
Shiga Toxin 2 Triggers C3a-Dependent Glomerular and Tubular Injury through Mitochondrial Dysfunction in Hemolytic Uremic Syndrome

 , ,
, , 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Experimental Model of Shiga Toxin-Associated HUS
2.2. Immunohistochemical Analyses in Renal Tissue
2.3. Histological and Ultrastructural Analyses
2.4. Estimation of Mitochondrial Size Measurements in Podocytes
2.5. Morphometric Analysis of Mitochondria in Proximal Tubular Cells
2.6. Cell Cultures and Incubation
2.7. Immunofluorescence Analysis in Cultured Cells
2.8. Mitochondrial Morphology in Cultured Cells
2.9. Western Blot Analysis
2.10. Statistics
3. Results
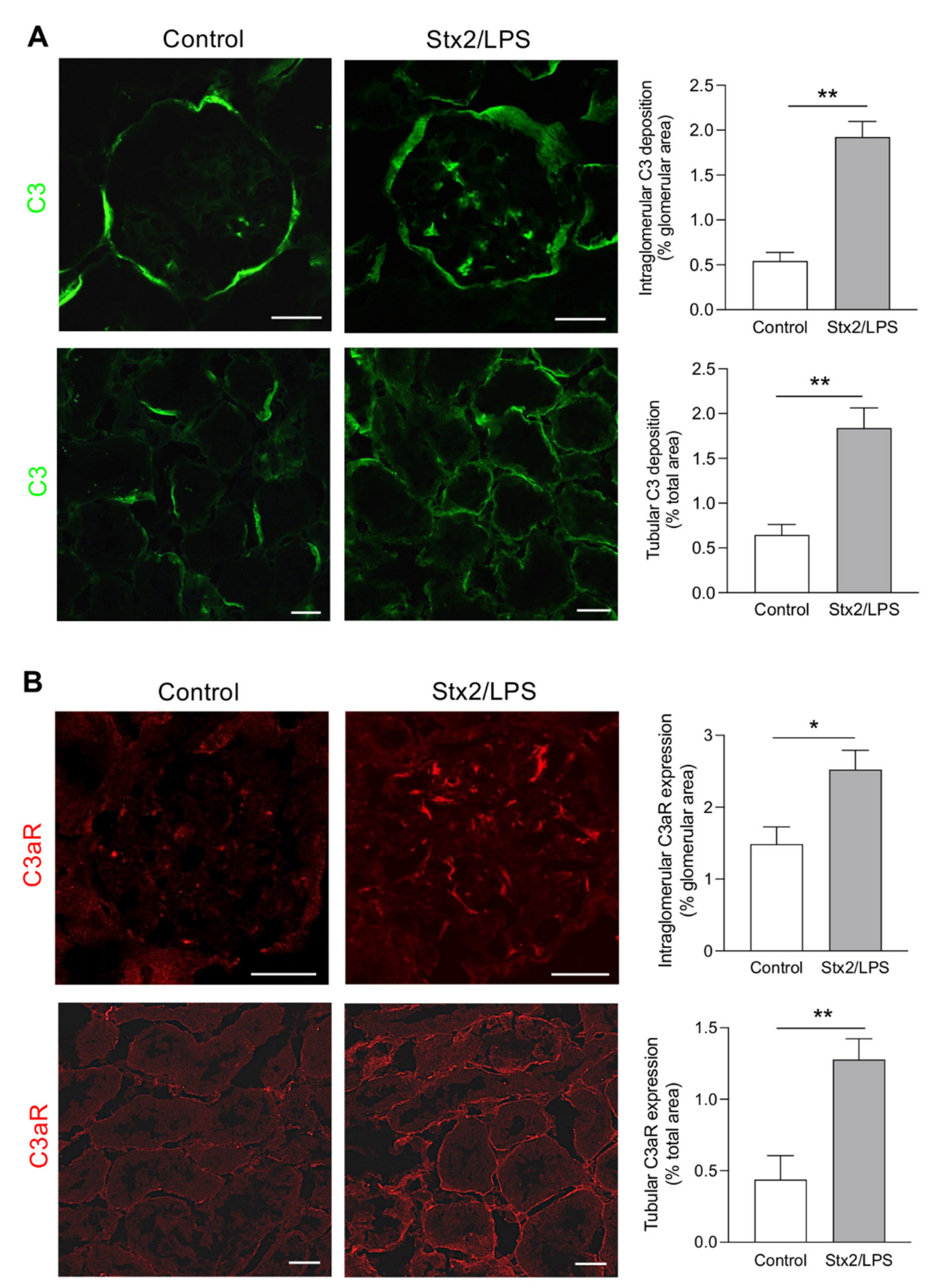
3.1. Activation of Complement System and C3a/C3aR Axis Occurs in the Glomerular and Tubular Compartments in Stx2/LPS-Coinjected Mice
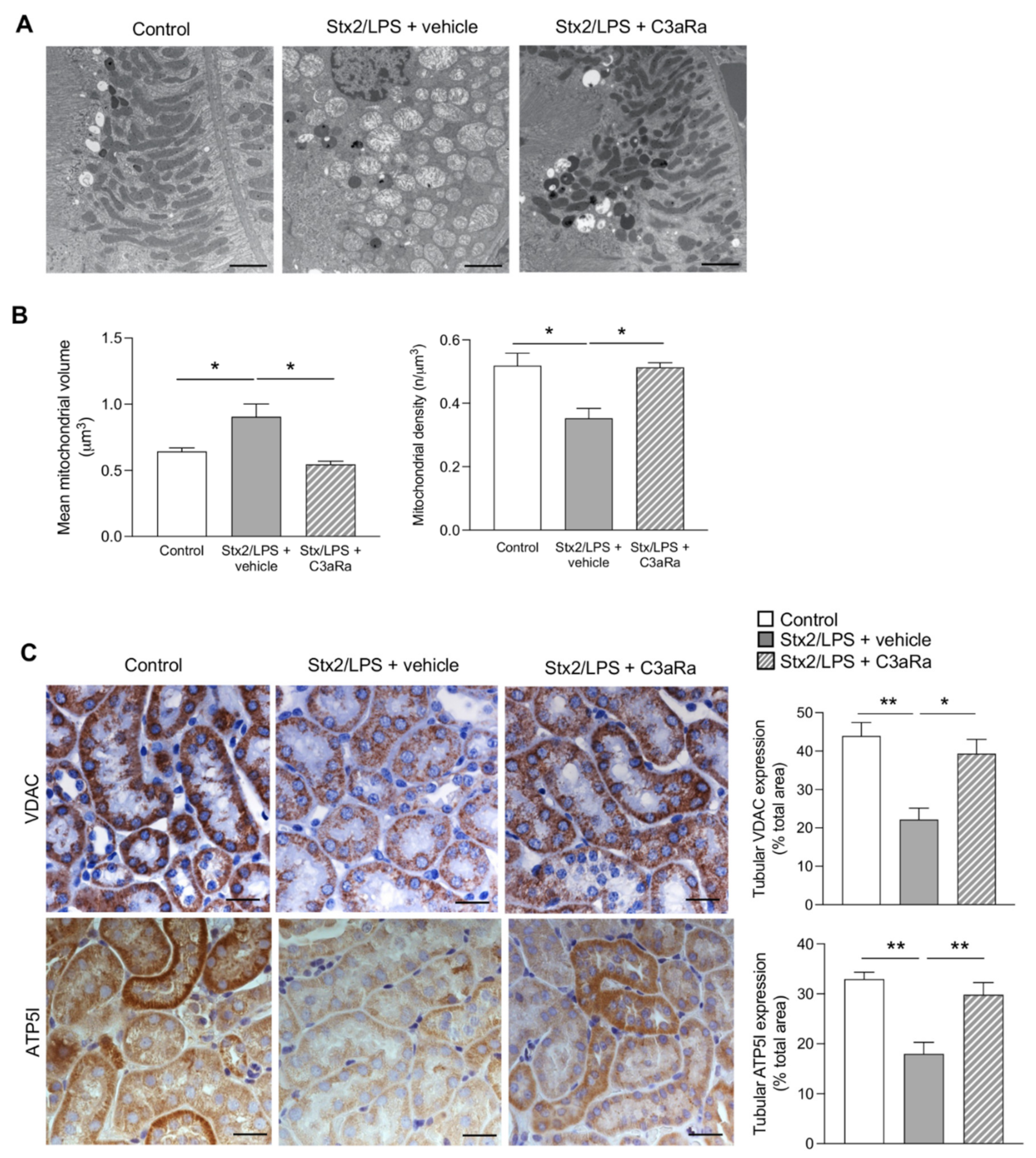
3.2. C3aR Antagonist Counteracts Podocyte Injury by Reducing Mitochondrial Alterations in Stx2/LPS-Coinjected Mice
3.3. C3aR Blockade Decreases Tubular Cell Injury by Limiting Mitochondrial Dysfunction in Stx2/LPS Mice
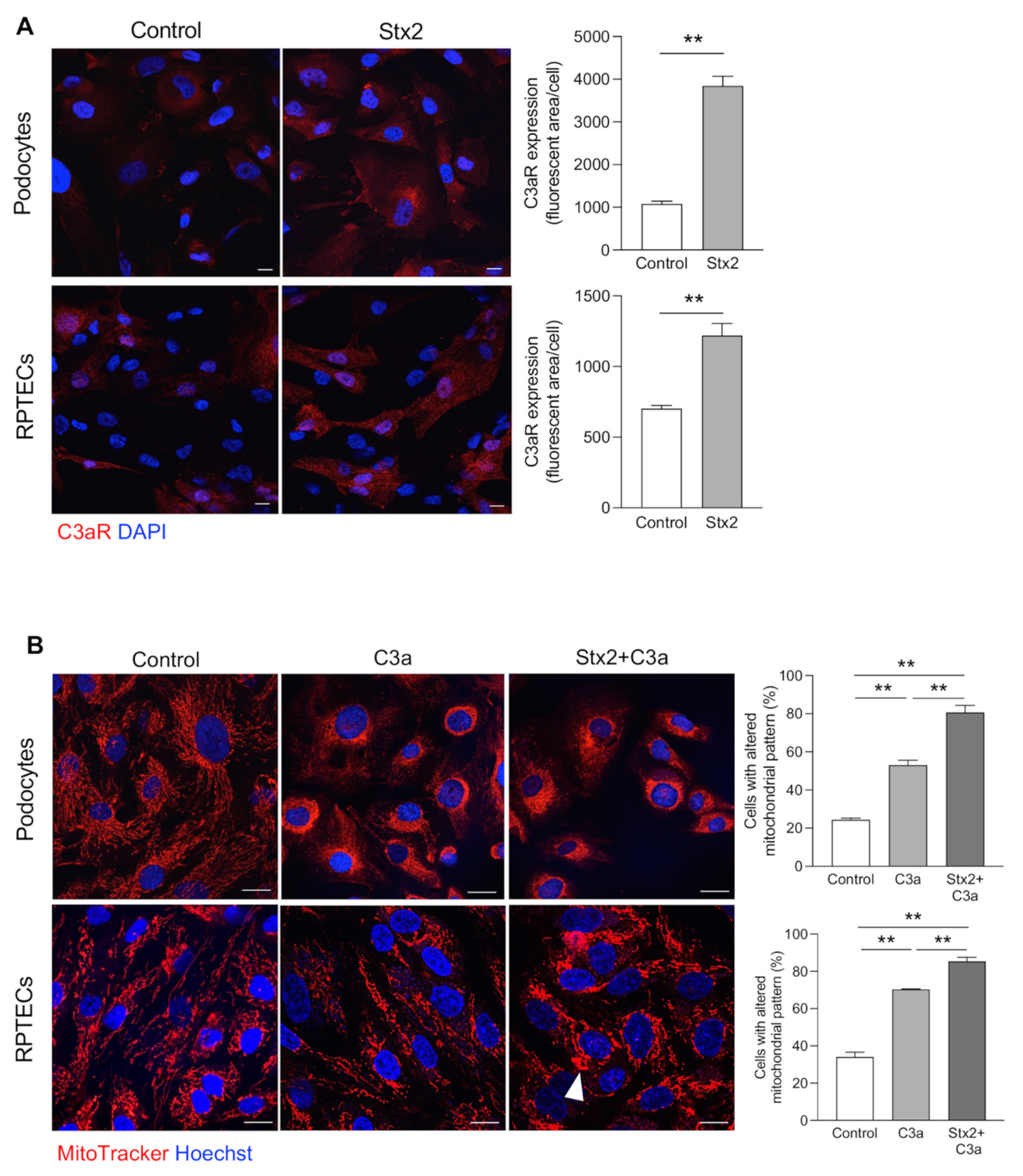
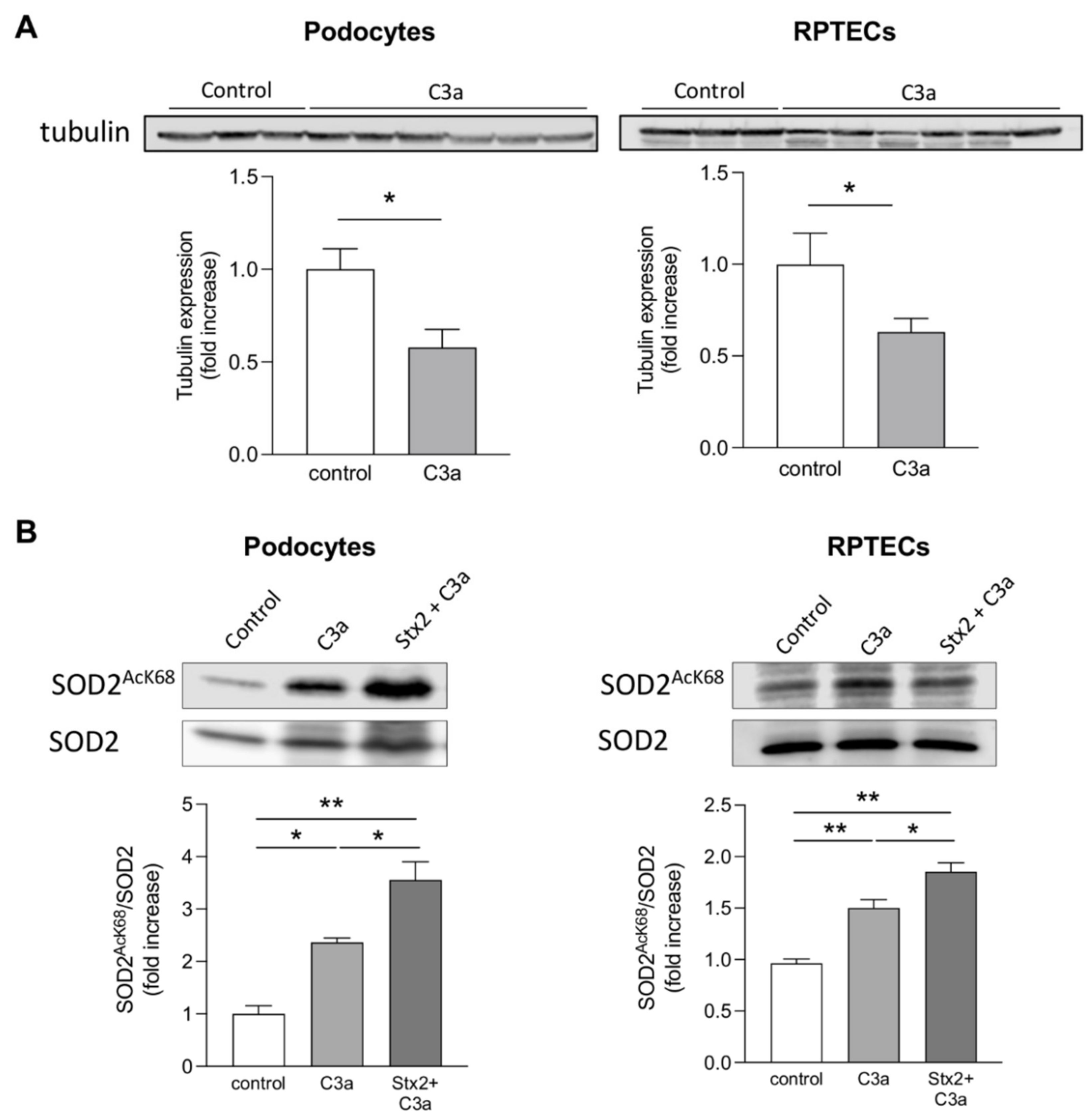
3.4. Stx2 Enhances Podocyte and Tubular Cell Susceptibility to the Toxic Effect of C3a on Mitochondria In Vitro
4. Discussion
- (a)
- increased intraglomerular and tubular staining for C3 and C3aR overexpression were accompanied by podocyte loss and tubular cell damage;
- (b)
- mitochondrial abnormalities, in terms of altered morphology and distribution, as well as the reduction in mitochondrial VDAC and ATP5I expression, occurred in podocytes and proximal tubular cells;
- (c)
- the inhibition of the C3a/C3aR axis by treatment with a C3aR antagonist reduced podocyte and tubular cell phenotypic changes having normalized the ultrastructural and functional mitochondrial abnormalities.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Noris, M.; Remuzzi, G. Hemolytic Uremic Syndrome. J. Am. Soc. Nephrol. 2005, 16, 1035–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoja, C.; Buelli, S.; Morigi, M. Shiga Toxin Triggers Endothelial and Podocyte Injury: The Role of Complement Activation. Pediatr. Nephrol. 2019, 34, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Fakhouri, F.; Zuber, J.; Frémeaux-Bacchi, V.; Loirat, C. Haemolytic Uraemic Syndrome. Lancet 2017, 390, 681–696. [Google Scholar] [CrossRef]
- Karpman, D.; Loos, S.; Tati, R.; Arvidsson, I. Haemolytic Uraemic Syndrome. J. Intern. Med. 2017, 281, 123–148. [Google Scholar] [CrossRef] [PubMed]
- Spinale, J.M.; Ruebner, R.L.; Copelovitch, L.; Kaplan, B.S. Long-Term Outcomes of Shiga Toxin Hemolytic Uremic Syndrome. Pediatr. Nephrol. 2013, 28, 2097–2105. [Google Scholar] [CrossRef] [PubMed]
- Vaterodt, L.; Holle, J.; Hüseman, D.; Müller, D.; Thumfart, J. Short- and Long-Term Renal Outcome of Hemolytic-Uremic Syndrome in Childhood. Front. Pediatr. 2018, 6, 220. [Google Scholar] [CrossRef]
- Hughes, A.K.; Ergonul, Z.; Stricklett, P.K.; Kohan, D.E.; Ergonal, Z. Molecular Basis for High Renal Cell Sensitivity to the Cytotoxic Effects of Shigatoxin-1: Upregulation of Globotriaosylceramide Expression. J. Am. Soc. Nephrol. JASN 2002, 13, 2239–2245. [Google Scholar] [CrossRef] [Green Version]
- Adler, S.; Bollu, R. Glomerular Endothelial Cell Injury Mediated by Shiga-like Toxin-1. Kidney Blood Press. Res. 1998, 21, 13–21. [Google Scholar] [CrossRef]
- Morigi, M.; Micheletti, G.; Figliuzzi, M.; Imberti, B.; Karmali, M.A.; Remuzzi, A.; Remuzzi, G.; Zoja, C. Verotoxin-1 Promotes Leukocyte Adhesion to Cultured Endothelial Cells under Physiologic Flow Conditions. Blood 1995, 86, 4553–4558. [Google Scholar] [CrossRef] [Green Version]
- Zoja, C.; Angioletti, S.; Donadelli, R.; Zanchi, C.; Tomasoni, S.; Binda, E.; Imberti, B.; te Loo, M.; Monnens, L.; Remuzzi, G.; et al. Shiga Toxin-2 Triggers Endothelial Leukocyte Adhesion and Transmigration via NF-KappaB Dependent up-Regulation of IL-8 and MCP-1. Kidney Int. 2002, 62, 846–856. [Google Scholar] [CrossRef] [Green Version]
- Dettmar, A.K.; Binder, E.; Greiner, F.R.; Liebau, M.C.; Kurschat, C.E.; Jungraithmayr, T.C.; Saleem, M.A.; Schmitt, C.-P.; Feifel, E.; Orth-Höller, D.; et al. Protection of Human Podocytes from Shiga Toxin 2-Induced Phosphorylation of Mitogen-Activated Protein Kinases and Apoptosis by Human Serum Amyloid P Component. Infect. Immun. 2014, 82, 1872–1879. [Google Scholar] [CrossRef] [Green Version]
- Hughes, A.K.; Stricklett, P.K.; Kohan, D.E. Shiga Toxin-1 Regulation of Cytokine Production by Human Proximal Tubule Cells. Kidney Int. 1998, 54, 1093–1106. [Google Scholar] [CrossRef] [Green Version]
- Buelli, S.; Zoja, C.; Remuzzi, G.; Morigi, M. Complement Activation Contributes to the Pathophysiology of Shiga Toxin-Associated Hemolytic Uremic Syndrome. Microorganisms 2019, 7, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monnens, L.; Molenaar, J.; Lambert, P.H.; Proesmans, W.; van Munster, P. The Complement System in Hemolytic-Uremic Syndrome in Childhood. Clin. Nephrol. 1980, 13, 168–171. [Google Scholar]
- Robson, W.L.; Leung, A.K.; Fick, G.H.; McKenna, A.I. Hypocomplementemia and Leukocytosis in Diarrhea-Associated Hemolytic Uremic Syndrome. Nephron 1992, 62, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Thurman, J.M.; Marians, R.; Emlen, W.; Wood, S.; Smith, C.; Akana, H.; Holers, V.M.; Lesser, M.; Kline, M.; Hoffman, C.; et al. Alternative Pathway of Complement in Children with Diarrhea-Associated Hemolytic Uremic Syndrome. Clin. J. Am. Soc. Nephrol. CJASN 2009, 4, 1920–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ståhl, A.; Sartz, L.; Karpman, D. Complement Activation on Platelet-Leukocyte Complexes and Microparticles in Enterohemorrhagic Escherichia Coli-Induced Hemolytic Uremic Syndrome. Blood 2011, 117, 5503–5513. [Google Scholar] [CrossRef] [Green Version]
- Arvidsson, I.; Ståhl, A.-L.; Hedström, M.M.; Kristoffersson, A.-C.; Rylander, C.; Westman, J.S.; Storry, J.R.; Olsson, M.L.; Karpman, D. Shiga Toxin-Induced Complement-Mediated Hemolysis and Release of Complement-Coated Red Blood Cell-Derived Microvesicles in Hemolytic Uremic Syndrome. J. Immunol. 2015, 194, 2309–2318. [Google Scholar] [CrossRef] [Green Version]
- Gonzalo, A.; Mampaso, F.; Gallego, N.; Bellas, C.; Segui, J.; Ortuño, J. Hemolytic Uremic Syndrome with Hypocomplementemia and Deposits of IgM and C3 in the Involved Renal Tissue. Clin. Nephrol. 1981, 16, 193–199. [Google Scholar]
- Cossio, P.M.; Laguens, R.P.; Pantin, D.J.; de Bracco, M.M.; Molinas, F.; Voyer, L.E.; Arana, R.M. Persistent Glomerulonephritis Following the Haemolytic-Uremic Syndrome. Immunopathological and Morphological Studies. Clin. Exp. Immunol. 1977, 29, 361–368. [Google Scholar]
- Koster, F.T.; Boonpucknavig, V.; Sujaho, S.; Gilman, R.H.; Rahaman, M.M. Renal Histopathology in the Hemolytic-Uremic Syndrome Following Shigellosis. Clin. Nephrol. 1984, 21, 126–133. [Google Scholar] [PubMed]
- Morigi, M.; Galbusera, M.; Gastoldi, S.; Locatelli, M.; Buelli, S.; Pezzotta, A.; Pagani, C.; Noris, M.; Gobbi, M.; Stravalaci, M.; et al. Alternative Pathway Activation of Complement by Shiga Toxin Promotes Exuberant C3a Formation That Triggers Microvascular Thrombosis. J. Immunol. 2011, 187, 172–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locatelli, M.; Buelli, S.; Pezzotta, A.; Corna, D.; Perico, L.; Tomasoni, S.; Rottoli, D.; Rizzo, P.; Conti, D.; Thurman, J.M.; et al. Shiga Toxin Promotes Podocyte Injury in Experimental Hemolytic Uremic Syndrome via Activation of the Alternative Pathway of Complement. J. Am. Soc. Nephrol. 2014, 25, 1786–1798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, A.K.; Stricklett, P.K.; Schmid, D.; Kohan, D.E. Cytotoxic Effect of Shiga Toxin-1 on Human Glomerular Epithelial Cells. Kidney Int. 2000, 57, 2350–2359. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Fazekasova, H.; Wang, N.; Peng, Q.; Sacks, S.H.; Lombardi, G.; Zhou, W. Functional Modulation of Human Monocytes Derived DCs by Anaphylatoxins C3a and C5a. Immunobiology 2012, 217, 65–73. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Anderson, K.J.; Peng, Q.; Noble, A.; Lu, B.; Kelly, A.P.; Wang, N.; Sacks, S.H.; Zhou, W. Cyclic AMP Plays a Critical Role in C3a-Receptor-Mediated Regulation of Dendritic Cells in Antigen Uptake and T-Cell Stimulation. Blood 2008, 112, 5084–5094. [Google Scholar] [CrossRef] [Green Version]
- Schraufstatter, I.U.; Trieu, K.; Sikora, L.; Sriramarao, P.; DiScipio, R. Complement C3a and C5a Induce Different Signal Transduction Cascades in Endothelial Cells. J. Immunol. 2002, 169, 2102–2110. [Google Scholar] [CrossRef] [Green Version]
- Thurman, J.M.; Lenderink, A.M.; Royer, P.A.; Coleman, K.E.; Zhou, J.; Lambris, J.D.; Nemenoff, R.A.; Quigg, R.J.; Holers, V.M. C3a Is Required for the Production of CXC Chemokines by Tubular Epithelial Cells after Renal Ishemia/Reperfusion. J. Immunol. 2007, 178, 1819–1828. [Google Scholar] [CrossRef] [Green Version]
- Ishii, M.; Beeson, G.; Beeson, C.; Rohrer, B. Mitochondrial C3a Receptor Activation in Oxidatively Stressed Epithelial Cells Reduces Mitochondrial Respiration and Metabolism. Front. Immunol. 2021, 12, 628062. [Google Scholar] [CrossRef]
- Brigotti, M.; Orth-Höller, D.; Carnicelli, D.; Porcellini, E.; Galassi, E.; Tazzari, P.L.; Ricci, F.; Manoli, F.; Manet, I.; Talasz, H.; et al. The Structure of the Shiga Toxin 2a A-subunit Dictates the Interactions of the Toxin with Blood Components. Cell. Microbiol. 2019, 21, e13000. [Google Scholar] [CrossRef] [Green Version]
- Matussek, A.; Lauber, J.; Bergau, A.; Hansen, W.; Rohde, M.; Dittmar, K.E.J.; Gunzer, M.; Mengel, M.; Gatzlaff, P.; Hartmann, M.; et al. Molecular and Functional Analysis of Shiga Toxin-Induced Response Patterns in Human Vascular Endothelial Cells. Blood 2003, 102, 1323–1332. [Google Scholar] [CrossRef] [PubMed]
- Morigi, M.; Perico, L.; Corna, D.; Locatelli, M.; Cassis, P.; Carminati, C.E.; Bolognini, S.; Zoja, C.; Remuzzi, G.; Benigni, A.; et al. C3a Receptor Blockade Protects Podocytes from Injury in Diabetic Nephropathy. JCI Insight 2020, 5, e131849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perico, L.; Morigi, M.; Rota, C.; Breno, M.; Mele, C.; Noris, M.; Introna, M.; Capelli, C.; Longaretti, L.; Rottoli, D.; et al. Human Mesenchymal Stromal Cells Transplanted into Mice Stimulate Renal Tubular Cells and Enhance Mitochondrial Function. Nat. Commun. 2017, 8, 983. [Google Scholar] [CrossRef] [PubMed]
- Weibel, E.R.; Kistler, G.S.; Scherle, W.F. Practical Stereological Methods for Morphometric Cytology. J. Cell Biol. 1966, 30, 23–38. [Google Scholar] [CrossRef]
- Morigi, M.; Buelli, S.; Zanchi, C.; Longaretti, L.; Macconi, D.; Benigni, A.; Moioli, D.; Remuzzi, G.; Zoja, C. Shigatoxin-Induced Endothelin-1 Expression in Cultured Podocytes Autocrinally Mediates Actin Remodeling. Am. J. Pathol. 2006, 169, 1965–1975. [Google Scholar] [CrossRef] [Green Version]
- Walport, M.J. Complement. First of Two Parts. N. Engl. J. Med. 2001, 344, 1058–1066. [Google Scholar] [CrossRef]
- Abu-Hamad, S.; Sivan, S.; Shoshan-Barmatz, V. The Expression Level of the Voltage-Dependent Anion Channel Controls Life and Death of the Cell. Proc. Natl. Acad. Sci. USA 2006, 103, 5787–5792. [Google Scholar] [CrossRef] [Green Version]
- Paumard, P.; Vaillier, J.; Coulary, B.; Schaeffer, J.; Soubannier, V.; Mueller, D.M.; Brèthes, D.; di Rago, J.-P.; Velours, J. The ATP Synthase Is Involved in Generating Mitochondrial Cristae Morphology. EMBO J. 2002, 21, 221–230. [Google Scholar] [CrossRef]
- Kaplan, B.S. Shiga Toxin-Induced Tubular Injury in Hemolytic Uremic Syndrome. Kidney Int. 1998, 54, 648–649. [Google Scholar] [CrossRef] [Green Version]
- Mahadevappa, R.; Nielsen, R.; Christensen, E.I.; Birn, H. Megalin in Acute Kidney Injury: Foe and Friend. Am. J. Physiol. Renal Physiol. 2014, 306, F147–F154. [Google Scholar] [CrossRef] [Green Version]
- Moore, A.S.; Holzbaur, E.L.F. Mitochondrial-Cytoskeletal Interactions: Dynamic Associations That Facilitate Network Function and Remodeling. Curr. Opin. Physiol. 2018, 3, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Tirichen, H.; Yaigoub, H.; Xu, W.; Wu, C.; Li, R.; Li, Y. Mitochondrial Reactive Oxygen Species and Their Contribution in Chronic Kidney Disease Progression Through Oxidative Stress. Front. Physiol. 2021, 12, 627837. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Branicky, R.; Noë, A.; Hekimi, S. Superoxide Dismutases: Dual Roles in Controlling ROS Damage and Regulating ROS Signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef]
- Lapeyraque, A.-L.; Malina, M.; Fremeaux-Bacchi, V.; Boppel, T.; Kirschfink, M.; Oualha, M.; Proulx, F.; Clermont, M.-J.; Le Deist, F.; Niaudet, P.; et al. Eculizumab in Severe Shiga-Toxin-Associated HUS. N. Engl. J. Med. 2011, 364, 2561–2563. [Google Scholar] [CrossRef] [PubMed]
- Menne, J.; Nitschke, M.; Stingele, R.; Abu-Tair, M.; Beneke, J.; Bramstedt, J.; Bremer, J.P.; Brunkhorst, R.; Busch, V.; Dengler, R.; et al. Validation of Treatment Strategies for Enterohaemorrhagic Escherichia Coli O104:H4 Induced Haemolytic Uraemic Syndrome: Case-Control Study. BMJ 2012, 345, e4565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pape, L.; Hartmann, H.; Bange, F.C.; Suerbaum, S.; Bueltmann, E.; Ahlenstiel-Grunow, T. Eculizumab in Typical Hemolytic Uremic Syndrome (HUS) With Neurological Involvement. Medicine 2016, 94, e1000. [Google Scholar] [CrossRef] [PubMed]
- Keenswijk, W.; Raes, A.; Vande Walle, J. Is Eculizumab Efficacious in Shigatoxin-Associated Hemolytic Uremic Syndrome? A Narrative Review of Current Evidence. Eur. J. Pediatr. 2018, 177, 311–318. [Google Scholar] [CrossRef]
- Ruggenenti, P.; Remuzzi, G. Thrombotic Microangiopathy: E. Coli O104:H4 German Outbreak: A Missed Opportunity. Nat. Rev. Nephrol. 2012, 8, 558–560. [Google Scholar] [CrossRef]
- Harkins, V.J.; McAllister, D.A.; Reynolds, B.C. Shiga-Toxin E. Coli Hemolytic Uremic Syndrome: Review of Management and Long-Term Outcome. Curr. Pediatr. Rep. 2020, 8, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Raghupathy, P.; Date, A.; Shastry, J.C.; Sudarsanam, A.; Jadhav, M. Haemolytic-Uraemic Syndrome Complicating Shigella Dystentery in South Indian Children. Br. Med. J. 1978, 1, 1518–1521. [Google Scholar] [CrossRef] [Green Version]
- Wagner, J. Acute Tubular Necrosis with Anaemia. Great Ormond Str. J. 1954, 7, 66–72. [Google Scholar]
- Porubsky, S.; Federico, G.; Müthing, J.; Jennemann, R.; Gretz, N.; Büttner, S.; Obermüller, N.; Jung, O.; Hauser, I.A.; Gröne, E.; et al. Direct Acute Tubular Damage Contributes to Shigatoxin-Mediated Kidney Failure. J. Pathol. 2014, 234, 120–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stearns-Kurosawa, D.J.; Oh, S.-Y.; Cherla, R.P.; Lee, M.-S.; Tesh, V.L.; Papin, J.; Henderson, J.; Kurosawa, S. Distinct Renal Pathology and a Chemotactic Phenotype after Enterohemorrhagic Escherichia Coli Shiga Toxins in Non-Human Primate Models of Hemolytic Uremic Syndrome. Am. J. Pathol. 2013, 182, 1227–1238. [Google Scholar] [CrossRef] [Green Version]
- Dennhardt, S.; Pirschel, W.; Wissuwa, B.; Daniel, C.; Gunzer, F.; Lindig, S.; Medyukhina, A.; Kiehntopf, M.; Rudolph, W.W.; Zipfel, P.F.; et al. Modeling Hemolytic-Uremic Syndrome: In-Depth Characterization of Distinct Murine Models Reflecting Different Features of Human Disease. Front. Immunol. 2018, 9, 1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, F.B.; Tesh, V.L.; DeBault, L.; Li, A.; Chang, A.C.; Kosanke, S.D.; Pysher, T.J.; Siegler, R.L. Characterization of the Baboon Responses to Shiga-like Toxin: Descriptive Study of a New Primate Model of Toxic Responses to Stx-1. Am. J. Pathol. 1999, 154, 1285–1299. [Google Scholar] [CrossRef]
- Siegler, R.L.; Pysher, T.J.; Tesh, V.L.; Taylor, F.B. Response to Single and Divided Doses of Shiga Toxin-1 in a Primate Model of Hemolytic Uremic Syndrome. J. Am. Soc. Nephrol. JASN 2001, 12, 1458–1467. [Google Scholar] [CrossRef]
- Karpman, D.; Håkansson, A.; Perez, M.T.; Isaksson, C.; Carlemalm, E.; Caprioli, A.; Svanborg, C. Apoptosis of Renal Cortical Cells in the Hemolytic-Uremic Syndrome: In Vivo and in Vitro Studies. Infect. Immun. 1998, 66, 636–644. [Google Scholar] [CrossRef] [Green Version]
- Thurman, J.M.; Lucia, M.S.; Ljubanovic, D.; Holers, V.M. Acute Tubular Necrosis Is Characterized by Activation of the Alternative Pathway of Complement. Kidney Int. 2005, 67, 524–530. [Google Scholar] [CrossRef] [Green Version]
- Stenson, E.K.; You, Z.; Reeder, R.; Norris, J.; Scott, H.F.; Dixon, B.P.; Thurman, J.M.; Frazer-Abel, A.; Mourani, P.; Kendrick, J. Complement Activation Fragments Are Increased in Critically Ill Pediatric Patients with Severe AKI. Kidney360 2021, 2, 1884–1891. [Google Scholar] [CrossRef]
- Peng, Q.; Li, K.; Smyth, L.A.; Xing, G.; Wang, N.; Meader, L.; Lu, B.; Sacks, S.H.; Zhou, W. C3a and C5a Promote Renal Ischemia-Reperfusion Injury. J. Am. Soc. Nephrol. JASN 2012, 23, 1474–1485. [Google Scholar] [CrossRef] [Green Version]
- Pfister, F.; Vonbrunn, E.; Ries, T.; Jäck, H.-M.; Überla, K.; Lochnit, G.; Sheriff, A.; Herrmann, M.; Büttner-Herold, M.; Amann, K.; et al. Complement Activation in Kidneys of Patients with COVID-19. Front. Immunol. 2020, 11, 594849. [Google Scholar] [CrossRef] [PubMed]
- Johansson, K.E.; Ståhl, A.-L.; Arvidsson, I.; Loos, S.; Tontanahal, A.; Rebetz, J.; Chromek, M.; Kristoffersson, A.-C.; Johannes, L.; Karpman, D. Shiga Toxin Signals via ATP and Its Effect Is Blocked by Purinergic Receptor Antagonism. Sci. Rep. 2019, 9, 14362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asgari, E.; Le Friec, G.; Yamamoto, H.; Perucha, E.; Sacks, S.S.; Köhl, J.; Cook, H.T.; Kemper, C. C3a Modulates IL-1β Secretion in Human Monocytes by Regulating ATP Efflux and Subsequent NLRP3 Inflammasome Activation. Blood 2013, 122, 3473–3481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, K.; Das, J.; Pal, P.B.; Sil, P.C. Oxidative Stress: The Mitochondria-Dependent and Mitochondria-Independent Pathways of Apoptosis. Arch. Toxicol. 2013, 87, 1157–1180. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buelli, S.; Locatelli, M.; Carminati, C.E.; Corna, D.; Cerullo, D.; Imberti, B.; Perico, L.; Brigotti, M.; Abbate, M.; Zoja, C.; et al. Shiga Toxin 2 Triggers C3a-Dependent Glomerular and Tubular Injury through Mitochondrial Dysfunction in Hemolytic Uremic Syndrome. Cells 2022, 11, 1755. https://doi.org/10.3390/cells11111755
Buelli S, Locatelli M, Carminati CE, Corna D, Cerullo D, Imberti B, Perico L, Brigotti M, Abbate M, Zoja C, et al. Shiga Toxin 2 Triggers C3a-Dependent Glomerular and Tubular Injury through Mitochondrial Dysfunction in Hemolytic Uremic Syndrome. Cells. 2022; 11(11):1755. https://doi.org/10.3390/cells11111755
Chicago/Turabian StyleBuelli, Simona, Monica Locatelli, Claudia Elisa Carminati, Daniela Corna, Domenico Cerullo, Barbara Imberti, Luca Perico, Maurizio Brigotti, Mauro Abbate, Carlamaria Zoja, and et al. 2022. "Shiga Toxin 2 Triggers C3a-Dependent Glomerular and Tubular Injury through Mitochondrial Dysfunction in Hemolytic Uremic Syndrome" Cells 11, no. 11: 1755. https://doi.org/10.3390/cells11111755
APA StyleBuelli, S., Locatelli, M., Carminati, C. E., Corna, D., Cerullo, D., Imberti, B., Perico, L., Brigotti, M., Abbate, M., Zoja, C., Benigni, A., Remuzzi, G., & Morigi, M. (2022). Shiga Toxin 2 Triggers C3a-Dependent Glomerular and Tubular Injury through Mitochondrial Dysfunction in Hemolytic Uremic Syndrome. Cells, 11(11), 1755. https://doi.org/10.3390/cells11111755