/SW480 co-culture models of colorectal cancer | ● | ||||||||
| Antonyak (2009), [77] | HeLa carcinoma cells, highly aggressive breast cancer cell line MDAMB231 | ● | ● | ● |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sima, L.E.; Matei, D.; Condello, S. The Outside-In Journey of Tissue Transglutaminase in Cancer. Cells 2022, 11, 1779. https://doi.org/10.3390/cells11111779
Sima LE, Matei D, Condello S. The Outside-In Journey of Tissue Transglutaminase in Cancer. Cells. 2022; 11(11):1779. https://doi.org/10.3390/cells11111779
Chicago/Turabian StyleSima, Livia Elena, Daniela Matei, and Salvatore Condello. 2022. "The Outside-In Journey of Tissue Transglutaminase in Cancer" Cells 11, no. 11: 1779. https://doi.org/10.3390/cells11111779
APA StyleSima, L. E., Matei, D., & Condello, S. (2022). The Outside-In Journey of Tissue Transglutaminase in Cancer. Cells, 11(11), 1779. https://doi.org/10.3390/cells11111779