
A Nuclear Belt Fastens on Neural Cell Fate


Abstract
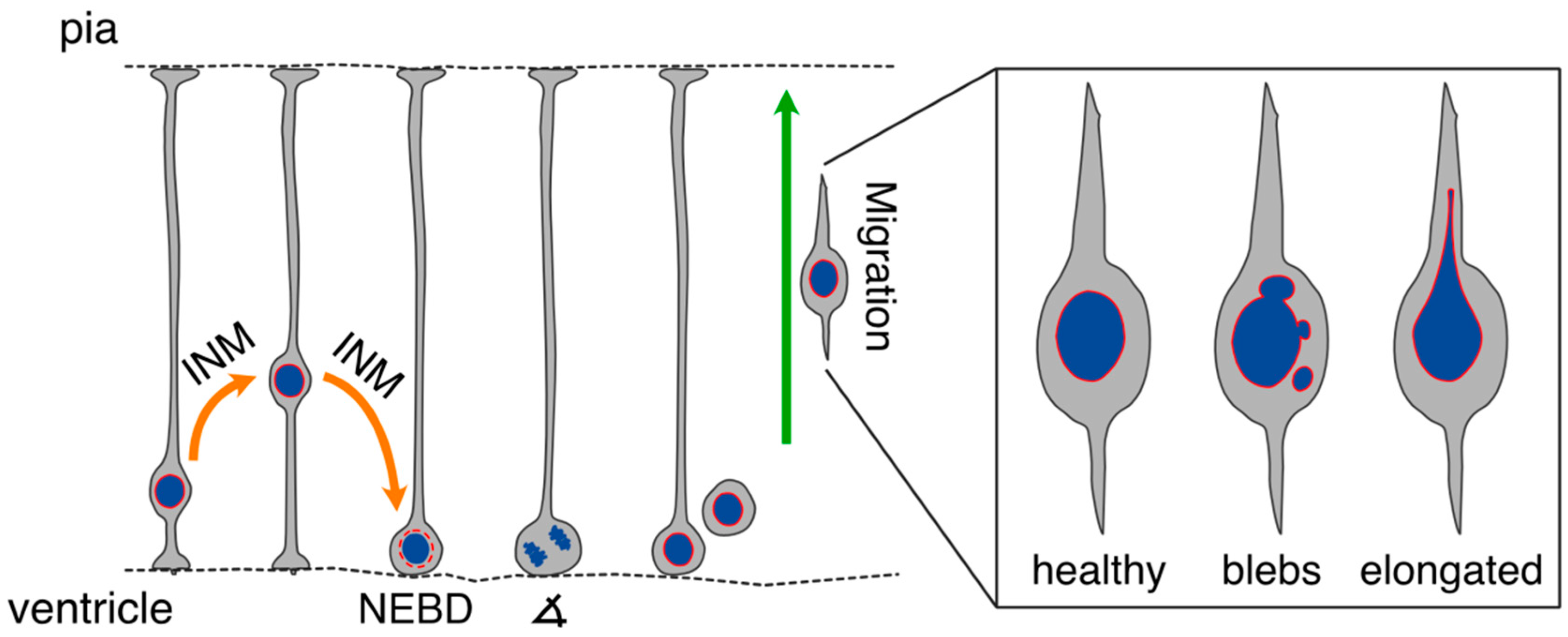
:1. Introduction
2. Nuclear Envelope during Nervous System Development and Maintenance
2.1. Nuclear Envelope and NSC Proliferation versus Differentiation
2.2. Nuclear Envelope and Migration
3. Nuclear Envelope in Chromatin Regulation
3.1. Nuclear Envelope and Transcriptional Inhibition
3.2. Nuclear Envelope in Transcriptional Activation/Bimodal Roles
3.3. Epigenetic Modifications
4. Nuclear Envelope in Aging
4.1. Cellular Aging
4.2. Aging of Adult Neural Stem Cells
4.3. NE-Dependent Biological Processes in Mature Neurons
5. Outlook on New Research Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Taverna, E.; Huttner, W.B. Neural Progenitor Nuclei IN Motion. Neuron 2010, 67, 906–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taverna, E.; Götz, M.; Huttner, W.B. The Cell Biology of Neurogenesis: Toward an Understanding of the Development and Evolution of the Neocortex. Annu. Rev. Cell Dev. Biol. 2014, 30, 465–502. [Google Scholar] [CrossRef] [PubMed]
- Urbán, N.; Guillemot, F. Neurogenesis in the Embryonic and Adult Brain: Same Regulators, Different Roles. Front. Cell. Neurosci. 2014, 8, 396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borrell, V.; Calegari, F. Mechanisms of Brain Evolution: Regulation of Neural Progenitor Cell Diversity and Cell Cycle Length. Neurosci. Res. 2014, 86, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Uzquiano, A.; Gladwyn-Ng, I.; Nguyen, L.; Reiner, O.; Götz, M.; Matsuzaki, F.; Francis, F. Cortical Progenitor Biology: Key Features Mediating Proliferation versus Differentiation. J. Neurochem. 2018, 146, 500–525. [Google Scholar] [CrossRef] [Green Version]
- Briscoe, J.; Marín, O. Looking at Neurodevelopment through a Big Data Lens. Science 2020, 369, eaaz8627. [Google Scholar] [CrossRef]
- Knoblich, J.A. Mechanisms of Asymmetric Stem Cell Division. Cell 2008, 132, 583–597. [Google Scholar] [CrossRef] [Green Version]
- Shitamukai, A.; Matsuzaki, F. Control of Asymmetric Cell Division of Mammalian Neural Progenitors. Dev. Growth Differ. 2012, 54, 277–286. [Google Scholar] [CrossRef]
- Hatanaka, Y.; Hisanaga, S.I.; Heizmann, C.W.; Murakami, F. Distinct Migratory Behavior of Early- and Late-Born Neurons Derived from the Cortical Ventricular Zone. J. Comp. Neurol. 2004, 479, 1–14. [Google Scholar] [CrossRef]
- Nadarajah, B.; Brunstrom, J.E.; Grutzendler, J.; Wong, R.O.L.; Pearlman, A.L. Two Modes of Radial Migration in Early Development of the Cerebral Cortex. Nat. Neurosci. 2001, 4, 143–150. [Google Scholar] [CrossRef]
- Lim, L.; Mi, D.; Llorca, A.; Marín, O. Development and Functional Diversification of Cortical Interneurons. Neuron 2018, 100, 294–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurkowski, M.P.; Bettio, L.; Woo, E.K.; Patten, A.; Yau, S.Y.; Gil-Mohapel, J. Beyond the Hippocampus and the SVZ: Adult Neurogenesis throughout the Brain. Front. Cell. Neurosci. 2020, 14, 293. [Google Scholar] [CrossRef] [PubMed]
- França, T.F.A.; Bitencourt, A.M.; Maximilla, N.R.; Barros, D.M.; Monserrat, J.M. Hippocampal Neurogenesis and Pattern Separation: A Meta-Analysis of Behavioral Data. Hippocampus 2017, 27, 937–950. [Google Scholar] [CrossRef] [PubMed]
- Toda, T.; Gage, F.H. Review: Adult Neurogenesis Contributes to Hippocampal Plasticity. Cell Tissue Res. 2018, 373, 693–709. [Google Scholar] [CrossRef]
- Encinas, J.M.; Michurina, T.V.; Peunova, N.; Park, J.-H.; Tordo, J.; Peterson, D.A.; Fishell, G.; Koulakov, A.; Enikolopov, G. Division-Coupled Astrocytic Differentiation and Age-Related Depletion of Neural Stem Cells in the Adult Hippocampus. Cell Stem Cell 2011, 8, 566–579. [Google Scholar] [CrossRef] [Green Version]
- Terreros-Roncal, J.; Moreno-Jiménez, E.P.; Flor-García, M.; Rodríguez-Moreno, C.B.; Trinchero, M.F.; Cafini, F.; Rábano, A.; Llorens-Martín, M. Impact of Neurodegenerative Diseases on Human Adult Hippocampal Neurogenesis. Science 2021, 374, 1106–1113. [Google Scholar] [CrossRef]
- Czapiewski, R.; Robson, M.I.; Schirmer, E.C. Anchoring a Leviathan: How the Nuclear Membrane Tethers the Genome. Front. Genetics 2016, 7, 82. [Google Scholar] [CrossRef] [Green Version]
- Dazzoni, R.; Grélard, A.; Morvan, E.; Bouter, A.; Applebee, C.J.; Loquet, A.; Larijani, B.; Dufourc, E.J. The Unprecedented Membrane Deformation of the Human Nuclear Envelope, in a Magnetic Field, Indicates Formation of Nuclear Membrane Invaginations. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Saito, M.; Sugiyama, K. Characterization of Nuclear Gangliosides in Rat Brain: Concentration, Composition, and Developmental Changes. Arch. Biochem. Biophys. 2002, 398, 153–159. [Google Scholar] [CrossRef]
- Dittmer, T.; Misteli, T. The Lamin Protein Family. Genome Biol. 2011, 12, 222. [Google Scholar] [CrossRef] [Green Version]
- Kitten, G.T.; Nigg, E.A. The CaaX Motif Is Required for Isoprenylation, Carboxyl Methylation, and Nuclear Membrane Association of Lamin B2. J. Cell Biol. 1991, 113, 13–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strambio-De-Castillia, C.; Niepel, M.; Rout, M.P. The Nuclear Pore Complex: Bridging Nuclear Transport and Gene Regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 490–501. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.H.; Hoelz, A. The Structure of the Nuclear Pore Complex (an Update). Annu. Rev. Biochem. 2019, 88, 725–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampoelz, B.; Andres-Pons, A.; Kastritis, P.; Beck, M. Structure and Assembly of the Nuclear Pore Complex. Annu. Rev. Biophys. 2019, 48, 515–536. [Google Scholar] [CrossRef]
- Ori, A.; Banterle, N.; Iskar, M.; Andrés-Pons, A.; Escher, C.; Khanh Bui, H.; Sparks, L.; Solis-Mezarino, V.; Rinner, O.; Bork, P.; et al. Cell Type-Specific Nuclear Pores: A Case in Point for Context-Dependent Stoichiometry of Molecular Machines. Mol. Syst. Biol. 2013, 9, 648. [Google Scholar] [CrossRef]
- Ori, A.; Iskar, M.; Buczak, K.; Kastritis, P.; Parca, L.; Andrés-Pons, A.; Singer, S.; Bork, P.; Beck, M. Spatiotemporal Variation of Mammalian Protein Complex Stoichiometries. Genome Biol. 2016, 17, 47. [Google Scholar] [CrossRef] [Green Version]
- Hetzer, M.W. The Nuclear Envelope. Cold Spring Harb. Perspect. Biol. 2010, 2, a000539. [Google Scholar] [CrossRef]
- Dey, G.; Baum, B. Nuclear Envelope Remodelling during Mitosis. Curr. Opin. Cell Biol. 2021, 70, 67–74. [Google Scholar] [CrossRef]
- Dauer, W.T.; Worman, H.J. The Nuclear Envelope as a Signaling Node in Development and Disease. Dev. Cell 2009, 17, 626–638. [Google Scholar] [CrossRef] [Green Version]
- Hebbar, S.; Mesngon, M.T.; Guillotte, A.M.; Desai, B.; Ayala, R.; Smith, D.S. Lis1 and Ndel1 Influence the Timing of Nuclear Envelope Breakdown in Neural Stem Cells. J. Cell Biol. 2008, 182, 1063–1071. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Sharov, A.A.; McDole, K.; Cheng, M.; Hao, H.; Fan, C.M.; Gaiano, N.; Ko, M.S.H.; Zheng, Y. Mouse B-Type Lamins Are Required for Proper Organogenesis but not by Embryonic Stem Cells. Science 2011, 334, 1706–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergnes, L.; Péterfy, M.; Bergo, M.O.; Young, S.G.; Reue, K. Lamin B1 Is Required for Mouse Development and Nuclear Integrity. Proc. Natl. Acad. Sci. USA 2004, 101, 10428–10433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahajani, S.; Giacomini, C.; Marinaro, F.; De Pietri Tonelli, D.; Contestabile, A.; Gasparini, L. Lamin B1 Levels Modulate Differentiation into Neurons during Embryonic Corticogenesis. Sci. Rep. 2017, 7, 4897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedrosian, T.A.; Houtman, J.; Eguiguren, J.S.; Ghassemzadeh, S.; Rund, N.; Novaresi, N.M.; Hu, L.; Parylak, S.L.; Denli, A.M.; Randolph-Moore, L.; et al. Lamin B1 Decline Underlies Age-related Loss of Adult Hippocampal Neurogenesis. EMBO J. 2021, 40, e105819. [Google Scholar] [CrossRef]
- bin Imtiaz, M.K.; Jaeger, B.N.; Bottes, S.; Machado, R.A.C.; Vidmar, M.; Moore, D.L.; Jessberger, S. Declining Lamin B1 Expression Mediates Age-Dependent Decreases of Hippocampal Stem Cell Activity. Cell Stem Cell 2021, 28, 967–977.e8. [Google Scholar] [CrossRef]
- Del Bene, F.; Wehman, A.M.; Link, B.A.; Baier, H. Regulation of Neurogenesis by Interkinetic Nuclear Migration through an Apical-Basal Notch Gradient. Cell 2008, 134, 1055–1065. [Google Scholar] [CrossRef] [Green Version]
- Swift, J.; Ivanovska, I.L.; Buxboim, A.; Harada, T.; Dingal, P.C.D.P.; Pinter, J.; Pajerowski, J.D.; Spinler, K.R.; Shin, J.W.; Tewari, M.; et al. Nuclear Lamin-A Scales with Tissue Stiffness and Enhances Matrix-Directed Differentiation. Science 2013, 341, 1240104. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.J.; Coffinier, C.; Choe, Y.; Beigneux, A.P.; Davies, B.S.J.; Yang, S.H.; Barnes, R.H.; Hong, J.; Sun, T.; Pleasure, S.J.; et al. Regulation of Prelamin A but not Lamin C by MiR-9, a Brain-Specific MicroRNA. Proc. Natl. Acad. Sci. USA 2012, 109, 423–431. [Google Scholar] [CrossRef] [Green Version]
- Nakazawa, N.; Kengaku, M. Mechanical Regulation of Nuclear Translocation in Migratory Neurons. Front. Cell Dev. Biol. 2020, 8, 150. [Google Scholar] [CrossRef] [Green Version]
- Rober, R.A.; Weber, K.; Osborn, M. Differential Timing of Nuclear Lamin A/C Expression in the Various Organs of the Mouse Embryo and the Young Animal: A Developmental Study. Development 1989, 105, 365–378. [Google Scholar] [CrossRef]
- Lammerding, J.; Fong, L.G.; Ji, J.Y.; Reue, K.; Stewart, C.L.; Young, S.G.; Lee, R.T. Lamins a and C but not Lamin B1 Regulate Nuclear Mechanics. J. Biol. Chem. 2006, 281, 25768–25780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takamori, Y.; Tamura, Y.; Kataoka, Y.; Cui, Y.; Seo, S.; Kanazawa, T.; Kurokawa, K.; Yamada, H. Differential Expression of Nuclear Lamin, the Major Component of Nuclear Lamina, during Neurogenesis in Two Germinal Regions of Adult Rat Brain. Eur. J. Neurosci. 2007, 25, 1653–1662. [Google Scholar] [CrossRef] [PubMed]
- Coffinier, C.; Chang, S.Y.; Nobumori, C.; Tu, Y.; Farber, E.A.; Toth, J.I.; Fong, L.G.; Young, S.G. Abnormal Development of the Cerebral Cortex and Cerebellum in the Setting of Lamin B2 Deficiency. Proc. Natl. Acad. Sci. USA 2010, 107, 5076–5081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coffinier, C.; Jung, H.J.; Nobumori, C.; Chang, S.; Tu, Y.; Barnes, R.H.; Yoshinaga, Y.; De Jong, P.J.; Vergnes, L.; Reue, K.; et al. Deficiencies in Lamin B1 and Lamin B2 Cause Neurodevelopmental Defects and Distinct Nuclear Shape Abnormalities in Neurons. Mol. Biol. Cell 2011, 22, 4683–4693. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.Y.; Yang, Y.; Weston, T.A.; Belling, J.N.; Heizer, P.; Tu, Y.; Kim, P.; Edillo, L.; Jonas, S.J.; Weiss, P.S.; et al. An Absence of Lamin B1 in Migrating Neurons Causes Nuclear Membrane Ruptures and Cell Death. Proc. Natl. Acad. Sci. USA 2019, 116, 25870–25879. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lei, K.; Yuan, X.; Wu, X.; Zhuang, Y.; Xu, T.; Xu, R.; Han, M. SUN1/2 and Syne/Nesprin-1/2 Complexes Connect Centrosome to the Nucleus during Neurogenesis and Neuronal Migration in Mice. Neuron 2009, 64, 173–187. [Google Scholar] [CrossRef] [Green Version]
- Kracklauer, M.P.; Banks, S.M.L.; Xie, X.; Wu, Y.; Fischer, J.A. Drosophila Klaroid Encodes a SUN Domain Protein Required for Klarsicht Localization to the Nuclear Envelope and Nuclear Migration in the Eye. Fly 2007, 1, 75–85. [Google Scholar] [CrossRef] [Green Version]
- Lupu, F.; Alves, A.; Anderson, K.; Doye, V.; Lacy, E. Nuclear Pore Composition Regulates Neural Stem/Progenitor Cell Differentiation in the Mouse Embryo. Dev. Cell 2008, 14, 831–842. [Google Scholar] [CrossRef] [Green Version]
- Hu, D.J.K.; Baffet, A.D.; Nayak, T.; Akhmanova, A.; Doye, V.; Vallee, R.B. Dynein Recruitment to Nuclear Pores Activates Apical Nuclear Migration and Mitotic Entry in Brain Progenitor Cells. Cell 2013, 154, 1300. [Google Scholar] [CrossRef] [Green Version]
- Kohwi, M.; Lupton, J.R.; Lai, S.L.; Miller, M.R.; Doe, C.Q. Developmentally Regulated Subnuclear Genome Reorganization Restricts Neural Progenitor Competence in Drosophila. Cell 2013, 152, 97–108. [Google Scholar] [CrossRef] [Green Version]
- Solovei, I.; Wang, A.S.; Thanisch, K.; Schmidt, C.S.; Krebs, S.; Zwerger, M.; Cohen, T.V.; Devys, D.; Foisner, R.; Peichl, L.; et al. LBR and Lamin A/C Sequentially Tether Peripheral Heterochromatin and Inversely Regulate Differentiation. Cell 2013, 152, 584–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clowney, E.J.; Legros, M.A.; Mosley, C.P.; Clowney, F.G.; Markenskoff-Papadimitriou, E.C.; Myllys, M.; Barnea, G.; Larabell, C.A.; Lomvardas, S. Nuclear Aggregation of Olfactory Receptor Genes Governs Their Monogenic Expression. Cell 2012, 151, 724–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Steensel, B.; Belmont, A.S. Lamina-Associated Domains: Links with Chromosome Architecture, Heterochromatin, and Gene Repression. Cell 2017, 169, 780–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniura, H.; Glass, C.; Gerace, L. A Chromatin Binding Site in the Tail Domain of Nuclear Lamins That Interacts with Core Histones. J. Cell Biol. 1995, 131, 33–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somech, R.; Shaklai, S.; Geller, O.; Amariglio, N.; Simon, A.J.; Rechavi, G.; Gal-Yam, E.N. The Nuclear-Envelope Protein and Transcriptional Repressor LAP2β Interacts with HDAC3 at the Nuclear Periphery, and Induces Histone H4 Deacetylation. J. Cell Sci. 2005, 118, 4017–4025. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.; Li, M.; Shao, S.; Li, C.; Ai, S.; Xue, B.; Hou, Y.; Zhang, Y.; Li, R.; Fan, X.; et al. Nuclear Peripheral Chromatin-Lamin B1 Interaction Is Required for Global Integrity of Chromatin Architecture and Dynamics in Human Cells. Protein Cell 2020, 13, 258–280. [Google Scholar] [CrossRef]
- Ahanger, S.H.; Delgado, R.N.; Gil, E.; Cole, M.A.; Zhao, J.; Hong, S.J.; Kriegstein, A.R.; Nowakowski, T.J.; Pollen, A.A.; Lim, D.A. Distinct Nuclear Compartment-Associated Genome Architecture in the Developing Mammalian Brain. Nat. Neurosci. 2021, 24, 1235–1242. [Google Scholar] [CrossRef]
- Goldowitz, D.; Mullen, R.J. Nuclear Morphology of Ichthyosis Mutant Mice as a Cell Marker in Chimeric Brain. Dev. Biol. 1982, 89, 261–267. [Google Scholar] [CrossRef]
- Schultz, L.D.; Lyons, B.L.; Burzenski, L.M.; Gott, B.; Samuels, R.; Schweitzer, P.A.; Dreger, C.; Herrmann, H.; Kalscheuer, V.; Olins, A.L.; et al. Mutations at the Mouse Ichthyosis Locus Are within the Lamin B Receptor Gene: A Single Gene Model for Human Pelger-Huët Anomaly. Hum. Mol. Genet. 2003, 12, 61–69. [Google Scholar] [CrossRef]
- Solovei, I.; Kreysing, M.; Lanctôt, C.; Kösem, S.; Peichl, L.; Cremer, T.; Guck, J.; Joffe, B. Nuclear Architecture of Rod Photoreceptor Cells Adapts to Vision in Mammalian Evolution. Cell 2009, 137, 356–368. [Google Scholar] [CrossRef] [Green Version]
- Finlan, L.E.; Sproul, D.; Thomson, I.; Boyle, S.; Kerr, E.; Perry, P.; Ylstra, B.; Chubb, J.R.; Bickmore, W.A. Recruitment to the Nuclear Periphery Can Alter Expression of Genes in Human Cells. PLoS Genet. 2008, 4, e1000039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falk, M.; Feodorova, Y.; Naumova, N.; Imakaev, M.; Lajoie, B.R.; Leonhardt, H.; Joffe, B.; Dekker, J.; Fudenberg, G.; Solovei, I.; et al. Heterochromatin Drives Compartmentalization of Inverted and Conventional Nuclei. Nature 2019, 570, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Peric-Hupkes, D.; Meuleman, W.; Pagie, L.; Bruggeman, S.W.M.; Solovei, I.; Brugman, W.; Gräf, S.; Flicek, P.; Kerkhoven, R.M.; van Lohuizen, M.; et al. Molecular Maps of the Reorganization of Genome-Nuclear Lamina Interactions during Differentiation. Mol. Cell 2010, 38, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Lucas, T.; Hafer, T.L.; Zhang, H.G.; Molotkova, N.; Kohwi, M. Discrete Cis-Acting Element Regulates Developmentally Timed Gene-Lamina Relocation and Neural Progenitor Competence in Vivo. Dev. Cell 2021, 56, 2649–2663.e6. [Google Scholar] [CrossRef]
- Capelson, M.; Hetzer, M.W. The Role of Nuclear Pores in Gene Regulation, Development and Disease. EMBO Rep. 2009, 10, 697–705. [Google Scholar] [CrossRef]
- Ibarra, A.; Hetzer, M.W. Nuclear Pore Proteins and the Control of Genome Functions. Genes Dev. 2015, 29, 337–349. [Google Scholar] [CrossRef] [Green Version]
- Jacinto, F.V.; Benner, C.; Hetzer, M.W. The Nucleoporin Nup153 Regulates Embryonic Stem Cell Pluripotency through Gene Silencing. Genes Dev. 2015, 29, 1224–1238. [Google Scholar] [CrossRef] [Green Version]
- Ikegami, K.; Secchia, S.; Almakki, O.; Lieb, J.D.; Moskowitz, I.P. Phosphorylated Lamin A/C in the Nuclear Interior Binds Active Enhancers Associated with Abnormal Transcription in Progeria. Dev. Cell 2020, 52, 699–713.e11. [Google Scholar] [CrossRef]
- D’Angelo, M.A.; Gomez-Cavazos, J.S.; Mei, A.; Lackner, D.H.; Hetzer, M.W. A Change in Nuclear Pore Complex Composition Regulates Cell Differentiation. Dev. Cell 2012, 22, 446–458. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Franks, T.M.; Marchetto, M.C.; Gage, F.H.; Hetzer, M.W. Dynamic Association of NUP98 with the Human Genome. PLoS Genet. 2013, 9, e1003308. [Google Scholar] [CrossRef]
- Ibarra, A.; Benner, C.; Tyagi, S.; Cool, J.; Hetzer, M.W. Nucleoporin-Mediated Regulation of Cell Identity Genes. Genes Dev. 2016, 30, 2253–2258. [Google Scholar] [CrossRef] [PubMed]
- Toda, T.; Hsu, J.Y.; Linker, S.B.; Hu, L.; Schafer, S.T.; Mertens, J.; Jacinto, F.V.; Hetzer, M.W.; Gage, F.H. Nup153 Interacts with Sox2 to Enable Bimodal Gene Regulation and Maintenance of Neural Progenitor Cells. Cell Stem Cell 2017, 21, 618–634.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takizawa, T.; Meshorer, E. Chromatin and Nuclear Architecture in the Nervous System. Trends Neurosci. 2008, 31, 343–352. [Google Scholar] [CrossRef]
- Stephens, A.D.; Liu, P.Z.; Banigan, E.J.; Almassalha, L.M.; Backman, V.; Adam, S.A.; Goldman, R.D.; Marko, J.F. Chromatin Histone Modifications and Rigidity Affect Nuclear Morphology Independent of Lamins. Mol. Biol. Cell 2018, 29, 220–233. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, M.A.; Raices, M.; Panowski, S.H.; Hetzer, M.W. Age-Dependent Deterioration of Nuclear Pore Complexes Causes a Loss of Nuclear Integrity in Postmitotic Cells. Cell 2009, 136, 284–295. [Google Scholar] [CrossRef] [Green Version]
- Mertens, J.; Paquola, A.C.M.; Ku, M.; Hatch, E.; Böhnke, L.; Ladjevardi, S.; McGrath, S.; Campbell, B.; Lee, H.; Herdy, J.R.; et al. Directly Reprogrammed Human Neurons Retain Aging-Associated Transcriptomic Signatures and Reveal Age-Related Nucleocytoplasmic Defects. Cell Stem Cell 2015, 17, 705–718. [Google Scholar] [CrossRef] [Green Version]
- Frost, B.; Bardai, F.H.; Feany, M.B. Lamin Dysfunction Mediates Neurodegeneration in Tauopathies. Curr. Biol. 2016, 26, 129–136. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.H.; Li, M.; Qu, J.; Belmonte, J.C.I. Gating Neural Development and Aging via Nuclear Pores. Cell Res. 2012, 22, 1212–1214. [Google Scholar] [CrossRef] [Green Version]
- Shani, V.; Safory, H.; Szargel, R.; Wang, N.; Cohen, T.; Elghani, F.A.; Hamza, H.; Savyon, M.; Radzishevsky, I.; Shaulov, L.; et al. Physiological and Pathological Roles of LRRK2 in the Nuclear Envelope Integrity. Hum. Mol. Genet. 2019, 28, 3982–3996. [Google Scholar] [CrossRef]
- Eftekharzadeh, B.; Daigle, J.G.; Kapinos, L.E.; Coyne, A.; Schiantarelli, J.; Carlomagno, Y.; Cook, C.; Miller, S.J.; Dujardin, S.; Amaral, A.S.; et al. Tau Protein Disrupts Nucleocytoplasmic Transport in Alzheimer’s Disease. Neuron 2018, 99, 925–940.e7. [Google Scholar] [CrossRef] [Green Version]
- Baron, O.; Boudi, A.; Dias, C.; Schilling, M.; Nölle, A.; Vizcay-Barrena, G.; Rattray, I.; Jungbluth, H.; Scheper, W.; Fleck, R.A.; et al. Stall in Canonical Autophagy-Lysosome Pathways Prompts Nucleophagy-Based Nuclear Breakdown in Neurodegeneration. Curr. Biol. 2017, 27, 3626–3642.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiLoreto, R.; Murphy, C.T. The Cell Biology of Aging. Mol. Biol. Cell 2015, 26, 4524–4531. [Google Scholar] [CrossRef] [PubMed]
- Kristiani, L.; Kim, M.; Kim, Y. Role of the Nuclear Lamina in Age-Associated Nuclear Reorganization and Inflammation. Cells 2020, 9, 718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regot, S.; de Nadal, E.; Rodríguez-Navarro, S.; González-Novo, A.; Pérez-Fernandez, J.; Gadal, O.; Seisenbacher, G.; Ammerer, G.; Posas, F. The Hog1 Stress-Activated Protein Kinase Targets Nucleoporins to Control MRNA Export upon Stress*. J. Biol. Chem. 2013, 288, 17384–17398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, T.; Pan, Y.; Kao, S.-Y.; Li, C.; Kohane, I.; Chan, J.; Yankner, B.A. Gene Regulation and DNA Damage in the Ageing Human Brain. Nature 2004, 429, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Tyler, J.K. Epigenetics and Aging. Sci. Adv. 2016, 2, e1600584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stegeman, R.; Weake, V.M. Transcriptional Signatures of Aging. J. Mol. Biol. 2017, 429, 2427–2437. [Google Scholar] [CrossRef]
- Toyama, B.H.; Savas, J.N.; Park, S.K.; Harris, M.S.; Ingolia, N.T.; Yates, J.R.; Hetzer, M.W. Identification of Long-Lived Proteins Reveals Exceptional Stability of Essential Cellular Structures. Cell 2013, 154, 971–982. [Google Scholar] [CrossRef] [Green Version]
- Dörrbaum, A.R.; Kochen, L.; Langer, J.D.; Schuman, E.M. Local and Global Influences on Protein Turnover in Neurons and Glia. Elife 2018, 7, e34202. [Google Scholar] [CrossRef]
- Moore, D.L.; Pilz, G.A.; Araúzo-Bravo, M.J.; Barral, Y.; Jessberger, S. A Mechanism for the Segregation of Age in Mammalian Neural Stem Cells. Science 2015, 349, 1334–1338. [Google Scholar] [CrossRef]
- Clay, L.; Caudron, F.; Denoth-Lippuner, A.; Boettcher, B.; Frei, S.B.; Snapp, E.L.; Barral, Y. A Sphingolipid-Dependent Diffusion Barrier Confines ER Stress to the Yeast Mother Cell. Elife 2014, 2014, e01883. [Google Scholar] [CrossRef]
- Fuentealba, L.C.; Eivers, E.; Geissert, D.; Taelman, V.; De Robertis, E.M. Asymmetric Mitosis: Unequal Segregation of Proteins Destined for Degradation. Proc. Natl. Acad. Sci. USA 2008, 105, 7732–7737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishioka, Y.; Imaizumi, H.; Imada, J.; Katahira, J.; Matsuura, N.; Hieda, M. SUN1 Splice Variants, SUN1_888, SUN1_785, and Predominant SUN1_916, Variably Function in Directional Cell Migration. Nucleus 2016, 7, 572–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabut, G.; Lénárt, P.; Ellenberg, J. Dynamics of Nuclear Pore Complex Organization through the Cell Cycle. Curr. Opin. Cell Biol. 2004, 16, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The Proteostasis Network and Its Decline in Ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef]
- Soto, C. Unfolding the Role of Protein Misfolding in Neurodegenerative Diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [Google Scholar] [CrossRef]
- Liu, G.H.; Qu, J.; Suzuki, K.; Nivet, E.; Li, M.; Montserrat, N.; Yi, F.; Xu, X.; Ruiz, S.; Zhang, W.; et al. Progressive Degeneration of Human Neural Stem Cells Caused by Pathogenic LRRK2. Nature 2012, 491, 603–607. [Google Scholar] [CrossRef] [Green Version]
- Gasset-Rosa, F.; Chillon-Marinas, C.; Goginashvili, A.; Atwal, R.S.; Artates, J.W.; Tabet, R.; Wheeler, V.C.; Bang, A.G.; Cleveland, D.W.; Lagier-Tourenne, C. Polyglutamine-Expanded Huntingtin Exacerbates Age-Related Disruption of Nuclear Integrity and Nucleocytoplasmic Transport. Neuron 2017, 94, 48–57.e4. [Google Scholar] [CrossRef] [Green Version]
- Grima, J.C.; Daigle, J.G.; Arbez, N.; Cunningham, K.C.; Zhang, K.; Ochaba, J.; Geater, C.; Morozko, E.; Stocksdale, J.; Glatzer, J.C.; et al. Mutant Huntingtin Disrupts the Nuclear Pore Complex. Neuron 2017, 94, 93–107.e6. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Donnelly, C.J.; Haeusler, A.R.; Grima, J.C.; Machamer, J.B.; Steinwald, P.; Daley, E.L.; Miller, S.J.; Cunningham, K.M.; Vidensky, S.; et al. The C9orf72 Repeat Expansion Disrupts Nucleocytoplasmic Transport. Nature 2015, 525, 56–61. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.I.; Nagakannan, P.; Shcholok, T.; Contu, F.; Mai, S.; Albensi, B.C.; Del Bigio, M.R.; Wang, J.-F.; Sharoar, M.G.; Yan, R.; et al. Regulatory Role of Cathepsin L in Induction of Nuclear Laminopathy in Alzheimer’s Disease. Aging Cell 2022, 21, e13531. [Google Scholar] [CrossRef] [PubMed]
- Rué, L.; Alcalá-Vida, R.; López-Soop, G.; Creus-Muncunill, J.; Alberch, J.; Pérez-Navarro, E. Early Down-Regulation of PKCδ as a Pro-Survival Mechanism in Huntington’s Disease. NeuroMolecular Med. 2014, 16, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Padiath, Q.S.; Saigoh, K.; Schiffmann, R.; Asahara, H.; Yamada, T.; Koeppen, A.; Hogan, K.; Ptáček, L.J.; Fu, Y.H. Lamin B1 Duplications Cause Autosomal Dominant Leukodystrophy. Nat. Genet. 2006, 38, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Ferrera, D.; Canale, C.; Marotta, R.; Mazzaro, N.; Gritti, M.; Mazzanti, M.; Capellari, S.; Cortelli, P.; Gasparini, L. Lamin B1 Overexpression Increases Nuclear Rigidity in Autosomal Dominant Leukodystrophy Fibroblasts. FASEB J. 2014, 28, 3906–3918. [Google Scholar] [CrossRef] [Green Version]
- Alcalá-Vida, R.; Garcia-Forn, M.; Castany-Pladevall, C.; Creus-Muncunill, J.; Ito, Y.; Blanco, E.; Golbano, A.; Crespí-Vázquez, K.; Parry, A.; Slater, G.; et al. Neuron Type-specific Increase in Lamin B1 Contributes to Nuclear Dysfunction in Huntington’s Disease. EMBO Mol. Med. 2021, 13, e12105. [Google Scholar] [CrossRef]
- Korfali, N.; Wilkie, G.S.; Swanson, S.K.; Srsen, V.; de las Heras, J.; Batrakou, D.G.; Malik, P.; Zuleger, N.; Kerr, A.R.W.; Florens, L.; et al. The Nuclear Envelope Proteome Differs Notably between Tissues. Nucleus 2012, 3, 552–564. [Google Scholar] [CrossRef] [Green Version]
- Worman, H.J.; Schirmer, E.C. Nuclear Membrane Diversity: Underlying Tissue-Specific Pathologies in Disease? Curr. Opin. Cell Biol. 2015, 34, 101–112. [Google Scholar] [CrossRef] [Green Version]
- Tullio-pelet, A.; Salomon, R.; Hadj-rabia, S.; Mugnier, C.; De Laet, M.; Bakiri, F.; Brottier, P.; Cattolico, L.; Penet, C.; Bégeot, M.; et al. Mutant WD-Repeat Protein in Triple-A Syndrome. Nat. Genet. 2000, 26, 332–335. [Google Scholar] [CrossRef]
- Ekström, A.B.; Hakenäs-Plate, L.; Samuelsson, L.; Tulinius, M.; Wentz, E. Autism Spectrum Conditons in Myotonic Dystrophy Type 1: A Study on 57 Individuals with Congenital and Childhood Forms. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2008, 147, 918–926. [Google Scholar] [CrossRef]
- Kenny, E.M.; Cormican, P.; Furlong, S.; Heron, E.; Kenny, G.; Fahey, C.; Kelleher, E.; Ennis, S.; Tropea, D.; Anney, R.; et al. Excess of Rare Novel Loss-of-Function Variants in Synaptic Genes in Schizophrenia and Autism Spectrum Disorders. Mol. Psychiatry 2014, 19, 872–879. [Google Scholar] [CrossRef] [Green Version]
- Bruel, A.L.; Vitobello, A.; Thiffault, I.; Manwaring, L.; Willing, M.; Agrawal, P.B.; Bayat, A.; Kitzler, T.M.; Brownstein, C.A.; Genetti, C.A.; et al. ITSN1: A Novel Candidate Gene Involved in Autosomal Dominant Neurodevelopmental Disorder Spectrum. Eur. J. Hum. Genet. 2022, 30, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Basel-Vanagaite, L.; Muncher, L.; Straussberg, R.; Pasmanik-Chor, M.; Yahav, M.; Rainshtein, L.; Walsh, C.A.; Magal, N.; Taub, E.; Drasinover, V.; et al. Mutated Nup62 Causes Autosomal Recessive Infantile Bilateral Striatal Necrosis. Ann. Neurol. 2006, 60, 214–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravindran, E.; Jühlen, R.; Vieira-Vieira, C.H.; Ha, T.; Salzberg, Y.; Fichtman, B.; Luise-Becker, L.; Martins, N.; Picker-Minh, S.; Bessa, P.; et al. Expanding the Phenotype of NUP85 Mutations beyond Nephrotic Syndrome to Primary Autosomal Recessive Microcephaly and Seckel Syndrome Spectrum Disorders. Hum. Mol. Genet. 2021, 30, 2068–2081. [Google Scholar] [CrossRef] [PubMed]
- Rosti, R.O.; Sotak, B.N.; Bielas, S.L.; Bhat, G.; Silhavy, J.L.; Aslanger, A.D.; Altunoglu, U.; Bilge, I.; Tasdemir, M.; Yzaguirrem, A.D.; et al. Homozygous Mutation in NUP107 Leads to Microcephaly with Steroid-Resistant Nephrotic Condition Similar to Galloway-Mowat Syndrome. J. Med. Genet. 2017, 54, 399–403. [Google Scholar] [CrossRef]
- Iossifov, I.; Ronemus, M.; Levy, D.; Wang, Z.; Hakker, I.; Rosenbaum, J.; Yamrom, B.; Lee, Y.-h.; Narzisi, G.; Leotta, A.; et al. De Novo Gene Disruptions in Children on the Autistic Spectrum. Neuron 2012, 74, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Fujita, A.; Tsukaguchi, H.; Koshimizu, E.; Nakazato, H.; Itoh, K.; Kuraoka, S.; Komohara, Y.; Shiina, M.; Nakamura, S.; Kitajima, M.; et al. Homozygous Splicing Mutation in NUP133 Causes Galloway–Mowat Syndrome. Ann. Neurol. 2018, 84, 814–828. [Google Scholar] [CrossRef]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584.e23. [Google Scholar] [CrossRef]
- Shamseldin, H.E.; Makhseed, N.; Ibrahim, N.; Al-Sheddi, T.; Alobeid, E.; Abdulwahab, F.; Alkuraya, F.S. NUP214 Deficiency Causes Severe Encephalopathy and Microcephaly in Humans. Hum. Genet. 2019, 138, 221–229. [Google Scholar] [CrossRef]
- Jiang, J.; Wang, Y.E.; Palazzo, A.F.; Shen, Q. Roles of Nucleoporin RanBP2/Nup358 in Acute Necrotizing Encephalopathy Type 1 (ANE1) and Viral Infection. Int. J. Mol. Sci. 2022, 23, 3548. [Google Scholar] [CrossRef]
- Talkowski, M.E.; Rosenfeld, J.A.; Blumenthal, I.; Pillalamarri, V.; Chiang, C.; Heilbut, A.; Ernst, C.; Hanscom, C.; Rossin, E.; Lindgren, A.M.; et al. Sequencing Chromosomal Abnormalities Reveals Neurodevelopmental Loci that Confer Risk across Diagnostic Boundaries. Cell 2012, 149, 525–537. [Google Scholar] [CrossRef] [Green Version]
- Gros-Louis, F.; Dupré, N.; Dion, P.; Fox, M.A.; Laurent, S.; Verreault, S.; Sanes, J.R.; Bouchard, J.P.; Rouleau, G.A. Mutations in SYNE1 Lead to a Newly Discovered Form of Autosomal Recessive Cerebellar Ataxia. Nat. Genet. 2007, 39, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Green, E.K.; Grozeva, D.; Forty, L.; Gordon-Smith, K.; Russell, E.; Farmer, A.; Hamshere, M.; Jones, I.R.; Jones, L.; McGuffin, P.; et al. Association at SYNE1 in Both Bipolar Disorder and Recurrent Major Depression. Mol. Psychiatry 2013, 18, 614–617. [Google Scholar] [CrossRef] [PubMed]
- O’Roak, B.J.; Deriziotis, P.; Lee, C.; Vives, L.; Schwartz, J.J.; Girirajan, S.; Karakoc, E.; MacKenzie, A.P.; Ng, S.B.; Baker, C.; et al. Exome Sequencing in Sporadic Autism Spectrum Disorders Identifies Severe de Novo Mutations. Nat. Genet. 2011, 43, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Young, N.; Asif, M.; Jackson, M.; Fernández-Mayoralas, D.M.; de la Peña, M.J.; Calleja-Pérez, B.; Álvarez, S.; Hunter-Featherstone, E.; Noegel, A.A.; Höhne, W.; et al. Biallelic SYNE2 Missense Mutations Leading to Nesprin-2 Giant Hypo-Expression Are Associated with Intellectual Disability and Autism. Genes 2021, 12, 1294. [Google Scholar] [CrossRef]
- Pinto, D.; Pagnamenta, A.T.; Klei, L.; Anney, R.; Merico, D.; Regan, R.; Conroy, J.; Magalhaes, T.R.; Correia, C.; Abrahams, B.S.; et al. Functional Impact of Global Rare Copy Number Variation in Autism Spectrum Disorders. Nature 2010, 466, 368–372. [Google Scholar] [CrossRef] [Green Version]
- Le Duc, D.; Giulivi, C.; Hiatt, S.M.; Napoli, E.; Panoutsopoulos, A.; de Crescenzo, A.H.; Kotzaeridou, U.; Syrbe, S.; Anagnostou, E.; Azage, M.; et al. Pathogenic WDFY3 Variants Cause Neurodevelopmental Disorders and Opposing Effects on Brain Size. Brain 2019, 142, 2617–2630. [Google Scholar] [CrossRef]
- Liu, X.; Malenfant, P.; Reesor, C.; Lee, A.; Hudson, M.L.; Harvard, C.; Qiao, Y.; Persico, A.M.; Cohen, I.L.; Chudley, A.E.; et al. 2p15-P16.1 Microdeletion Syndrome: Molecular Characterization and Association of the OTX1 and XPO1 Genes with Autism Spectrum Disorders. Eur. J. Hum. Genet. 2011, 19, 1264–1270. [Google Scholar] [CrossRef]
- Karoutas, A.; Szymanski, W.; Rausch, T.; Guhathakurta, S.; Rog-Zielinska, E.A.; Peyronnet, R.; Seyfferth, J.; Chen, H.R.; de Leeuw, R.; Herquel, B.; et al. The NSL Complex Maintains Nuclear Architecture Stability via Lamin A/C Acetylation. Nat. Cell Biol. 2019, 21, 1248–1260. [Google Scholar] [CrossRef]
- Malhas, A.; Goulbourne, C.; Vaux, D.J. The Nucleoplasmic Reticulum: Form and Function. Trends Cell Biol. 2011, 21, 362–373. [Google Scholar] [CrossRef]
- Meyer, K.; Feldman, H.M.; Lu, T.; Drake, D.; Lim, E.T.; Ling, K.H.; Bishop, N.A.; Pan, Y.; Seo, J.; Lin, Y.T.; et al. REST and Neural Gene Network Dysregulation in IPSC Models of Alzheimer’s Disease. Cell Rep. 2019, 26, 1112–1127.e9. [Google Scholar] [CrossRef] [Green Version]
- Biedzinski, S.; Agsu, G.; Vianay, B.; Delord, M.; Blanchoin, L.; Larghero, J.; Faivre, L.; Théry, M.; Brunet, S. Microtubules Control Nuclear Shape and Gene Expression during Early Stages of Hematopoietic Differentiation. EMBO J. 2020, 39, e103957. [Google Scholar] [CrossRef] [PubMed]
- Cebrián-Silla, A.; Alfaro-Cervelló, C.; Herranz-Pérez, V.; Kaneko, N.; Park, D.H.; Sawamoto, K.; Alvarez-Buylla, A.; Lim, D.A.; García-Verdugo, J.M. Unique Organization of the Nuclear Envelope in the Post-Natal Quiescent Neural Stem Cells. Stem Cell Rep. 2017, 9, 203–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| NE Component | Species | Cell Type | Phenotype | Reference |
|---|---|---|---|---|
| Klaroid or klarsicht | Drosophila melanogaster | Photoreceptor | Rough eyes | [47] |
| Lamin | Drosophila melanogaster | Neuroblast | Extended neurogenesis | [50] |
| Lamin B1 or B2 | Mus musculus | Radial glial cells and neurons | Disorganized cortex, smaller cerebellum | [31,43,44] |
| Lbr | Nocturnal mammals | Rods | Heterochromatin inversion | [51] |
| Lbr | Mus musculus | Olfactory neurons | Co-expression of multiple olfactory genes, and glomeruli mistargeting | [52] |
| Lis1 | Mus musculus | Radial glial cells | Lissencephalic brain | [30] |
| Nesprin 2 | Danio rerio | Retinal ganglion cells | Smaller eyes | [36] |
| Nup133 | Mus musculus, Rattus norvegicus | Radial glial cells and neurons | Exencephalic neural tube | [48,49] |
| Sun 1/2 or nesprin 1/2 | Mus musculus | Radial glial cells and neurons | Inverted cortex and enlarged ventricles | [46] |
| NE Component | Species | Cell Type | Change from Physiological State | NE Component Changes | Phenotype | Refs. |
|---|---|---|---|---|---|---|
| RanBP17 | Homo sapiens | iN | Aging | Age-dependent transcription level decrease | Decline in nucleocytoplasmic compartmentalization, age-associated transcriptome | [76] |
| RanGAP1, Gle1, Nup62 | Mus musculus | Cortical neurons | mHtt (HD) | Interaction with mHtt, subsequent accumulation and perinuclear mislocalization | Disrupted nucleocytoplasmic transport, leaky and compromised nuclear pore | [98,99] |
| RanGAP1, Nup107, Nup205 | Homo sapiens | Neurons from patient-derived iPSC | C9orf72 mutation (ALS/FTD) | Accumulation and perinuclear mislocalization | Nucleocytoplasmic transport defects | [100] |
| Nup98 | Homo sapiens | Hippocampal neurons | pTau (AD) | Interacts with pTau, accumulation and perinuclear mislocalization | Hampers nucleocytoplasmic transport, disrupts NPC distribution, accelerates and stabilizes pTau aggregation | [80] |
| Msp300 and koi (homologs of human Nesprin and SUN1) | Drosophila melanogaster | Cortical neurons | pTau (AD) | Interaction with pTau subsequent induction filamentous actin, mislocalization in foci and nuclear blebs | Lamin B loss, nuclear invaginations, heterochromatin relaxation and DNA damage | [77] |
| Lamin B1 | Homo sapiens | Frontal cortex neurons | pTau (AD) | Reduction in protein level | Nuclear invagination, heterochromatin relaxation | [77] |
| Lamin B1 | Homo sapiens | Neuroblastoma cells | Aβ42 (AD) | Protein cleavage after Aβ42 dependent release of Cathepsin L from lysosomes | Nuclear invaginations | [101] |
| Lamin B1 | Mus musculus | Hippocampal CA1 & striatal neurons | Downregulated PKCδ kinase (HD) | Protein level increase up to 4× wildtype level | Altered nuclear morphology, transcriptional changes | [102] |
| Lamin B1 | Homo sapiens | Neurons/patient-derived fibroblasts | PolyQ ataxin (DRPLA) | Protein level decrease and co-localization with ataxin aggregates | Cytoplasmic localization and degradation, nuclear invagination | [81] |
| Lamin B1 | Homo sapiens | HEK293 | ADLD | Extra copy of LMNB1 | Altered nuclear morphology | [103] |
| Lamin B1 | Homo sapiens | Patient-derived fibroblasts | ADLD | Extra copy of LMNB1 | Enhanced nuclear stiffness, reduced ion channel opening capacity, reduced proliferation | [104] |
| Lamin B1, B2, C | Homo sapiens | Neurons from patient-derived iPSC, brain sections | LRRK2 G2019S mutation (PD) | Abolished interaction between LRRK2 and lamins | Decline in nucleocytoplasmic compartmentalization, nuclear envelope disorganization | [79,97] |
| Gene | Disorder/Condition | Localization in Addition to the NE | References |
|---|---|---|---|
| AAAS | Triple-A syndrome | [108] | |
| DMPK | ASD | mitochondria, cytoplasm | [109] |
| DST | SCZ, ASD | cytoplasm | [110] |
| ITSN1 | ASD, ID, EPI | vesicles, cytoplasm | [111] |
| NUP62 | IBSN | [112] | |
| NUP85 | Microcephaly | [113] | |
| NUP107 | Microcephaly | [114] | |
| NUP133 | ASD, Microcephaly | [115,116] | |
| NUP155 | ASD | [117] | |
| NUP214 | Microcephaly | [118] | |
| RANBP2 | ANE1 | [119] | |
| SPAST | ASD | ER, cytoplasm | [120] |
| SYNE1 | ASD, CA, B, D | nucleus, cytoplasm | [121,122,123] |
| SYNE2 | ASD, ID | nucleus, cytoplasm | [124] |
| TERB2 | ASD | [125] | |
| WDFY3 | ASD, ADHD | vesicles, cytoplasm | [126] |
| XPO1 | ASD | nucleoplasm, cytoplasm | [127] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mestres, I.; Houtman, J.; Calegari, F.; Toda, T. A Nuclear Belt Fastens on Neural Cell Fate. Cells 2022, 11, 1761. https://doi.org/10.3390/cells11111761
Mestres I, Houtman J, Calegari F, Toda T. A Nuclear Belt Fastens on Neural Cell Fate. Cells. 2022; 11(11):1761. https://doi.org/10.3390/cells11111761
Chicago/Turabian StyleMestres, Ivan, Judith Houtman, Federico Calegari, and Tomohisa Toda. 2022. "A Nuclear Belt Fastens on Neural Cell Fate" Cells 11, no. 11: 1761. https://doi.org/10.3390/cells11111761
APA StyleMestres, I., Houtman, J., Calegari, F., & Toda, T. (2022). A Nuclear Belt Fastens on Neural Cell Fate. Cells, 11(11), 1761. https://doi.org/10.3390/cells11111761