
Angiogenesis, Lymphangiogenesis, and Inflammation in Chronic Obstructive Pulmonary Disease (COPD): Few Certainties and Many Outstanding Questions

 ,
,  , ,
, ,  , and
, and 
Abstract
1. Introduction
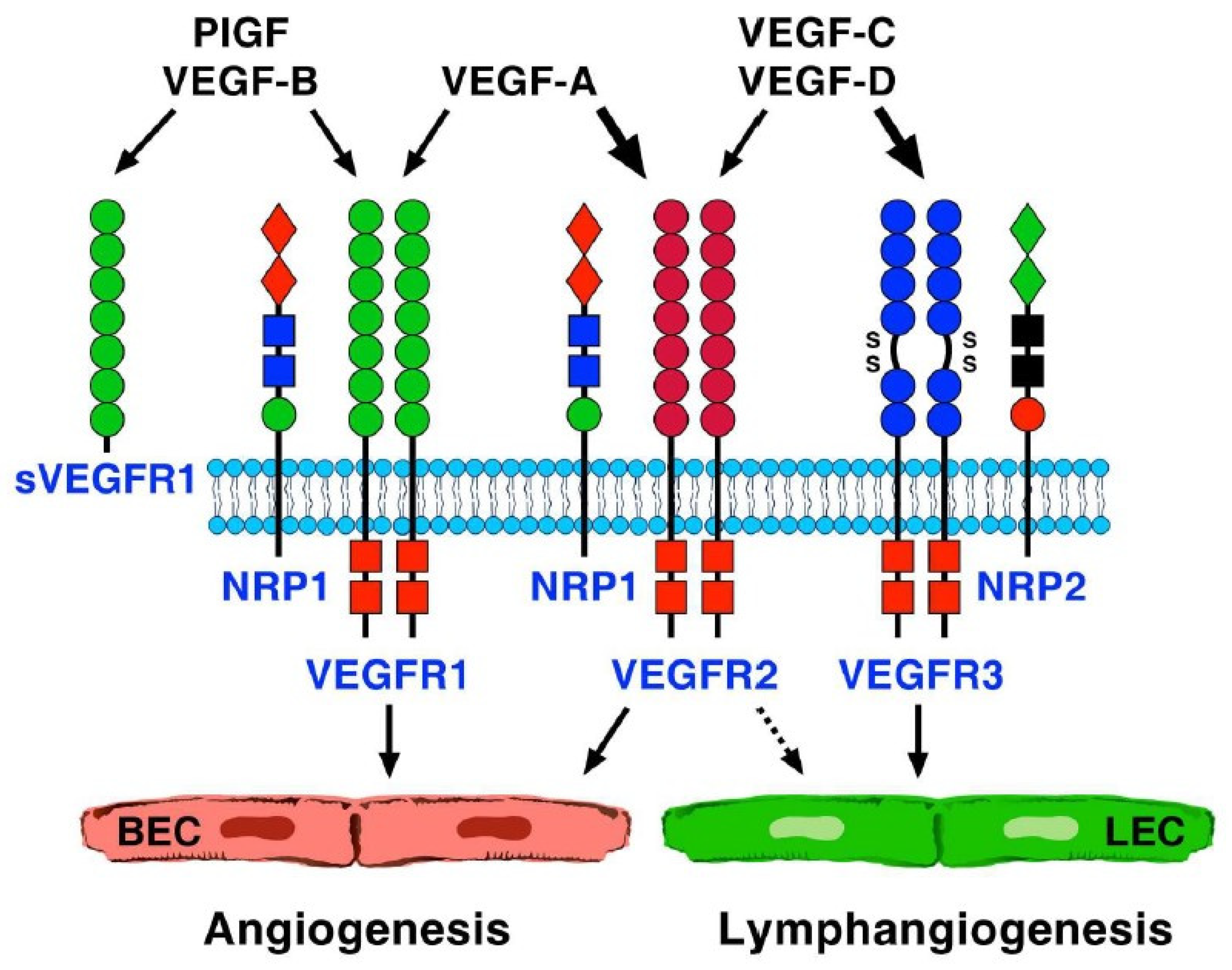
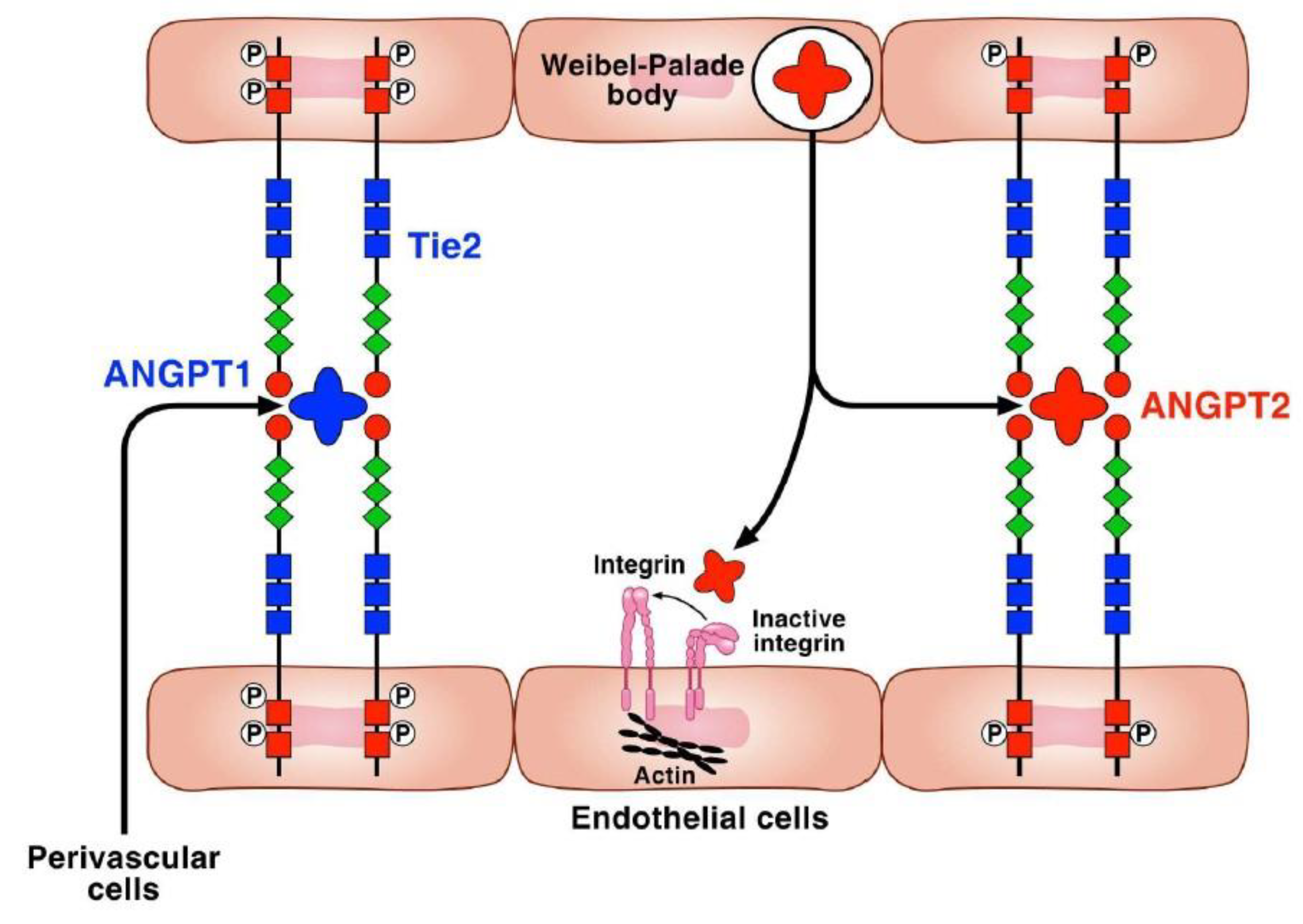
2. Proangiogenic and Antiangiogenic Factors
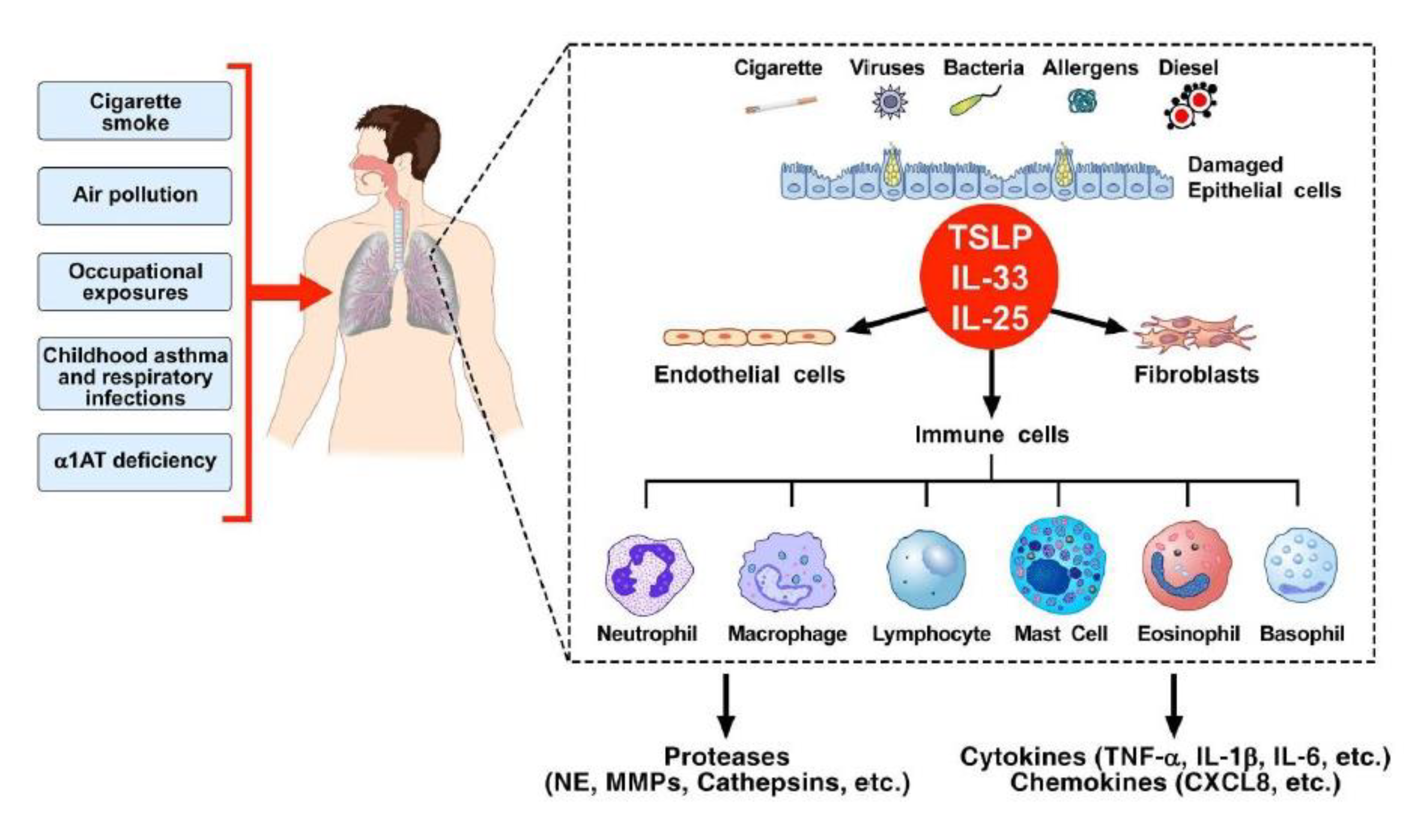
3. Chronic Obstructive Pulmonary Disease (COPD) and Inflammation
4. Epithelial-Derived Cytokines
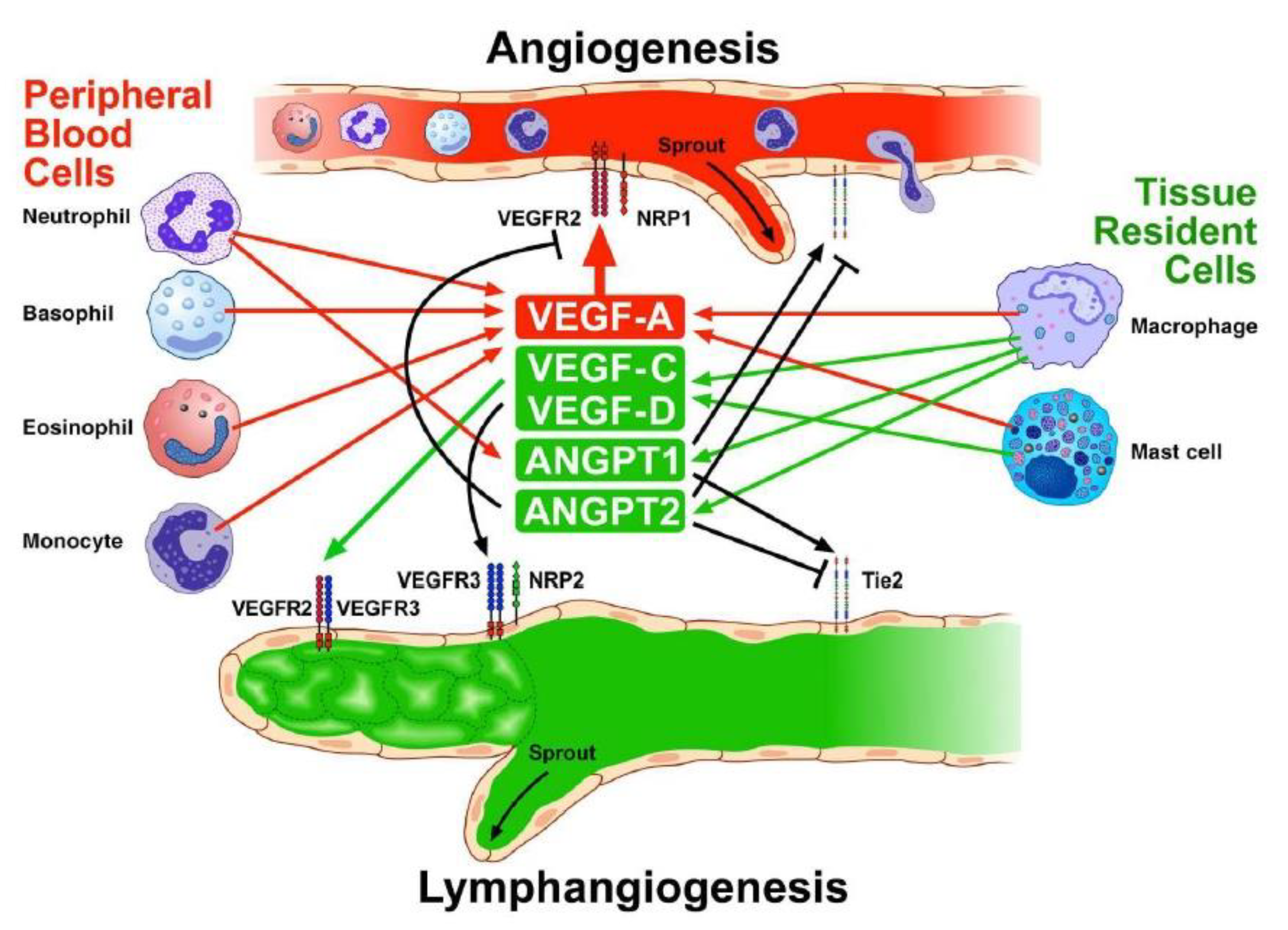
5. Inflammatory Cells, Angiogenesis, and Lymphangiogenesis
5.1. Macrophages
5.2. Mast Cells
5.3. Neutrophils
5.4. Eosinophils
5.5. Basophils
5.6. Lymphocytes
5.7. Dendritic Cells
5.8. NK Cells
6. Structural Cells and Angiogenesis
Epithelial Cells
7. Angiogenesis and Lymphangiogenesis in Experimental Models of Chronic Airway Inflammation
8. Angiogenesis and Lymphangiogenesis in COPD
9. Therapeutic Opportunities
10. Closing Thoughts
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Carmeliet, P. Angiogenesis in Life, Disease and Medicine. Nature 2005, 438, 932–936. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Angiogenesis in Cancer, Vascular, Rheumatoid and Other Disease. Nat. Med. 1995, 1, 27–31. [Google Scholar] [CrossRef]
- Lee, H.J.; Hong, Y.J.; Kim, M. Angiogenesis in Chronic Inflammatory Skin Disorders. Int. J. Mol. Sci. 2021, 22, 12035. [Google Scholar] [CrossRef] [PubMed]
- Tas, S.W.; Maracle, C.X.; Balogh, E.; Szekanecz, Z. Targeting of Proangiogenic Signalling Pathways in Chronic Inflammation. Nat. Rev. Rheumatol. 2016, 12, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Oliver, G. Development of the Mammalian Lymphatic Vasculature. J. Clin. Investig. 2014, 124, 888–897. [Google Scholar] [CrossRef]
- Ny, A.; Koch, M.; Schneider, M.; Neven, E.; Tong, R.T.; Maity, S.; Fischer, C.; Plaisance, S.; Lambrechts, D.; Heligon, C.; et al. A Genetic Xenopus laevis Tadpole Model to Study Lymphangiogenesis. Nat. Med. 2005, 11, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular Mechanisms and Clinical Applications of Angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Poto, R.; Cristinziano, L.; Modestino, L.; de Paulis, A.; Marone, G.; Loffredo, S.; Galdiero, M.R.; Varricchi, G. Neutrophil Extracellular Traps, Angiogenesis and Cancer. Biomedicines 2022, 10, 431. [Google Scholar] [CrossRef] [PubMed]
- Marcella, S.; Petraroli, A.; Braile, M.; Parente, R.; Ferrara, A.L.; Galdiero, M.R.; Modestino, L.; Cristinziano, L.; Rossi, F.W.; Varricchi, G.; et al. Vascular Endothelial Growth Factors and Angiopoietins as New Players in Mastocytosis. Clin. Exp. Med. 2021, 21, 415–427. [Google Scholar] [CrossRef]
- Sammarco, G.; Varricchi, G.; Ferraro, V.; Ammendola, M.; de Fazio, M.; Altomare, D.F.; Luposella, M.; Maltese, L.; Curro, G.; Marone, G.; et al. Mast Cells, Angiogenesis and Lymphangiogenesis in Human Gastric Cancer. Int. J. Mol. Sci. 2019, 20, 2106. [Google Scholar] [CrossRef]
- Varricchi, G.; Loffredo, S.; Bencivenga, L.; Ferrara, A.L.; Gambino, G.; Ferrara, N.; de Paulis, A.; Marone, G.; Rengo, G. Angiopoietins, Vascular Endothelial Growth Factors and Secretory Phospholipase A2 in Ischemic and Non-Ischemic Heart Failure. J. Clin. Med. 2020, 9, 1928. [Google Scholar] [CrossRef] [PubMed]
- Varricchi, G.; Loffredo, S.; Borriello, F.; Pecoraro, A.; Rivellese, F.; Genovese, A.; Spadaro, G.; Marone, G. Superantigenic Activation of Human Cardiac Mast Cells. Int. J. Mol. Sci. 2019, 20, 1828. [Google Scholar] [CrossRef] [PubMed]
- Brakenhielm, E.; Alitalo, K. Cardiac Lymphatics in Health and Disease. Nat. Rev. Cardiol. 2019, 16, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Sainz-Jaspeado, M.; Claesson-Welsh, L. Cytokines Regulating Lymphangiogenesis. Curr. Opin. Immunol. 2018, 53, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Senger, D.R.; Galli, S.J.; Dvorak, A.M.; Perruzzi, C.A.; Harvey, V.S.; Dvorak, H.F. Tumor Cells Secrete a Vascular Permeability Factor That Promotes Accumulation of Ascites Fluid. Science 1983, 219, 983–985. [Google Scholar] [CrossRef] [PubMed]
- Gerber, H.P.; Malik, A.K.; Solar, G.P.; Sherman, D.; Liang, X.H.; Meng, G.; Hong, K.; Marsters, J.C.; Ferrara, N. Vegf Regulates Haematopoietic Stem Cell Survival by an Internal Autocrine Loop Mechanism. Nature 2002, 417, 954–958. [Google Scholar] [CrossRef]
- De Falco, S.; Gigante, B.; Persico, M.G. Structure and Function of Placental Growth Factor. Trends Cardiovasc. Med. 2002, 12, 241–246. [Google Scholar] [CrossRef]
- Alitalo, K. The Lymphatic Vasculature in Disease. Nat. Med. 2011, 17, 1371–1380. [Google Scholar] [CrossRef]
- Zheng, W.; Aspelund, A.; Alitalo, K. Lymphangiogenic Factors, Mechanisms, and Applications. J. Clin. Investig. 2014, 124, 878–887. [Google Scholar] [CrossRef]
- Braile, M.; Fiorelli, A.; Sorriento, D.; di Crescenzo, R.M.; Galdiero, M.R.; Marone, G.; Santini, M.; Varricchi, G.; Loffredo, S. Human Lung-Resident Macrophages Express and Are Targets of Thymic Stromal Lymphopoietin in the Tumor Microenvironment. Cells 2021, 10, 2012. [Google Scholar] [CrossRef]
- Keyt, B.A.; Berleau, L.T.; Nguyen, H.V.; Chen, H.; Heinsohn, H.; Vandlen, R.; Ferrara, N. The Carboxyl-Terminal Domain (111–165) of Vascular Endothelial Growth Factor Is Critical for Its Mitogenic Potency. J. Biol. Chem. 1996, 271, 7788–7795. [Google Scholar] [CrossRef] [PubMed]
- Maglione, D.; Guerriero, V.; Viglietto, G.; Delli-Bovi, P.; Persico, M.G. Isolation of a Human Placenta Cdna Coding for a Protein Related to the Vascular Permeability Factor. Proc. Natl. Acad. Sci. USA 1991, 88, 9267–9271. [Google Scholar] [CrossRef] [PubMed]
- Gluzman-Poltorak, Z.; Cohen, T.; Herzog, Y.; Neufeld, G. Neuropilin-2 Is a Receptor for the Vascular Endothelial Growth Factor (Vegf) Forms Vegf-145 and Vegf-165 [Corrected]. J. Biol. Chem. 2000, 275, 18040–18045. [Google Scholar] [CrossRef] [PubMed]
- Moyon, D.; Pardanaud, L.; Yuan, L.; Breant, C.; Eichmann, A. Plasticity of Endothelial Cells During Arterial-Venous Differentiation in the Avian Embryo. Development 2001, 128, 3359–3370. [Google Scholar] [CrossRef] [PubMed]
- Herzog, Y.; Kalcheim, C.; Kahane, N.; Reshef, R.; Neufeld, G. Differential Expression of Neuropilin-1 and Neuropilin-2 in Arteries and Veins. Mech. Dev. 2001, 109, 115–119. [Google Scholar] [CrossRef]
- Becker, P.M.; Waltenberger, J.; Yachechko, R.; Mirzapoiazova, T.; Sham, J.S.; Lee, C.G.; Elias, J.A.; Verin, A.D. Neuropilin-1 Regulates Vascular Endothelial Growth Factor-Mediated Endothelial Permeability. Circ. Res. 2005, 96, 1257–1265. [Google Scholar] [CrossRef]
- Akwii, R.G.; Mikelis, C.M. Targeting the Angiopoietin/Tie Pathway: Prospects for Treatment of Retinal and Respiratory Disorders. Drugs 2021, 81, 1731–1749. [Google Scholar] [CrossRef]
- Augustin, H.G.; Koh, G.Y.; Thurston, G.; Alitalo, K. Control of Vascular Morphogenesis and Homeostasis through the Angiopoietin-Tie System. Nat. Rev. Mol. Cell Biol. 2009, 10, 165–177. [Google Scholar] [CrossRef]
- Hu, B.; Cheng, S.Y. Angiopoietin-2: Development of Inhibitors for Cancer Therapy. Curr. Oncol. Rep. 2009, 11, 111–116. [Google Scholar] [CrossRef]
- Prevete, N.; Staiano, R.I.; Granata, F.; Detoraki, A.; Necchi, V.; Ricci, V.; Triggiani, M.; de Paulis, A.; Marone, G.; Genovese, A. Expression and Function of Angiopoietins and Their Tie Receptors in Human Basophils and Mast Cells. J. Biol. Regul. Homeost. Agents 2013, 27, 827–839. [Google Scholar]
- Ismail, H.; Mofarrahi, M.; Echavarria, R.; Harel, S.; Verdin, E.; Lim, H.W.; Jin, Z.G.; Sun, J.; Zeng, H.; Hussain, S.N. Angiopoietin-1 and Vascular Endothelial Growth Factor Regulation of Leukocyte Adhesion to Endothelial Cells: Role of Nuclear Receptor-77. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1707–1716. [Google Scholar] [CrossRef] [PubMed]
- Thurston, G.; Suri, C.; Smith, K.; McClain, J.; Sato, T.N.; Yancopoulos, G.D.; McDonald, D.M. Leakage-Resistant Blood Vessels in Mice Transgenically Overexpressing Angiopoietin-1. Science 1999, 286, 2511–2514. [Google Scholar] [CrossRef] [PubMed]
- Loffredo, S.; Bova, M.; Suffritti, C.; Borriello, F.; Zanichelli, A.; Petraroli, A.; Varricchi, G.; Triggiani, M.; Cicardi, M.; Marone, G. Elevated Plasma Levels of Vascular Permeability Factors in C1 Inhibitor-Deficient Hereditary Angioedema. Allergy 2016, 71, 989–996. [Google Scholar] [CrossRef] [PubMed]
- Daly, C.; Eichten, A.; Castanaro, C.; Pasnikowski, E.; Adler, A.; Lalani, A.S.; Papadopoulos, N.; Kyle, A.H.; Minchinton, A.I.; Yancopoulos, G.D.; et al. Angiopoietin-2 Functions as a Tie2 Agonist in Tumor Models, Where It Limits the Effects of Vegf Inhibition. Cancer Res. 2013, 73, 108–118. [Google Scholar] [CrossRef]
- Fiedler, U.; Reiss, Y.; Scharpfenecker, M.; Grunow, V.; Koidl, S.; Thurston, G.; Gale, N.W.; Witzenrath, M.; Rosseau, S.; Suttorp, N.; et al. Angiopoietin-2 Sensitizes Endothelial Cells to Tnf-Alpha and Has a Crucial Role in the Induction of Inflammation. Nat. Med. 2006, 12, 235–239. [Google Scholar] [CrossRef]
- Roviezzo, F.; Tsigkos, S.; Kotanidou, A.; Bucci, M.; Brancaleone, V.; Cirino, G.; Papapetropoulos, A. Angiopoietin-2 Causes Inflammation in Vivo by Promoting Vascular Leakage. J. Pharmacol. Exp. Ther. 2005, 314, 738–744. [Google Scholar] [CrossRef]
- Bhandari, V.; Choo-Wing, R.; Lee, C.G.; Zhu, Z.; Nedrelow, J.H.; Chupp, G.L.; Zhang, X.; Matthay, M.A.; Ware, L.B.; Homer, R.J.; et al. Hyperoxia Causes Angiopoietin 2-Mediated Acute Lung Injury and Necrotic Cell Death. Nat. Med. 2006, 12, 1286–1293. [Google Scholar] [CrossRef]
- Tammela, T.; Saaristo, A.; Lohela, M.; Morisada, T.; Tornberg, J.; Norrmen, C.; Oike, Y.; Pajusola, K.; Thurston, G.; Suda, T.; et al. Angiopoietin-1 Promotes Lymphatic Sprouting and Hyperplasia. Blood 2005, 105, 4642–4648. [Google Scholar] [CrossRef]
- Daneman, R.; Zhou, L.; Kebede, A.A.; Barres, B.A. Pericytes Are Required for Blood-Brain Barrier Integrity During Embryogenesis. Nature 2010, 468, 562–566. [Google Scholar] [CrossRef]
- Yin, J.; Gong, G.; Liu, X. Angiopoietin: A Novel Neuroprotective/Neurotrophic Agent. Neuroscience 2019, 411, 177–184. [Google Scholar] [CrossRef]
- Fiedler, U.; Augustin, H.G. Angiopoietins: A Link between Angiogenesis and Inflammation. Trends Immunol. 2006, 27, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, U.; Scharpfenecker, M.; Koidl, S.; Hegen, A.; Grunow, V.; Schmidt, J.M.; Kriz, W.; Thurston, G.; Augustin, H.G. The Tie-2 Ligand Angiopoietin-2 Is Stored in and Rapidly Released Upon Stimulation from Endothelial Cell Weibel-Palade Bodies. Blood 2004, 103, 4150–4156. [Google Scholar] [CrossRef] [PubMed]
- Maisonpierre, P.C.; Suri, C.; Jones, P.F.; Bartunkova, S.; Wiegand, S.J.; Radziejewski, C.; Compton, D.; McClain, J.; Aldrich, T.H.; Papadopoulos, N.; et al. Angiopoietin-2, a Natural Antagonist for Tie2 That Disrupts in Vivo Angiogenesis. Science 1997, 277, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.Y.; Hughes, R.; Murdoch, C.; Coffelt, S.B.; Biswas, S.K.; Harris, A.L.; Johnson, R.S.; Imityaz, H.Z.; Simon, M.C.; Fredlund, E.; et al. Hypoxia-Inducible Factors 1 and 2 Are Important Transcriptional Effectors in Primary Macrophages Experiencing Hypoxia. Blood 2009, 114, 844–859. [Google Scholar] [CrossRef]
- Staiano, R.I.; Loffredo, S.; Borriello, F.; Iannotti, F.A.; Piscitelli, F.; Orlando, P.; Secondo, A.; Granata, F.; Lepore, M.T.; Fiorelli, A.; et al. Human Lung-Resident Macrophages Express Cb1 and Cb2 Receptors Whose Activation Inhibits the Release of Angiogenic and Lymphangiogenic Factors. J. Leukoc. Biol. 2016, 99, 531–540. [Google Scholar] [CrossRef]
- Loffredo, S.; Borriello, F.; Iannone, R.; Ferrara, A.L.; Galdiero, M.R.; Gigantino, V.; Esposito, P.; Varricchi, G.; Lambeau, G.; Cassatella, M.A.; et al. Group V Secreted Phospholipase A2 Induces the Release of Proangiogenic and Antiangiogenic Factors by Human Neutrophils. Front. Immunol. 2017, 8, 443. [Google Scholar] [CrossRef]
- Hakanpaa, L.; Sipila, T.; Leppanen, V.M.; Gautam, P.; Nurmi, H.; Jacquemet, G.; Eklund, L.; Ivaska, J.; Alitalo, K.; Saharinen, P. Endothelial Destabilization by Angiopoietin-2 Via Integrin Beta1 Activation. Nat. Commun. 2015, 6, 5962. [Google Scholar] [CrossRef]
- Felcht, M.; Luck, R.; Schering, A.; Seidel, P.; Srivastava, K.; Hu, J.; Bartol, A.; Kienast, Y.; Vettel, C.; Loos, E.K.; et al. Angiopoietin-2 Differentially Regulates Angiogenesis through Tie2 and Integrin Signaling. J. Clin. Investig. 2012, 122, 1991–2005. [Google Scholar] [CrossRef]
- Joosten, S.P.J.; Spaargaren, M.; Clevers, H.; Pals, S.T. Hepatocyte Growth Factor/Met and Cd44 in Colorectal Cancer: Partners in Tumorigenesis and Therapy Resistance. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188437. [Google Scholar] [CrossRef]
- Fett, J.W.; Strydom, D.J.; Lobb, R.R.; Alderman, E.M.; Bethune, J.L.; Riordan, J.F.; Vallee, B.L. Isolation and Characterization of Angiogenin, an Angiogenic Protein from Human Carcinoma Cells. Biochemistry 1985, 24, 5480–5486. [Google Scholar] [CrossRef]
- Garnett, E.R.; Raines, R.T. Emerging Biological Functions of Ribonuclease 1 and Angiogenin. Crit. Rev. Biochem. Mol. Biol. 2021, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Cai, Y.; Zhou, W.; Sheng, J.; Xu, Z. The Potential of Angiogenin as a Serum Biomarker for Diseases: Systematic Review and Meta-Analysis. Dis. Markers 2018, 2018, 1984718. [Google Scholar] [CrossRef] [PubMed]
- Geerts, L.; Jorens, P.G.; Willems, J.; de Ley, M.; Slegers, H. Natural Inhibitors of Neutrophil Function in Acute Respiratory Distress Syndrome. Crit. Care Med. 2001, 29, 1920–1924. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wen, D.; Jiang, W.; Song, J.; Yang, J.; Gao, X.; Xue, H.; Wang, L. Angiogenin Negatively Regulates the Expression of Basic Fibroblast Growth Factor (Bfgf) and Inhibits Bfgf Promoter Activity. Int. J. Clin. Exp. Pathol. 2018, 11, 3277–3285. [Google Scholar] [PubMed]
- Broadley, K.N.; Aquino, A.M.; Woodward, S.C.; Buckley-Sturrock, A.; Sato, Y.; Rifkin, D.B.; Davidson, J.M. Monospecific Antibodies Implicate Basic Fibroblast Growth Factor in Normal Wound Repair. Lab. Investig. 1989, 61, 571–575. [Google Scholar] [PubMed]
- Henke, C.; Marineili, W.; Jessurun, J.; Fox, J.; Harms, D.; Peterson, M.; Chiang, L.; Doran, P. Macrophage Production of Basic Fibroblast Growth Factor in the Fibroproliferative Disorder of Alveolar Fibrosis after Lung Injury. Am. J. Pathol. 1993, 143, 1189–1199. [Google Scholar]
- Cassatella, M.A.; Ostberg, N.K.; Tamassia, N.; Soehnlein, O. Biological Roles of Neutrophil-Derived Granule Proteins and Cytokines. Trends Immunol. 2019, 40, 648–664. [Google Scholar] [CrossRef]
- Braile, M.; Cristinziano, L.; Marcella, S.; Varricchi, G.; Marone, G.; Modestino, L.; Ferrara, A.L.; de Ciuceis, A.; Scala, S.; Galdiero, M.R.; et al. Lps-Mediated Neutrophil Vegf-a Release Is Modulated by Cannabinoid Receptor Activation. J. Leukoc. Biol. 2021, 109, 621–631. [Google Scholar] [CrossRef]
- Meniailo, M.E.; Malashchenko, V.V.; Shmarov, V.A.; Gazatova, N.D.; Melashchenko, O.B.; Goncharov, A.G.; Seledtsova, G.V.; Seledtsov, V.I. Interleukin-8 Favors Pro-Inflammatory Activity of Human Monocytes/Macrophages. Int. Immunopharmacol. 2018, 56, 217–221. [Google Scholar] [CrossRef]
- Gross, A.R.; Theoharides, T.C. Chondroitin Sulfate Inhibits Secretion of Tnf and Cxcl8 from Human Mast Cells Stimulated by Il-33. Biofactors 2019, 45, 49–61. [Google Scholar] [CrossRef]
- Asokan, S.; Bandapalli, O.R. Cxcl8 Signaling in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2021, 1302, 25–39. [Google Scholar] [PubMed]
- Li, A.; Dubey, S.; Varney, M.L.; Dave, B.J.; Singh, R.K. Il-8 Directly Enhanced Endothelial Cell Survival, Proliferation, and Matrix Metalloproteinases Production and Regulated Angiogenesis. J. Immunol. 2003, 170, 3369–3376. [Google Scholar] [CrossRef] [PubMed]
- Brat, D.J.; Bellail, A.C.; van Meir, E.G. The Role of Interleukin-8 and Its Receptors in Gliomagenesis and Tumoral Angiogenesis. Neuro-Oncology 2005, 7, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Ritzmann, F.; Beisswenger, C. Preclinical Studies and the Function of Il-17 Cytokines in Copd. Ann. Anat. 2021, 237, 151729. [Google Scholar] [CrossRef]
- McGeachy, M.J.; Cua, D.J.; Gaffen, S.L. The Il-17 Family of Cytokines in Health and Disease. Immunity 2019, 50, 892–906. [Google Scholar] [CrossRef]
- Numasaki, M.; Fukushi, J.; Ono, M.; Narula, S.K.; Zavodny, P.J.; Kudo, T.; Robbins, P.D.; Tahara, H.; Lotze, M.T. Interleukin-17 Promotes Angiogenesis and Tumor Growth. Blood 2003, 101, 2620–2627. [Google Scholar] [CrossRef]
- Numasaki, M.; Watanabe, M.; Suzuki, T.; Takahashi, H.; Nakamura, A.; McAllister, F.; Hishinuma, T.; Goto, J.; Lotze, M.T.; Kolls, J.K.; et al. Il-17 Enhances the Net Angiogenic Activity and in Vivo Growth of Human Non-Small Cell Lung Cancer in Scid Mice through Promoting Cxcr-2-Dependent Angiogenesis. J. Immunol. 2005, 175, 6177–6189. [Google Scholar] [CrossRef]
- Takahashi, H.; Numasaki, M.; Lotze, M.T.; Sasaki, H. Interleukin-17 Enhances Bfgf-, Hgf- and Vegf-Induced Growth of Vascular Endothelial Cells. Immunol. Lett. 2005, 98, 189–193. [Google Scholar] [CrossRef]
- Varricchi, G.; de Paulis, A.; Marone, G.; Galli, S.J. Future Needs in Mast Cell Biology. Int. J. Mol. Sci. 2019, 20, 4397. [Google Scholar] [CrossRef]
- Varricchi, G.; Rossi, F.W.; Galdiero, M.R.; Granata, F.; Criscuolo, G.; Spadaro, G.; de Paulis, A.; Marone, G. Physiological Roles of Mast Cells: Collegium Internationale Allergologicum Update 2019. Int. Arch. Allergy Immunol. 2019, 179, 247–261. [Google Scholar] [CrossRef]
- Marone, G.; Borriello, F.; Varricchi, G.; Genovese, A.; Granata, F. Basophils: Historical Reflections and Perspectives. Chem. Immunol. Allergy 2014, 100, 172–192. [Google Scholar] [PubMed]
- Kanaoka, Y.; Austen, K.F. Roles of Cysteinyl Leukotrienes and Their Receptors in Immune Cell-Related Functions. Adv. Immunol. 2019, 142, 65–84. [Google Scholar] [PubMed]
- Tsopanoglou, N.E.; Pipili-Synetos, E.; Maragoudakis, M.E. Leukotrienes C4 and D4 Promote Angiogenesis Via a Receptor-Mediated Interaction. Eur. J. Pharmacol. 1994, 258, 151–154. [Google Scholar] [CrossRef]
- Kanayasu, T.; Nakao-Hayashi, J.; Asuwa, N.; Morita, I.; Ishii, T.; Ito, H.; Murota, S. Leukotriene C4 Stimulates Angiogenesis in Bovine Carotid Artery Endothelial Cells in Vitro. Biochem. Biophys. Res. Commun. 1989, 159, 572–578. [Google Scholar] [CrossRef]
- Duah, E.; Teegala, L.R.; Kondeti, V.; Adapala, R.K.; Keshamouni, V.G.; Kanaoka, Y.; Austen, K.F.; Thodeti, C.K.; Paruchuri, S. Cysteinyl Leukotriene 2 Receptor Promotes Endothelial Permeability, Tumor Angiogenesis, and Metastasis. Proc. Natl. Acad. Sci. USA 2019, 116, 199–204. [Google Scholar] [CrossRef]
- Gupta, K.; Gupta, P.; Wild, R.; Ramakrishnan, S.; Hebbel, R.P. Binding and Displacement of Vascular Endothelial Growth Factor (Vegf) by Thrombospondin: Effect on Human Microvascular Endothelial Cell Proliferation and Angiogenesis. Angiogenesis 1999, 3, 147–158. [Google Scholar] [CrossRef]
- Kaur, S.; Bronson, S.M.; Pal-Nath, D.; Miller, T.W.; Soto-Pantoja, D.R.; Roberts, D.D. Functions of Thrombospondin-1 in the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 4570. [Google Scholar] [CrossRef]
- O’Reilly, M.S.; Boehm, T.; Shing, Y.; Fukai, N.; Vasios, G.; Lane, W.S.; Flynn, E.; Birkhead, J.R.; Olsen, B.R.; Folkman, J. Endostatin: An Endogenous Inhibitor of Angiogenesis and Tumor Growth. Cell 1997, 88, 277–285. [Google Scholar] [CrossRef]
- Yamaguchi, N.; Anand-Apte, B.; Lee, M.; Sasaki, T.; Fukai, N.; Shapiro, R.; Que, I.; Lowik, C.; Timpl, R.; Olsen, B.R. Endostatin Inhibits Vegf-Induced Endothelial Cell Migration and Tumor Growth Independently of Zinc Binding. EMBO J. 1999, 18, 4414–4423. [Google Scholar] [CrossRef]
- Liu, L.; Qiao, Y.; Hu, C.; Liu, Y.; Xia, Y.; Wang, L.; Liu, B.; Chen, H.; Jiang, X. Endostatin Exerts Radiosensitizing Effect in Non-Small Cell Lung Cancer Cells by Inhibiting Vegfr2 Expression. Clin. Transl. Oncol. 2016, 18, 18–26. [Google Scholar] [CrossRef]
- Harper, S.J.; Bates, D.O. Vegf-a Splicing: The Key to Anti-Angiogenic Therapeutics? Nat. Rev. Cancer 2008, 8, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Manetti, M.; Guiducci, S.; Romano, E.; Ceccarelli, C.; Bellando-Randone, S.; Conforti, M.L.; Ibba-Manneschi, L.; Matucci-Cerinic, M. Overexpression of Vegf165b, an Inhibitory Splice Variant of Vascular Endothelial Growth Factor, Leads to Insufficient Angiogenesis in Patients with Systemic Sclerosis. Circ. Res. 2011, 109, e14–e26. [Google Scholar] [CrossRef] [PubMed]
- Peiris-Pages, M. The Role of Vegf 165b in Pathophysiology. Cell Adh. Migr. 2012, 6, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Bowler, E.; Oltean, S. Alternative Splicing in Angiogenesis. Int. J. Mol. Sci. 2019, 20, 2067. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Inflammatory Mechanisms in Patients with Chronic Obstructive Pulmonary Disease. J. Allergy Clin. Immunol. 2016, 138, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Celli, B.R.; Fabbri, L.M.; Aaron, S.D.; Agusti, A.; Brook, R.; Criner, G.J.; Franssen, F.M.E.; Humbert, M.; Hurst, J.R.; O’Donnell, D.; et al. An Updated Definition and Severity Classification of Chronic Obstructive Pulmonary Disease Exacerbations: The Rome Proposal. Am. J. Respir. Crit. Care Med. 2021, 204, 1251–1258. [Google Scholar] [CrossRef]
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and Regional Mortality from 235 Causes of Death for 20 Age Groups in 1990 and 2010: A Systematic Analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef]
- Jasper, A.E.; McIver, W.J.; Sapey, E.; Walton, G.M. Understanding the Role of Neutrophils in Chronic Inflammatory Airway Disease. F1000Research 2019, 8. [Google Scholar] [CrossRef]
- Barnes, P.J. Inflammatory Endotypes in Copd. Allergy 2019, 74, 1249–1256. [Google Scholar] [CrossRef]
- Kim, V.L.; Coombs, N.A.; Staples, K.J.; Ostridge, K.K.; Williams, N.P.; Wootton, S.A.; Devaster, J.M.; Aris, E.; Clarke, S.C.; Tuck, A.C.; et al. Impact and Associations of Eosinophilic Inflammation in Copd: Analysis of the Aeris Cohort. Eur. Respir. J. 2017, 50, 1700853. [Google Scholar] [CrossRef]
- Wang, Z.; Locantore, N.; Haldar, K.; Ramsheh, M.Y.; Beech, A.S.; Ma, W.; Brown, J.R.; Tal-Singer, R.; Barer, M.R.; Bafadhel, M.; et al. Inflammatory Endotype-Associated Airway Microbiome in Chronic Obstructive Pulmonary Disease Clinical Stability and Exacerbations: A Multicohort Longitudinal Analysis. Am. J. Respir. Crit. Care Med. 2021, 203, 1488–1502. [Google Scholar] [CrossRef] [PubMed]
- Brightling, C.E.; Monteiro, W.; Ward, R.; Parker, D.; Morgan, M.D.; Wardlaw, A.J.; Pavord, I.D. Sputum Eosinophilia and Short-Term Response to Prednisolone in Chronic Obstructive Pulmonary Disease: A Randomised Controlled Trial. Lancet 2000, 356, 1480–1485. [Google Scholar] [CrossRef]
- Kolsum, U.; Donaldson, G.C.; Singh, R.; Barker, B.L.; Gupta, V.; George, L.; Webb, A.J.; Thurston, S.; Brookes, A.J.; McHugh, T.D.; et al. Blood and Sputum Eosinophils in Copd; Relationship with Bacterial Load. Respir. Res. 2017, 18, 88. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Bafadhel, M.; Brightling, C.E.; Sciurba, F.C.; Curtis, J.L.; Martinez, F.J.; Pasquale, C.B.; Merrill, D.D.; Metzdorf, N.; Petruzzelli, S.; et al. Blood Eosinophil Counts in Clinical Trials for Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2020, 202, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Cellular and Molecular Mechanisms of Chronic Obstructive Pulmonary Disease. Clin. Chest Med. 2014, 35, 71–86. [Google Scholar] [CrossRef]
- Tan, W.C.; Sin, D.D.; Bourbeau, J.; Hernandez, P.; Chapman, K.R.; Cowie, R.; FitzGerald, J.M.; Marciniuk, D.D.; Maltais, F.; Buist, A.S.; et al. Characteristics of Copd in Never-Smokers and Ever-Smokers in the General Population: Results from the Cancold Study. Thorax 2015, 70, 822–829. [Google Scholar] [CrossRef]
- Aloufi, N.; Alluli, A.; Eidelman, D.H.; Baglole, C.J. Aberrant Post-Transcriptional Regulation of Protein Expression in the Development of Chronic Obstructive Pulmonary Disease. Int. J. Mol. Sci. 2021, 22, 11963. [Google Scholar] [CrossRef]
- Cosio, M.G.; Saetta, M.; Agusti, A. Immunologic Aspects of Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2009, 360, 2445–2454. [Google Scholar] [CrossRef]
- Soltani, A.; Ewe, Y.P.; Lim, Z.S.; Sohal, S.S.; Reid, D.; Weston, S.; Wood-Baker, R.; Walters, E.H. Mast Cells in Copd Airways: Relationship to Bronchodilator Responsiveness and Angiogenesis. Eur. Respir. J. 2012, 39, 1361–1367. [Google Scholar] [CrossRef]
- Jogdand, P.; Siddhuraj, P.; Mori, M.; Sanden, C.; Jonsson, J.; Walls, A.F.; Kearley, J.; Humbles, A.A.; Kolbeck, R.; Bjermer, L.; et al. Eosinophils, Basophils and Type 2 Immune Microenvironments in Copd-Affected Lung Tissue. Eur. Respir. J. 2020, 55, 1900110. [Google Scholar] [CrossRef]
- Barbera, J.A.; Riverola, A.; Roca, J.; Ramirez, J.; Wagner, P.D.; Ros, D.; Wiggs, B.R.; Rodriguez-Roisin, R. Pulmonary Vascular Abnormalities and Ventilation-Perfusion Relationships in Mild Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 1994, 149, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, C.; Bocchino, V.; Vatrella, A.; Marzo, C.; Guarino, C.; Mascitti, S.; Tranfa, C.M.; Cazzola, M.; Micheli, P.; Caputi, M.; et al. Evidence of Angiogenesis in Bronchial Biopsies of Smokers with and without Airway Obstruction. Respir. Med. 2006, 100, 1415–1422. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, H.; Asai, K.; Hirata, K.; Yoshikawa, J. Possible Effects of Vascular Endothelial Growth Factor in the Pathogenesis of Chronic Obstructive Pulmonary Disease. Am. J. Med. 2003, 114, 354–358. [Google Scholar] [CrossRef]
- Kierszniewska-Stepien, D.; Pietras, T.; Gorski, P.; Stepien, H. Serum Vascular Endothelial Growth Factor and Its Receptor Level in Patients with Chronic Obstructive Pulmonary Disease. Eur. Cytokine Netw. 2006, 17, 75–79. [Google Scholar]
- Kranenburg, A.R.; de Boer, W.I.; Alagappan, V.K.; Sterk, P.J.; Sharma, H.S. Enhanced Bronchial Expression of Vascular Endothelial Growth Factor and Receptors (Flk-1 and Flt-1) in Patients with Chronic Obstructive Pulmonary Disease. Thorax 2005, 60, 106–113. [Google Scholar] [CrossRef]
- Santos, S.; Peinado, V.I.; Ramirez, J.; Morales-Blanhir, J.; Bastos, R.; Roca, J.; Rodriguez-Roisin, R.; Barbera, J.A. Enhanced Expression of Vascular Endothelial Growth Factor in Pulmonary Arteries of Smokers and Patients with Moderate Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2003, 167, 1250–1256. [Google Scholar] [CrossRef]
- Ying, S.; O’Connor, B.; Ratoff, J.; Meng, Q.; Fang, C.; Cousins, D.; Zhang, G.; Gu, S.; Gao, Z.; Shamji, Z.B.; et al. Expression and Cellular Provenance of Thymic Stromal Lymphopoietin and Chemokines in Patients with Severe Asthma and Chronic Obstructive Pulmonary Disease. J. Immunol. 2008, 181, 2790–2798. [Google Scholar] [CrossRef]
- Varricchi, G.; Pecoraro, A.; Marone, G.; Criscuolo, G.; Spadaro, G.; Genovese, A. Thymic Stromal Lymphopoietin Isoforms, Inflammatory Disorders, and Cancer. Front. Immunol. 2018, 9, 1595. [Google Scholar] [CrossRef]
- Marone, G.; Spadaro, G.; Braile, M.; Poto, R.; Criscuolo, G.; Pahima, H.; Loffredo, S.; Levi-Schaffer, F.; Varricchi, G. Tezepelumab: A Novel Biological Therapy for the Treatment of Severe Uncontrolled Asthma. Expert Opin. Investig. Drugs 2019, 28, 931–940. [Google Scholar] [CrossRef]
- Gauvreau, G.M.; Sehmi, R.; Ambrose, C.S.; Griffiths, J.M. Thymic Stromal Lymphopoietin: Its Role and Potential as a Therapeutic Target in Asthma. Expert Opin. Ther. Targets 2020, 24, 777–792. [Google Scholar] [CrossRef]
- Zhang, K.; Shan, L.; Rahman, M.S.; Unruh, H.; Halayko, A.J.; Gounni, A.S. Constitutive and Inducible Thymic Stromal Lymphopoietin Expression in Human Airway Smooth Muscle Cells: Role in Chronic Obstructive Pulmonary Disease. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 293, L375–L382. [Google Scholar] [CrossRef] [PubMed]
- Kienhofer, D.; Hahn, J.; Stoof, J.; Csepregi, J.Z.; Reinwald, C.; Urbonaviciute, V.; Johnsson, C.; Maueroder, C.; Podolska, M.J.; Biermann, M.H.; et al. Experimental Lupus Is Aggravated in Mouse Strains with Impaired Induction of Neutrophil Extracellular Traps. JCI Insight 2017, 2, e92920. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Miyata, M.; Ohba, T.; Ando, T.; Hatsushika, K.; Suenaga, F.; Shimokawa, N.; Ohnuma, Y.; Katoh, R.; Ogawa, H.; et al. Cigarette Smoke Extract Induces Thymic Stromal Lymphopoietin Expression, Leading to T(H)2-Type Immune Responses and Airway Inflammation. J. Allergy Clin. Immunol. 2008, 122, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Smelter, D.F.; Sathish, V.; Thompson, M.A.; Pabelick, C.M.; Vassallo, R.; Prakash, Y.S. Thymic Stromal Lymphopoietin in Cigarette Smoke-Exposed Human Airway Smooth Muscle. J. Immunol. 2010, 185, 3035–3040. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Tao, S.; Zhang, S.; Wang, J.; Zhang, F.; Li, F.; Ding, J. Type 2 Innate Lymphoid Cells Participate in Il-33-Stimulated Th2-Associated Immune Response in Chronic Obstructive Pulmonary Disease. Exp. Ther. Med. 2019, 18, 3109–3116. [Google Scholar] [CrossRef]
- Kearley, J.; Silver, J.S.; Sanden, C.; Liu, Z.; Berlin, A.A.; White, N.; Mori, M.; Pham, T.H.; Ward, C.K.; Criner, G.J.; et al. Cigarette Smoke Silences Innate Lymphoid Cell Function and Facilitates an Exacerbated Type I Interleukin-33-Dependent Response to Infection. Immunity 2015, 42, 566–579. [Google Scholar] [CrossRef]
- Gorska, K.; Nejman-Gryz, P.; Paplinska-Goryca, M.; Korczynski, P.; Prochorec-Sobieszek, M.; Krenke, R. Comparative Study of Il-33 and Il-6 Levels in Different Respiratory Samples in Mild-to-Moderate Asthma and Copd. COPD 2018, 15, 36–45. [Google Scholar] [CrossRef]
- Kim, S.W.; Rhee, C.K.; Kim, K.U.; Lee, S.H.; Hwang, H.G.; Kim, Y.I.; Kim, D.K.; Lee, S.D.; Oh, Y.M.; Yoon, H.K. Factors Associated with Plasma Il-33 Levels in Patients with Chronic Obstructive Pulmonary Disease. Int. J. Chron. Obstruct. Pulmon. Dis. 2017, 12, 395–402. [Google Scholar] [CrossRef]
- Byers, D.E.; Alexander-Brett, J.; Patel, A.C.; Agapov, E.; Dang-Vu, G.; Jin, X.; Wu, K.; You, Y.; Alevy, Y.; Girard, J.P.; et al. Long-Term Il-33-Producing Epithelial Progenitor Cells in Chronic Obstructive Lung Disease. J. Clin. Investig. 2013, 123, 3967–3982. [Google Scholar] [CrossRef]
- Choi, J.; Lee, H.C.; Yoo, K.; Jung, K.; Rhee, C. Abstract: Serum Interleukin-33 Level Was Associated with Previous Acute Exacerbation History in Chronic Obstructive Pulmonary Disease Patients. Am. J. Respir. Crit. Care Med. 2020, 201, A2863. [Google Scholar]
- De Falco, G.; Colarusso, C.; Terlizzi, M.; Popolo, A.; Pecoraro, M.; Commodo, M.; Minutolo, P.; Sirignano, M.; D’Anna, A.; Aquino, R.P.; et al. Chronic Obstructive Pulmonary Disease-Derived Circulating Cells Release Il-18 and Il-33 under Ultrafine Particulate Matter Exposure in a Caspase-1/8-Independent Manner. Front. Immunol. 2017, 8, 1415. [Google Scholar] [CrossRef] [PubMed]
- Fahey, E.; Doyle, S.L. Il-1 Family Cytokine Regulation of Vascular Permeability and Angiogenesis. Front. Immunol. 2019, 10, 1426. [Google Scholar] [CrossRef] [PubMed]
- Afferni, C.; Buccione, C.; Andreone, S.; Galdiero, M.R.; Varricchi, G.; Marone, G.; Mattei, F.; Schiavoni, G. The Pleiotropic Immunomodulatory Functions of Il-33 and Its Implications in Tumor Immunity. Front. Immunol. 2018, 9, 2601. [Google Scholar] [CrossRef]
- Hams, E.; Armstrong, M.E.; Barlow, J.L.; Saunders, S.P.; Schwartz, C.; Cooke, G.; Fahy, R.J.; Crotty, T.B.; Hirani, N.; Flynn, R.J.; et al. Il-25 and Type 2 Innate Lymphoid Cells Induce Pulmonary Fibrosis. Proc. Natl. Acad. Sci. USA 2014, 111, 367–372. [Google Scholar] [CrossRef]
- Nejman-Gryz, P.; Górska, K.; Paplińska-Goryca, M.; Proboszcz, M.; Krenke, R. Epithelial Derived Cytokines Il-25, Il-33 and Tslp in Obstructive Lung Diseases. Eur. Respir. J. 2017, 50, PA571. [Google Scholar]
- Katoh, S.; Ikeda, M.; Shirai, R.; Abe, M.; Ohue, Y.; Kobashi, Y.; Oka, M. Biomarkers for Differentiation of Patients with Asthma and Chronic Obstructive Pulmonary Disease. J. Asthma 2018, 55, 1052–1058. [Google Scholar] [CrossRef] [PubMed]
- Corrigan, C.J.; Wang, W.; Meng, Q.; Fang, C.; Wu, H.; Reay, V.; Lv, Z.; Fan, Y.; An, Y.; Wang, Y.H.; et al. T-Helper Cell Type 2 (Th2) Memory T Cell-Potentiating Cytokine Il-25 Has the Potential to Promote Angiogenesis in Asthma. Proc. Natl. Acad. Sci. USA 2011, 108, 1579–1584. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.E.; Thorley, A.; Culpitt, S.V.; Dodd, S.; Donnelly, L.E.; Demattos, C.; Fitzgerald, M.; Barnes, P.J. Alveolar Macrophage-Mediated Elastolysis: Roles of Matrix Metalloproteinases, Cysteine, and Serine Proteases. Am. J. Physiol. Lung Cell Mol. Physiol. 2002, 283, L867–L873. [Google Scholar] [CrossRef]
- Chevrier, S.; Levine, J.H.; Zanotelli, V.R.T.; Silina, K.; Schulz, D.; Bacac, M.; Ries, C.H.; Ailles, L.; Jewett, M.A.S.; Moch, H.; et al. An Immune Atlas of Clear Cell Renal Cell Carcinoma. Cell 2017, 169, 736–749.e18. [Google Scholar] [CrossRef]
- Lavin, Y.; Kobayashi, S.; Leader, A.; Amir, E.D.; Elefant, N.; Bigenwald, C.; Remark, R.; Sweeney, R.; Becker, C.D.; Levine, J.H.; et al. Innate Immune Landscape in Early Lung Adenocarcinoma by Paired Single-Cell Analyses. Cell 2017, 169, 750–765.e17. [Google Scholar] [CrossRef]
- Zilionis, R.; Engblom, C.; Pfirschke, C.; Savova, V.; Zemmour, D.; Saatcioglu, H.D.; Krishnan, I.; Maroni, G.; Meyerovitz, C.V.; Kerwin, C.M.; et al. Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations across Individuals and Species. Immunity 2019, 50, 1317–1334.e10. [Google Scholar] [CrossRef] [PubMed]
- Vieira Braga, F.A.; Kar, G.; Berg, M.; Carpaij, O.A.; Polanski, K.; Simon, L.M.; Brouwer, S.; Gomes, T.; Hesse, L.; Jiang, J.; et al. A Cellular Census of Human Lungs Identifies Novel Cell States in Health and in Asthma. Nat. Med. 2019, 25, 1153–1163. [Google Scholar] [CrossRef] [PubMed]
- Hua, X.; Hu, G.; Hu, Q.; Chang, Y.; Hu, Y.; Gao, L.; Chen, X.; Yang, P.C.; Zhang, Y.; Li, M.; et al. Single-Cell Rna Sequencing to Dissect the Immunological Network of Autoimmune Myocarditis. Circulation 2020, 142, 384–400. [Google Scholar] [CrossRef] [PubMed]
- Balestrieri, B.; Granata, F.; Loffredo, S.; Petraroli, A.; Scalia, G.; Morabito, P.; Cardamone, C.; Varricchi, G.; Triggiani, M. Phenotypic and Functional Heterogeneity of Low-Density and High-Density Human Lung Macrophages. Biomedicines 2021, 9, 505. [Google Scholar] [CrossRef] [PubMed]
- Chana, K.K.; Fenwick, P.S.; Nicholson, A.G.; Barnes, P.J.; Donnelly, L.E. Identification of a Distinct Glucocorticosteroid-Insensitive Pulmonary Macrophage Phenotype in Patients with Chronic Obstructive Pulmonary Disease. J. Allergy Clin. Immunol. 2014, 133, 207–216.e11. [Google Scholar] [CrossRef]
- Finney, L.J.; Belchamber, K.B.R.; Fenwick, P.S.; Kemp, S.V.; Edwards, M.R.; Mallia, P.; Donaldson, G.; Johnston, S.L.; Donnelly, L.E.; Wedzicha, J.A. Human Rhinovirus Impairs the Innate Immune Response to Bacteria in Alveolar Macrophages in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2019, 199, 1496–1507. [Google Scholar] [CrossRef]
- Granata, F.; Frattini, A.; Loffredo, S.; Staiano, R.I.; Petraroli, A.; Ribatti, D.; Oslund, R.; Gelb, M.H.; Lambeau, G.; Marone, G.; et al. Production of Vascular Endothelial Growth Factors from Human Lung Macrophages Induced by Group Iia and Group X Secreted Phospholipases A2. J. Immunol. 2010, 184, 5232–5241. [Google Scholar] [CrossRef]
- Granata, F.; Balestrieri, B.; Petraroli, A.; Giannattasio, G.; Marone, G.; Triggiani, M. Secretory Phospholipases A2 as Multivalent Mediators of Inflammatory and Allergic Disorders. Int. Arch. Allergy Immunol. 2003, 131, 153–163. [Google Scholar] [CrossRef]
- Ferrara, A.L.; Galdiero, M.R.; Fiorelli, A.; Cristinziano, L.; Granata, F.; Marone, G.; Crescenzo, R.M.D.; Braile, M.; Marcella, S.; Modestino, L.; et al. Macrophage-Polarizing Stimuli Differentially Modulate the Inflammatory Profile Induced by the Secreted Phospholipase A2 Group Ia in Human Lung Macrophages. Cytokine 2021, 138, 155378. [Google Scholar] [CrossRef]
- Plum, T.; Wang, X.; Rettel, M.; Krijgsveld, J.; Feyerabend, T.B.; Rodewald, H.R. Human Mast Cell Proteome Reveals Unique Lineage, Putative Functions, and Structural Basis for Cell Ablation. Immunity 2020, 52, 404–416.e5. [Google Scholar] [CrossRef]
- Boesiger, J.; Tsai, M.; Maurer, M.; Yamaguchi, M.; Brown, L.F.; Claffey, K.P.; Dvorak, H.F.; Galli, S.J. Mast Cells Can Secrete Vascular Permeability Factor/Vascular Endothelial Cell Growth Factor and Exhibit Enhanced Release after Immunoglobulin E-Dependent Upregulation of Fc Epsilon Receptor I Expression. J. Exp. Med. 1998, 188, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Cosio, B.G.; Soriano, J.B.; Lopez-Campos, J.L.; Calle-Rubio, M.; Soler-Cataluna, J.J.; de-Torres, J.P.; Marin, J.M.; Martinez-Gonzalez, C.; de Lucas, P.; Mir, I.; et al. Defining the Asthma-Copd Overlap Syndrome in a Copd Cohort. Chest 2016, 149, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Crivellato, E.; Candussio, L.; Nico, B.; Vacca, A.; Roncali, L.; Dammacco, F. Mast Cells and Their Secretory Granules Are Angiogenic in the Chick Embryo Chorioallantoic Membrane. Clin. Exp. Allergy 2001, 31, 602–608. [Google Scholar] [CrossRef]
- Detoraki, A.; Staiano, R.I.; Granata, F.; Giannattasio, G.; Prevete, N.; de Paulis, A.; Ribatti, D.; Genovese, A.; Triggiani, M.; Marone, G. Vascular Endothelial Growth Factors Synthesized by Human Lung Mast Cells Exert Angiogenic Effects. J. Allergy Clin. Immunol. 2009, 123, 1142–1149.e5. [Google Scholar] [CrossRef]
- Cristinziano, L.; Poto, R.; Criscuolo, G.; Ferrara, A.L.; Galdiero, M.R.; Modestino, L.; Loffredo, S.; de Paulis, A.; Marone, G.; Spadaro, G.; et al. Il-33 and Superantigenic Activation of Human Lung Mast Cells Induce the Release of Angiogenic and Lymphangiogenic Factors. Cells 2021, 10, 145. [Google Scholar] [CrossRef]
- Miralda, I.; Uriarte, S.M.; McLeish, K.R. Multiple Phenotypic Changes Define Neutrophil Priming. Front. Cell Infect. Microbiol. 2017, 7, 217. [Google Scholar] [CrossRef] [PubMed]
- Keatings, V.M.; Barnes, P.J. Granulocyte Activation Markers in Induced Sputum: Comparison between Chronic Obstructive Pulmonary Disease, Asthma, and Normal Subjects. Am. J. Respir. Crit. Care Med. 1997, 155, 449–453. [Google Scholar] [CrossRef]
- Fahy, J.V.; Dickey, B.F. Airway Mucus Function and Dysfunction. N. Engl. J. Med. 2010, 363, 2233–2247. [Google Scholar] [CrossRef]
- Biernacki, W.A.; Kharitonov, S.A.; Barnes, P.J. Increased Leukotriene B4 and 8-Isoprostane in Exhaled Breath Condensate of Patients with Exacerbations of Copd. Thorax 2003, 58, 294–298. [Google Scholar] [CrossRef]
- Cristinziano, L.; Modestino, L.; Loffredo, S.; Varricchi, G.; Braile, M.; Ferrara, A.L.; de Paulis, A.; Antonelli, A.; Marone, G.; Galdiero, M.R. Anaplastic Thyroid Cancer Cells Induce the Release of Mitochondrial Extracellular DNA Traps by Viable Neutrophils. J. Immunol. 2020, 204, 1362–1372. [Google Scholar] [CrossRef]
- Varricchi, G.; Modestino, L.; Poto, R.; Cristinziano, L.; Gentile, L.; Postiglione, L.; Spadaro, G.; Galdiero, M.R. Neutrophil Extracellular Traps and Neutrophil-Derived Mediators as Possible Biomarkers in Bronchial Asthma. Clin. Exp. Med. 2021, 22, 285–300. [Google Scholar] [CrossRef] [PubMed]
- Grabcanovic-Musija, F.; Obermayer, A.; Stoiber, W.; Krautgartner, W.D.; Steinbacher, P.; Winterberg, N.; Bathke, A.C.; Klappacher, M.; Studnicka, M. Neutrophil Extracellular Trap (Net) Formation Characterises Stable and Exacerbated Copd and Correlates with Airflow Limitation. Respir. Res. 2015, 16, 59. [Google Scholar] [CrossRef] [PubMed]
- Obermayer, A.; Stoiber, W.; Krautgartner, W.D.; Klappacher, M.; Kofler, B.; Steinbacher, P.; Vitkov, L.; Grabcanovic-Musija, F.; Studnicka, M. New Aspects on the Structure of Neutrophil Extracellular Traps from Chronic Obstructive Pulmonary Disease and in Vitro Generation. PLoS ONE 2014, 9, e97784. [Google Scholar]
- Dicker, A.J.; Crichton, M.L.; Pumphrey, E.G.; Cassidy, A.J.; Suarez-Cuartin, G.; Sibila, O.; Furrie, E.; Fong, C.J.; Ibrahim, W.; Brady, G.; et al. Neutrophil Extracellular Traps Are Associated with Disease Severity and Microbiota Diversity in Patients with Chronic Obstructive Pulmonary Disease. J. Allergy Clin. Immunol. 2018, 141, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Pullan, J.; Greenwood, H.; Walton, G.M.; Stockley, R.A.; Sapey, E. Neutrophil Extracellular Traps (Nets) in Copd: A Potential Novel Mechanism for Host Damage in Acute Exacerbations. Eur. Respir. J. 2015, 46, PA5055. [Google Scholar]
- Ilangumaran, S.; Villalobos-Hernandez, A.; Bobbala, D.; Ramanathan, S. The Hepatocyte Growth Factor (Hgf)-Met Receptor Tyrosine Kinase Signaling Pathway: Diverse Roles in Modulating Immune Cell Functions. Cytokine 2016, 82, 125–139. [Google Scholar] [CrossRef]
- De Paulis, A.; Prevete, N.; Fiorentino, I.; Rossi, F.W.; Staibano, S.; Montuori, N.; Ragno, P.; Longobardi, A.; Liccardo, B.; Genovese, A.; et al. Expression and Functions of the Vascular Endothelial Growth Factors and Their Receptors in Human Basophils. J. Immunol. 2006, 177, 7322–7331. [Google Scholar] [CrossRef]
- Barnes, P.J. Role of Hdac2 in the Pathophysiology of Copd. Annu. Rev. Physiol. 2009, 71, 451–464. [Google Scholar] [CrossRef]
- Postma, D.S.; Rabe, K.F. The Asthma-Copd Overlap Syndrome. N. Engl. J. Med. 2015, 373, 1241–1249. [Google Scholar] [CrossRef]
- Bateman, E.D.; Reddel, H.K.; van Zyl-Smit, R.N.; Agusti, A. The Asthma-Copd Overlap Syndrome: Towards a Revised Taxonomy of Chronic Airways Diseases? Lancet Respir. Med. 2015, 3, 719–728. [Google Scholar] [CrossRef]
- Barnes, P.J. Asthma-Copd Overlap. Chest 2016, 149, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.Y.; Hew, G.S.Y.; Mehta, M.; Shukla, S.D.; Satija, S.; Khurana, N.; Anand, K.; Dureja, H.; Singh, S.K.; Mishra, V.; et al. Targeting Eosinophils in Respiratory Diseases: Biological Axis, Emerging Therapeutics and Treatment Modalities. Life Sci. 2021, 267, 118973. [Google Scholar] [CrossRef] [PubMed]
- De Kleer, I.M.; Kool, M.; de Bruijn, M.J.; Willart, M.; van Moorleghem, J.; Schuijs, M.J.; Plantinga, M.; Beyaert, R.; Hams, E.; Fallon, P.G.; et al. Perinatal Activation of the Interleukin-33 Pathway Promotes Type 2 Immunity in the Developing Lung. Immunity 2016, 45, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Zhao, J.; Shang, J.; Li, M.; Zeng, Z.; Zhao, J.; Wang, J.; Xu, Y.; Xie, J. Increased Il-33 Expression in Chronic Obstructive Pulmonary Disease. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L619–L627. [Google Scholar] [CrossRef] [PubMed]
- Pavord, I.D.; Chanez, P.; Criner, G.J.; Kerstjens, H.A.M.; Korn, S.; Lugogo, N.; Martinot, J.B.; Sagara, H.; Albers, F.C.; Bradford, E.S.; et al. Mepolizumab for Eosinophilic Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2017, 377, 1613–1629. [Google Scholar] [CrossRef] [PubMed]
- Criner, G.J.; Celli, B.R.; Brightling, C.E.; Agusti, A.; Papi, A.; Singh, D.; Sin, D.D.; Vogelmeier, C.F.; Sciurba, F.C.; Bafadhel, M.; et al. Benralizumab for the Prevention of Copd Exacerbations. N. Engl. J. Med. 2019, 381, 1023–1034. [Google Scholar] [CrossRef]
- Puxeddu, I.; Berkman, N.; Efraim, A.H.N.B.; Davies, D.E.; Ribatti, D.; Gleich, G.J.; Levi-Schaffer, F. The Role of Eosinophil Major Basic Protein in Angiogenesis. Allergy 2009, 64, 368–374. [Google Scholar] [CrossRef]
- Puxeddu, I.; Alian, A.; Piliponsky, A.M.; Ribatti, D.; Panet, A.; Levi-Schaffer, F. Human Peripheral Blood Eosinophils Induce Angiogenesis. Int. J. Biochem. Cell Biol. 2005, 37, 628–636. [Google Scholar] [CrossRef]
- Rivellese, F.; Suurmond, J.; de Paulis, A.; Marone, G.; Huizinga, T.W.; Toes, R.E. Ige and Il-33-Mediated Triggering of Human Basophils Inhibits Tlr4-Induced Monocyte Activation. Eur. J. Immunol. 2014, 44, 3045–3055. [Google Scholar] [CrossRef]
- Galli, S.J. Mast Cells and Basophils. Curr. Opin. Hematol. 2000, 7, 32–39. [Google Scholar] [CrossRef]
- Siracusa, M.C.; Kim, B.S.; Spergel, J.M.; Artis, D. Basophils and Allergic Inflammation. J. Allergy Clin. Immunol. 2013, 132, 789–801. [Google Scholar] [CrossRef] [PubMed]
- Varricchi, G.; Raap, U.; Rivellese, F.; Marone, G.; Gibbs, B.F. Human Mast Cells and Basophils-How Are They Similar How Are They Different? Immunol. Rev. 2018, 282, 8–34. [Google Scholar] [CrossRef] [PubMed]
- Cerny-Reiterer, S.; Ghanim, V.; Hoermann, G.; Aichberger, K.J.; Herrmann, H.; Muellauer, L.; Repa, A.; Sillaber, C.; Walls, A.F.; Mayerhofer, M.; et al. Identification of Basophils as a Major Source of Hepatocyte Growth Factor in Chronic Myeloid Leukemia: A Novel Mechanism of Bcr-Abl1-Independent Disease Progression. Neoplasia 2012, 14, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Saetta, M.; di Stefano, A.; Turato, G.; Facchini, F.M.; Corbino, L.; Mapp, C.E.; Maestrelli, P.; Ciaccia, A.; Fabbri, L.M. Cd8+ T-Lymphocytes in Peripheral Airways of Smokers with Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 1998, 157, 822–826. [Google Scholar] [CrossRef]
- Williams, M.; Todd, I.; Fairclough, L.C. The Role of Cd8+ T Lymphocytes in Chronic Obstructive Pulmonary Disease: A Systematic Review. Inflamm. Res. 2021, 70, 11–18. [Google Scholar] [CrossRef]
- Saetta, M.; Mariani, M.; Panina-Bordignon, P.; Turato, G.; Buonsanti, C.; Baraldo, S.; Bellettato, C.M.; Papi, A.; Corbetta, L.; Zuin, R.; et al. Increased Expression of the Chemokine Receptor Cxcr3 and Its Ligand Cxcl10 in Peripheral Airways of Smokers with Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2002, 165, 1404–1409. [Google Scholar] [CrossRef]
- Ebbo, M.; Crinier, A.; Vely, F.; Vivier, E. Innate Lymphoid Cells: Major Players in Inflammatory Diseases. Nat. Rev. Immunol. 2017, 17, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Artis, D.; Spits, H. The Biology of Innate Lymphoid Cells. Nature 2015, 517, 293–301. [Google Scholar] [CrossRef]
- De Grove, K.C.; Provoost, S.; Verhamme, F.M.; Bracke, K.R.; Joos, G.F.; Maes, T.; Brusselle, G.G. Characterization and Quantification of Innate Lymphoid Cell Subsets in Human Lung. PLoS ONE 2016, 11, e0145961. [Google Scholar] [CrossRef]
- Silver, J.S.; Kearley, J.; Copenhaver, A.M.; Sanden, C.; Mori, M.; Yu, L.; Pritchard, G.H.; Berlin, A.A.; Hunter, C.A.; Bowler, R.; et al. Inflammatory Triggers Associated with Exacerbations of Copd Orchestrate Plasticity of Group 2 Innate Lymphoid Cells in the Lungs. Nat. Immunol. 2016, 17, 626–635. [Google Scholar] [CrossRef]
- Bal, S.M.; Bernink, J.H.; Nagasawa, M.; Groot, J.; Shikhagaie, M.M.; Golebski, K.; van Drunen, C.M.; Lutter, R.; Jonkers, R.E.; Hombrink, P.; et al. Il-1beta, Il-4 and Il-12 Control the Fate of Group 2 Innate Lymphoid Cells in Human Airway Inflammation in the Lungs. Nat. Immunol. 2016, 17, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, A.; Caramori, G.; Gnemmi, I.; Contoli, M.; Vicari, C.; Capelli, A.; Magno, F.; D’Anna, S.E.; Zanini, A.; Brun, P.; et al. T Helper Type 17-Related Cytokine Expression Is Increased in the Bronchial Mucosa of Stable Chronic Obstructive Pulmonary Disease Patients. Clin. Exp. Immunol. 2009, 157, 316–324. [Google Scholar] [CrossRef]
- Pridgeon, C.; Bugeon, L.; Donnelly, L.; Straschil, U.; Tudhope, S.J.; Fenwick, P.; Lamb, J.R.; Barnes, P.J.; Dallman, M.J. Regulation of Il-17 in Chronic Inflammation in the Human Lung. Clin. Sci. 2011, 120, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, S.K.; Jin, Y.; Goyal, S.; Lee, H.S.; Fuchsluger, T.A.; Lee, H.K.; Dana, R. A Novel Pro-Lymphangiogenic Function for Th17/Il-17. Blood 2011, 118, 4630–4634. [Google Scholar] [CrossRef] [PubMed]
- Panariti, A.; Baglole, C.J.; Sanchez, V.; Eidelman, D.H.; Hussain, S.; Olivenstein, R.; Martin, J.G.; Hamid, Q. Interleukin-17a and Vascular Remodelling in Severe Asthma; Lack of Evidence for a Direct Role. Clin. Exp. Allergy 2018, 48, 365–378. [Google Scholar] [CrossRef]
- Wang, W.; Li, Y.; Lv, Z.; Chen, Y.; Huang, K.; Corrigan, C.J.; Ying, S. Bronchial Allergen Challenge of Patients with Atopic Asthma Triggers an Alarmin (Il-33, Tslp, and Il-25) Response in the Airways Epithelium and Submucosa. J. Immunol. 2018, 201, 2221–2231. [Google Scholar] [CrossRef]
- Kohanski, M.A.; Workman, A.D.; Patel, N.N.; Hung, L.Y.; Shtraks, J.P.; Chen, B.; Blasetti, M.; Doghramji, L.; Kennedy, D.W.; Adappa, N.D.; et al. Solitary Chemosensory Cells Are a Primary Epithelial Source of Il-25 in Patients with Chronic Rhinosinusitis with Nasal Polyps. J. Allergy Clin. Immunol. 2018, 142, 460–469.e7. [Google Scholar] [CrossRef]
- Beale, J.; Jayaraman, A.; Jackson, D.J.; Macintyre, J.D.R.; Edwards, M.R.; Walton, R.P.; Zhu, J.; Ching, Y.M.; Shamji, B.; Edwards, M.; et al. Rhinovirus-Induced Il-25 in Asthma Exacerbation Drives Type 2 Immunity and Allergic Pulmonary Inflammation. Sci. Transl. Med. 2014, 6, 256ra134. [Google Scholar] [CrossRef]
- Polverino, F.; Baraldo, S.; Bazzan, E.; Agostini, S.; Turato, G.; Lunardi, F.; Balestro, E.; Damin, M.; Papi, A.; Maestrelli, P.; et al. A Novel Insight into Adaptive Immunity in Chronic Obstructive Pulmonary Disease: B Cell Activating Factor Belonging to the Tumor Necrosis Factor Family. Am. J. Respir. Crit. Care Med. 2010, 182, 1011–1019. [Google Scholar] [CrossRef]
- Hogg, J.C.; Chu, F.; Utokaparch, S.; Woods, R.; Elliott, W.M.; Buzatu, L.; Cherniack, R.M.; Rogers, R.M.; Sciurba, F.C.; Coxson, H.O.; et al. The Nature of Small-Airway Obstruction in Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2004, 350, 2645–2653. [Google Scholar] [CrossRef]
- Seys, L.J.; Verhamme, F.M.; Schinwald, A.; Hammad, H.; Cunoosamy, D.M.; Bantsimba-Malanda, C.; Sabirsh, A.; McCall, E.; Flavell, L.; Herbst, R.; et al. Role of B Cell-Activating Factor in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2015, 192, 706–718. [Google Scholar] [CrossRef] [PubMed]
- Polverino, F.; Cosio, B.G.; Pons, J.; Laucho-Contreras, M.; Tejera, P.; Iglesias, A.; Rios, A.; Jahn, A.; Sauleda, J.; Divo, M.; et al. B Cell-Activating Factor. An Orchestrator of Lymphoid Follicles in Severe Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2015, 192, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Ma, Z.; Huang, Y.; Sun, Y.; Yi, H. Chronic Obstructive Pulmonary Disease Is Characterized by Reduced Levels and Defective Suppressive Function of Regulatory B Cells in Peripheral Blood. Mol. Immunol. 2022, 141, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Bosisio, D.; Ronca, R.; Salvi, V.; Presta, M.; Sozzani, S. Dendritic Cells in Inflammatory Angiogenesis and Lymphangiogenesis. Curr. Opin. Immunol. 2018, 53, 180–186. [Google Scholar] [CrossRef]
- Givi, M.E.; Redegeld, F.A.; Folkerts, G.; Mortaz, E. Dendritic Cells in Pathogenesis of Copd. Curr. Pharm. Des. 2012, 18, 2329–2335. [Google Scholar] [CrossRef]
- Paplinska-Goryca, M.; Misiukiewicz-Stepien, P.; Proboszcz, M.; Nejman-Gryz, P.; Gorska, K.; Krenke, R. The Expressions of Tslp, Il-33, and Il-17a in Monocyte Derived Dendritic Cells from Asthma and Copd Patients Are Related to Epithelial-Macrophage Interactions. Cells 2020, 9, 1944. [Google Scholar] [CrossRef]
- Kitajima, M.; Lee, H.C.; Nakayama, T.; Ziegler, S.F. Tslp Enhances the Function of Helper Type 2 Cells. Eur. J. Immunol. 2011, 41, 1862–1871. [Google Scholar] [CrossRef]
- Elder, M.J.; Webster, S.J.; Fitzmaurice, T.J.; Shaunak, A.S.D.; Steinmetz, M.; Chee, R.; Mallat, Z.; Cohen, E.S.; Williams, D.L.; Gaston, J.S.H.; et al. Dendritic Cell-Derived Tslp Negatively Regulates Hif-1alpha and Il-1beta During Dectin-1 Signaling. Front. Immunol. 2019, 10, 921. [Google Scholar] [CrossRef]
- Su, Z.; Lin, J.; Lu, F.; Zhang, X.; Zhang, L.; Gandhi, N.B.; de Paiva, C.S.; Pflugfelder, S.C.; Li, D.Q. Potential Autocrine Regulation of Interleukin-33/St2 Signaling of Dendritic Cells in Allergic Inflammation. Mucosal Immunol. 2013, 6, 921–930. [Google Scholar] [CrossRef]
- Paplinska-Goryca, M.; Misiukiewicz-Stepien, P.; Proboszcz, M.; Nejman-Gryz, P.; Gorska, K.; Zajusz-Zubek, E.; Krenke, R. Interactions of Nasal Epithelium with Macrophages and Dendritic Cells Variously Alter Urban Pm-Induced Inflammation in Healthy, Asthma and Copd. Sci. Rep. 2021, 11, 13259. [Google Scholar] [CrossRef]
- Van Pottelberge, G.R.; Bracke, K.R.; Demedts, I.K.; de Rijck, K.; Reinartz, S.M.; van Drunen, C.M.; Verleden, G.M.; Vermassen, F.E.; Joos, G.F.; Brusselle, G.G. Selective Accumulation of Langerhans-Type Dendritic Cells in Small Airways of Patients with Copd. Respir. Res. 2010, 11, 35. [Google Scholar] [CrossRef] [PubMed]
- Freeman, C.M.; Martinez, F.J.; Han, M.K.; Ames, T.M.; Chensue, S.W.; Todt, J.C.; Arenberg, D.A.; Meldrum, C.A.; Getty, C.; McCloskey, L.; et al. Lung Dendritic Cell Expression of Maturation Molecules Increases with Worsening Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2009, 180, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Vassallo, R.; Walters, P.R.; Lamont, J.; Kottom, T.J.; Yi, E.S.; Limper, A.H. Cigarette Smoke Promotes Dendritic Cell Accumulation in Copd; A Lung Tissue Research Consortium Study. Respir. Res. 2010, 11, 45. [Google Scholar] [CrossRef] [PubMed]
- Janela, B.; Patel, A.A.; Lau, M.C.; Goh, C.C.; Msallam, R.; Kong, W.T.; Fehlings, M.; Hubert, S.; Lum, J.; Simoni, Y.; et al. A Subset of Type I Conventional Dendritic Cells Controls Cutaneous Bacterial Infections through Vegfalpha-Mediated Recruitment of Neutrophils. Immunity 2019, 50, 1069–1083.e8. [Google Scholar] [CrossRef]
- Quatrini, L.; della Chiesa, M.; Sivori, S.; Mingari, M.C.; Pende, D.; Moretta, L. Human Nk Cells, Their Receptors and Function. Eur. J. Immunol. 2021, 51, 1566–1579. [Google Scholar] [CrossRef]
- Cong, J.; Wei, H. Natural Killer Cells in the Lungs. Front. Immunol. 2019, 10, 1416. [Google Scholar] [CrossRef]
- Hervier, B.; Russick, J.; Cremer, I.; Vieillard, V. Nk Cells in the Human Lungs. Front. Immunol. 2019, 10, 1263. [Google Scholar] [CrossRef]
- Rao, Y.; Le, Y.; Xiong, J.; Pei, Y.; Sun, Y. Nk Cells in the Pathogenesis of Chronic Obstructive Pulmonary Disease. Front. Immunol. 2021, 12, 666045. [Google Scholar] [CrossRef]
- Marquardt, N.; Kekalainen, E.; Chen, P.; Kvedaraite, E.; Wilson, J.N.; Ivarsson, M.A.; Mjosberg, J.; Berglin, L.; Safholm, J.; Manson, M.L.; et al. Human Lung Natural Killer Cells Are Predominantly Comprised of Highly Differentiated Hypofunctional Cd69(-)Cd56(Dim) Cells. J. Allergy Clin. Immunol. 2017, 139, 1321–1330.e4. [Google Scholar] [CrossRef]
- Radomska-Lesniewska, D.M.; Bialoszewska, A.; Kaminski, P. Angiogenic Properties of Nk Cells in Cancer and Other Angiogenesis-Dependent Diseases. Cells 2021, 10, 1621. [Google Scholar] [CrossRef]
- Gao, W.; Li, L.; Wang, Y.; Zhang, S.; Adcock, I.M.; Barnes, P.J.; Huang, M.; Yao, X. Bronchial Epithelial Cells: The Key Effector Cells in the Pathogenesis of Chronic Obstructive Pulmonary Disease? Respirology 2015, 20, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Moorehead, A.; Hanna, R.; Heroux, D.; Neighbour, H.; Sandford, A.; Gauvreau, G.M.; Sommer, D.D.; Denburg, J.A.; Akhabir, L. A Thymic Stromal Lymphopoietin Polymorphism May Provide Protection from Asthma by Altering Gene Expression. Clin. Exp. Allergy 2020, 50, 471–478. [Google Scholar] [CrossRef]
- Kato, A.; Favoreto, S., Jr.; Avila, P.C.; Schleimer, R.P. Tlr3- and Th2 Cytokine-Dependent Production of Thymic Stromal Lymphopoietin in Human Airway Epithelial Cells. J. Immunol. 2007, 179, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- Nagarkar, D.R.; Poposki, J.A.; Comeau, M.R.; Biyasheva, A.; Avila, P.C.; Schleimer, R.P.; Kato, A. Airway Epithelial Cells Activate Th2 Cytokine Production in Mast Cells through Il-1 and Thymic Stromal Lymphopoietin. J. Allergy Clin. Immunol. 2012, 130, 225–232.e4. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Headley, M.B.; Loo, Y.M.; Berlin, A.; Gale, M., Jr.; Debley, J.S.; Lukacs, N.W.; Ziegler, S.F. Thymic Stromal Lymphopoietin Is Induced by Respiratory Syncytial Virus-Infected Airway Epithelial Cells and Promotes a Type 2 Response to Infection. J. Allergy Clin. Immunol. 2012, 130, 1187–1196.e5. [Google Scholar] [CrossRef] [PubMed]
- Weng, C.M.; Wang, C.H.; Lee, M.J.; He, J.R.; Huang, H.Y.; Chao, M.W.; Chung, K.F.; Kuo, H.P. Aryl Hydrocarbon Receptor Activation by Diesel Exhaust Particles Mediates Epithelium-Derived Cytokines Expression in Severe Allergic Asthma. Allergy 2018, 73, 2192–2204. [Google Scholar] [CrossRef]
- Semlali, A.; Jacques, E.; Koussih, L.; Gounni, A.S.; Chakir, J. Thymic Stromal Lymphopoietin-Induced Human Asthmatic Airway Epithelial Cell Proliferation through an Il-13-Dependent Pathway. J. Allergy Clin. Immunol. 2010, 125, 844–850. [Google Scholar] [CrossRef]
- Uller, L.; Leino, M.; Bedke, N.; Sammut, D.; Green, B.; Lau, L.; Howarth, P.H.; Holgate, S.T.; Davies, D.E. Double-Stranded Rna Induces Disproportionate Expression of Thymic Stromal Lymphopoietin Versus Interferon-Beta in Bronchial Epithelial Cells from Donors with Asthma. Thorax 2010, 65, 626–632. [Google Scholar] [CrossRef]
- Perez-Padilla, R.; Schilmann, A.; Riojas-Rodriguez, H. Respiratory Health Effects of Indoor Air Pollution. Int. J. Tuberc. Lung Dis. 2010, 14, 1079–1086. [Google Scholar]
- Kanazawa, H.; Tochino, Y.; Asai, K.; Hirata, K. Simultaneous Assessment of Hepatocyte Growth Factor and Vascular Endothelial Growth Factor in Epithelial Lining Fluid from Patients with Copd. Chest 2014, 146, 1159–1165. [Google Scholar] [CrossRef]
- De Boer, W.I.; Hau, C.M.; van Schadewijk, A.; Stolk, J.; van Krieken, J.H.; Hiemstra, P.S. Expression of Epidermal Growth Factors and Their Receptors in the Bronchial Epithelium of Subjects with Chronic Obstructive Pulmonary Disease. Am. J. Clin. Pathol. 2006, 125, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Burgel, P.R.; Nadel, J.A. Roles of Epidermal Growth Factor Receptor Activation in Epithelial Cell Repair and Mucin Production in Airway Epithelium. Thorax 2004, 59, 992–996. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.X.; Nakanaga, T.; Nadel, J.A. Cigarette Smoke Induces Muc5ac Mucin Overproduction Via Tumor Necrosis Factor-Alpha-Converting Enzyme in Human Airway Epithelial (Nci-H292) Cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 287, L420–L427. [Google Scholar] [CrossRef]
- Boussat, S.; Eddahibi, S.; Coste, A.; Fataccioli, V.; Gouge, M.; Housset, B.; Adnot, S.; Maitre, B. Expression and Regulation of Vascular Endothelial Growth Factor in Human Pulmonary Epithelial Cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 279, L371–L378. [Google Scholar] [CrossRef] [PubMed]
- Ishida, A.; Murray, J.; Saito, Y.; Kanthou, C.; Benzakour, O.; Shibuya, M.; Wijelath, E.S. Expression of Vascular Endothelial Growth Factor Receptors in Smooth Muscle Cells. J. Cell Physiol. 2001, 188, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Knox, A.J.; Corbett, L.; Stocks, J.; Holland, E.; Zhu, Y.M.; Pang, L. Human Airway Smooth Muscle Cells Secrete Vascular Endothelial Growth Factor: Up-Regulation by Bradykinin Via a Protein Kinase C and Prostanoid-Dependent Mechanism. FASEB J. 2001, 15, 2480–2488. [Google Scholar] [CrossRef]
- Wen, F.Q.; Liu, X.; Manda, W.; Terasaki, Y.; Kobayashi, T.; Abe, S.; Fang, Q.; Ertl, R.; Manouilova, L.; Rennard, S.I. Th2 Cytokine-Enhanced and Tgf-Beta-Enhanced Vascular Endothelial Growth Factor Production by Cultured Human Airway Smooth Muscle Cells Is Attenuated by Ifn-Gamma and Corticosteroids. J. Allergy Clin. Immunol. 2003, 111, 1307–1318. [Google Scholar] [CrossRef]
- Wang, H.; Keiser, J.A. Vascular Endothelial Growth Factor Upregulates the Expression of Matrix Metalloproteinases in Vascular Smooth Muscle Cells: Role of Flt-1. Circ. Res. 1998, 83, 832–840. [Google Scholar] [CrossRef]
- Polverino, F.; Laucho-Contreras, M.E.; Petersen, H.; Bijol, V.; Sholl, L.M.; Choi, M.E.; Divo, M.; Pinto-Plata, V.; Chetta, A.; Tesfaigzi, Y.; et al. A Pilot Study Linking Endothelial Injury in Lungs and Kidneys in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2017, 195, 1464–1476. [Google Scholar] [CrossRef]
- Eickhoff, P.; Valipour, A.; Kiss, D.; Schreder, M.; Cekici, L.; Geyer, K.; Kohansal, R.; Burghuber, O.C. Determinants of Systemic Vascular Function in Patients with Stable Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2008, 178, 1211–1218. [Google Scholar] [CrossRef]
- Piccari, L.; del Pozo, R.; Blanco, I.; Garcia-Lucio, J.; Torralba, Y.; Tura-Ceide, O.; Moises, J.; Sitges, M.; Peinado, V.I.; Barbera, J.A. Association between Systemic and Pulmonary Vascular Dysfunction in Copd. Int. J. Chron. Obstruct. Pulmon. Dis. 2020, 15, 2037–2047. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of Microrna Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed]
- Ezzie, M.E.; Crawford, M.; Cho, J.H.; Orellana, R.; Zhang, S.; Gelinas, R.; Batte, K.; Yu, L.; Nuovo, G.; Galas, D.; et al. Gene Expression Networks in Copd: Microrna and Mrna Regulation. Thorax 2012, 67, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Green, C.E.; Clarke, J.; Bicknell, R.; Turner, A.M. Pulmonary Microrna Changes Alter Angiogenesis in Chronic Obstructive Pulmonary Disease and Lung Cancer. Biomedicines 2021, 9, 830. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.L.; Tai, H.; Churg, A. Cigarette Smoke Induces Persisting Increases of Vasoactive Mediators in Pulmonary Arteries. Am. J. Respir. Cell Mol. Biol. 2004, 31, 501–509. [Google Scholar] [CrossRef]
- Nilsson, I.; Shibuya, M.; Wennstrom, S. Differential Activation of Vascular Genes by Hypoxia in Primary Endothelial Cells. Exp. Cell Res. 2004, 299, 476–485. [Google Scholar] [CrossRef]
- Tuder, R.M.; Flook, B.E.; Voelkel, N.F. Increased Gene Expression for Vegf and the Vegf Receptors Kdr/Flk and Flt in Lungs Exposed to Acute or to Chronic Hypoxia. Modulation of Gene Expression by Nitric Oxide. J. Clin. Investig. 1995, 95, 1798–1807. [Google Scholar] [CrossRef]
- Tuder, R.M.; Zhen, L.; Cho, C.Y.; Taraseviciene-Stewart, L.; Kasahara, Y.; Salvemini, D.; Voelkel, N.F.; Flores, S.C. Oxidative Stress and Apoptosis Interact and Cause Emphysema Due to Vascular Endothelial Growth Factor Receptor Blockade. Am. J. Respir. Cell Mol. Biol. 2003, 29, 88–97. [Google Scholar] [CrossRef]
- Alon, T.; Hemo, I.; Itin, A.; Pe’er, J.; Stone, J.; Keshet, E. Vascular Endothelial Growth Factor Acts as a Survival Factor for Newly Formed Retinal Vessels and Has Implications for Retinopathy of Prematurity. Nat. Med. 1995, 1, 1024–1028. [Google Scholar] [CrossRef]
- Macnee, W.; Rahman, I. Oxidants and Antioxidants as Therapeutic Targets in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 1999, 160, S58–S65. [Google Scholar] [CrossRef]
- Kasahara, Y.; Tuder, R.M.; Taraseviciene-Stewart, L.; le Cras, T.D.; Abman, S.; Hirth, P.K.; Waltenberger, J.; Voelkel, N.F. Inhibition of Vegf Receptors Causes Lung Cell Apoptosis and Emphysema. J. Clin. Investig. 2000, 106, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, B.; Pajusola, K.; Kaipainen, A.; von Euler, G.; Joukov, V.; Saksela, O.; Orpana, A.; Pettersson, R.F.; Alitalo, K.; Eriksson, U. Vascular Endothelial Growth Factor B, a Novel Growth Factor for Endothelial Cells. Proc. Natl. Acad. Sci. USA 1996, 93, 2576–2581. [Google Scholar] [CrossRef] [PubMed]
- Louzier, V.; Raffestin, B.; Leroux, A.; Branellec, D.; Caillaud, J.M.; Levame, M.; Eddahibi, S.; Adnot, S. Role of Vegf-B in the Lung During Development of Chronic Hypoxic Pulmonary Hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2003, 284, L926–L937. [Google Scholar] [CrossRef]
- Baluk, P.; Tammela, T.; Ator, E.; Lyubynska, N.; Achen, M.G.; Hicklin, D.J.; Jeltsch, M.; Petrova, T.V.; Pytowski, B.; Stacker, S.A.; et al. Pathogenesis of Persistent Lymphatic Vessel Hyperplasia in Chronic Airway Inflammation. J. Clin. Investig. 2005, 115, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Tanaka, H.; Abe, S. Quantitative Analysis of Bronchial Wall Vascularity in the Medium and Small Airways of Patients with Asthma and Copd. Chest 2005, 127, 965–972. [Google Scholar] [CrossRef]
- Tanaka, H.; Yamada, G.; Saikai, T.; Hashimoto, M.; Tanaka, S.; Suzuki, K.; Fujii, M.; Takahashi, H.; Abe, S. Increased Airway Vascularity in Newly Diagnosed Asthma Using a High-Magnification Bronchovideoscope. Am. J. Respir. Crit. Care Med. 2003, 168, 1495–1499. [Google Scholar] [CrossRef]
- Kristan, S.S.; Marc, M.M.; Kern, I.; Flezar, M.; Suskovic, S.; Kosnik, M.; Korosec, P. Airway Angiogenesis in Stable and Exacerbated Chronic Obstructive Pulmonary Disease. Scand. J. Immunol. 2012, 75, 109–114. [Google Scholar] [CrossRef][Green Version]
- Yu, Z.G.; Wang, B.Z.; Cheng, Z.Z. The Association of Genetic Polymorphisms of Hypoxia Inducible Factor-1 Alpha and Vascular Endothelial Growth Factor with Increased Risk of Chronic Obstructive Pulmonary Disease: A Case-Control Study. Kaohsiung J. Med. Sci. 2017, 33, 433–441. [Google Scholar] [CrossRef]
- Valipour, A.; Schreder, M.; Wolzt, M.; Saliba, S.; Kapiotis, S.; Eickhoff, P.; Burghuber, O.C. Circulating Vascular Endothelial Growth Factor and Systemic Inflammatory Markers in Patients with Stable and Exacerbated Chronic Obstructive Pulmonary Disease. Clin. Sci. 2008, 115, 225–232. [Google Scholar] [CrossRef]
- Kasahara, Y.; Tuder, R.M.; Cool, C.D.; Lynch, D.A.; Flores, S.C.; Voelkel, N.F. Endothelial Cell Death and Decreased Expression of Vascular Endothelial Growth Factor and Vascular Endothelial Growth Factor Receptor 2 in Emphysema. Am. J. Respir. Crit. Care Med. 2001, 163, 737–744. [Google Scholar] [CrossRef]
- Nagai, K.; Betsuyaku, T.; Ito, Y.; Nasuhara, Y.; Nishimura, M. Decrease of Vascular Endothelial Growth Factor in Macrophages from Long-Term Smokers. Eur. Respir. J. 2005, 25, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.L.; Wang, H.C.; Yu, C.J.; Yang, P.C. Increased Expression of Placenta Growth Factor in Copd. Thorax 2008, 63, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Guddo, F.; Vignola, A.M.; Saetta, M.; Baraldo, S.; Siena, L.; Balestro, E.; Zuin, R.; Papi, A.; Maestrelli, P.; Fabbri, L.M.; et al. Upregulation of Basic Fibroblast Growth Factor in Smokers with Chronic Bronchitis. Eur. Respir. J. 2006, 27, 957–963. [Google Scholar] [CrossRef]
- Vanfleteren, L.E.; Spruit, M.A.; Groenen, M.; Gaffron, S.; van Empel, V.P.; Bruijnzeel, P.L.; Rutten, E.P.; Op’t Roodt, J.; Wouters, E.F.; Franssen, F.M. Clusters of Comorbidities Based on Validated Objective Measurements and Systemic Inflammation in Patients with Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2013, 187, 728–735. [Google Scholar] [CrossRef]
- Oga, T.; Nishimura, K.; Tsukino, M.; Sato, S.; Hajiro, T. Analysis of the Factors Related to Mortality in Chronic Obstructive Pulmonary Disease: Role of Exercise Capacity and Health Status. Am. J. Respir. Crit. Care Med. 2003, 167, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Baum, O.; Gubeli, J.; Frese, S.; Torchetti, E.; Malik, C.; Odriozola, A.; Graber, F.; Hoppeler, H.; Tschanz, S.A. Angiogenesis-Related Ultrastructural Changes to Capillaries in Human Skeletal Muscle in Response to Endurance Exercise. J. Appl. Physiol. 2015, 119, 1118–1126. [Google Scholar] [CrossRef] [PubMed]
- Blervaque, L.; Passerieux, E.; Pomies, P.; Catteau, M.; Heraud, N.; Blaquiere, M.; Bughin, F.; Ayoub, B.; Molinari, N.; Cristol, J.P.; et al. Impaired Training-Induced Angiogenesis Process with Loss of Pericyte-Endothelium Interactions Is Associated with an Abnormal Capillary Remodelling in the Skeletal Muscle of Copd Patients. Respir. Res. 2019, 20, 278. [Google Scholar] [CrossRef]
- Montano, M.; Perez-Bautista, O.; Velasco-Torres, Y.; Gonzalez-Avila, G.; Ramos, C. Women with Copd from Biomass Smoke Have Reduced Serum Levels of Biomarkers of Angiogenesis and Cancer, with Egfr Predominating, Compared to Women with Copd from Smoking. Chron. Respir. Dis. 2021, 18, 14799731211005023. [Google Scholar] [CrossRef]
- Wang, L.; Xu, Z.; Chen, B.; He, W.; Hu, J.; Zhang, L.; Liu, X.; Chen, F. The Role of Vascular Endothelial Growth Factor in Small-Airway Remodelling in a Rat Model of Chronic Obstructive Pulmonary Disease. Sci. Rep. 2017, 7, 41202. [Google Scholar] [CrossRef]
- Lanza, G.M.; Jenkins, J.; Schmieder, A.H.; Moldobaeva, A.; Cui, G.; Zhang, H.; Yang, X.; Zhong, Q.; Keupp, J.; Sergin, I.; et al. Anti-Angiogenic Nanotherapy Inhibits Airway Remodeling and Hyper-Responsiveness of Dust Mite Triggered Asthma in the Brown Norway Rat. Theranostics 2017, 7, 377–389. [Google Scholar] [CrossRef]
- Feltis, B.N.; Wignarajah, D.; Reid, D.W.; Ward, C.; Harding, R.; Walters, E.H. Effects of Inhaled Fluticasone on Angiogenesis and Vascular Endothelial Growth Factor in Asthma. Thorax 2007, 62, 314–319. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Menzies-Gow, A.; Corren, J.; Bourdin, A.; Chupp, G.; Israel, E.; Wechsler, M.E.; Brightling, C.E.; Griffiths, J.M.; Hellqvist, A.; Bowen, K.; et al. Tezepelumab in Adults and Adolescents with Severe, Uncontrolled Asthma. N. Engl. J. Med. 2021, 384, 1800–1809. [Google Scholar] [CrossRef] [PubMed]
- Corren, J.; Parnes, J.R.; Wang, L.; Mo, M.; Roseti, S.L.; Griffiths, J.M.; van der Merwe, R. Tezepelumab in Adults with Uncontrolled Asthma. N. Engl. J. Med. 2017, 377, 936–946. [Google Scholar] [CrossRef]
- Wechsler, M.E.; Ruddy, M.K.; Pavord, I.D.; Israel, E.; Rabe, K.F.; Ford, L.B.; Maspero, J.F.; Abdulai, R.M.; Hu, C.C.; Martincova, R.; et al. Efficacy and Safety of Itepekimab in Patients with Moderate-to-Severe Asthma. N. Engl. J. Med. 2021, 385, 1656–1668. [Google Scholar] [CrossRef] [PubMed]
- Parmar, D.; Apte, M. Angiopoietin Inhibitors: A Review on Targeting Tumor Angiogenesis. Eur. J. Pharmacol. 2021, 899, 174021. [Google Scholar] [CrossRef]
- Cantelmo, A.R.; Dejos, C.; Kocher, F.; Hilbe, W.; Wolf, D.; Pircher, A. Angiogenesis Inhibition in Non-Small Cell Lung Cancer: A Critical Appraisal, Basic Concepts and Updates from American Society for Clinical Oncology 2019. Curr. Opin. Oncol. 2020, 32, 44–53. [Google Scholar] [CrossRef]
- Abdel-Majid, R.M.; Marshall, J.S. Prostaglandin E2 Induces Degranulation-Independent Production of Vascular Endothelial Growth Factor by Human Mast Cells. J. Immunol. 2004, 172, 1227–1236. [Google Scholar] [CrossRef]
- Kazi, A.S.; Lotfi, S.; Goncharova, E.A.; Tliba, O.; Amrani, Y.; Krymskaya, V.P.; Lazaar, A.L. Vascular Endothelial Growth Factor-Induced Secretion of Fibronectin Is Erk Dependent. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 286, L539–L545. [Google Scholar] [CrossRef]
- Sitkovsky, M.V.; Lukashev, D.; Apasov, S.; Kojima, H.; Koshiba, M.; Caldwell, C.; Ohta, A.; Thiel, M. Physiological Control of Immune Response and Inflammatory Tissue Damage by Hypoxia-Inducible Factors and Adenosine A2a Receptors. Annu. Rev. Immunol. 2004, 22, 657–682. [Google Scholar] [CrossRef]
- Walters, E.H.; Reid, D.; Soltani, A.; Ward, C. Angiogenesis: A Potentially Critical Part of Remodelling in Chronic Airway Diseases? Pharmacol. Ther. 2008, 118, 128–137. [Google Scholar] [CrossRef]
- Magee, F.; Wright, J.L.; Wiggs, B.R.; Pare, P.D.; Hogg, J.C. Pulmonary Vascular Structure and Function in Chronic Obstructive Pulmonary Disease. Thorax 1988, 43, 183–189. [Google Scholar] [CrossRef] [PubMed]
- D’Alessio, S.; Correale, C.; Tacconi, C.; Gandelli, A.; Pietrogrande, G.; Vetrano, S.; Genua, M.; Arena, V.; Spinelli, A.; Peyrin-Biroulet, L.; et al. Vegf-C-Dependent Stimulation of Lymphatic Function Ameliorates Experimental Inflammatory Bowel Disease. J. Clin. Investig. 2014, 124, 3863–3878. [Google Scholar] [CrossRef] [PubMed]
- Turkeli, A.; Yilmaz, O.; Karaman, M.; Kanik, E.T.; Firinci, F.; Inan, S.; Yuksel, H. Anti-Vegf Treatment Suppresses Remodeling Factors and Restores Epithelial Barrier Function through the E-Cadherin/Beta-Catenin Signaling Axis in Experimental Asthma Models. Exp. Ther. Med. 2021, 22, 689. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, H. Vegf, Angiopoietin-1 and -2 in Bronchial Asthma: New Molecular Targets in Airway Angiogenesis and Microvascular Remodeling. Recent Pat. Inflamm. Allergy Drug Discov. 2007, 1, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Detoraki, A.; Granata, F.; Staibano, S.; Rossi, F.W.; Marone, G.; Genovese, A. Angiogenesis and Lymphangiogenesis in Bronchial Asthma. Allergy 2010, 65, 946–958. [Google Scholar] [CrossRef]
- Asai, K.; Kanazawa, H.; Otani, K.; Shiraishi, S.; Hirata, K.; Yoshikawa, J. Imbalance between Vascular Endothelial Growth Factor and Endostatin Levels in Induced Sputum from Asthmatic Subjects. J. Allergy Clin. Immunol. 2002, 110, 571–575. [Google Scholar] [CrossRef]
- Lee, C.G.; Link, H.; Baluk, P.; Homer, R.J.; Chapoval, S.; Bhandari, V.; Kang, M.J.; Cohn, L.; Kim, Y.K.; McDonald, D.M.; et al. Vascular Endothelial Growth Factor (Vegf) Induces Remodeling and Enhances Th2-Mediated Sensitization and Inflammation in the Lung. Nat. Med. 2004, 10, 1095–1103. [Google Scholar] [CrossRef]
- Asai, K.; Kanazawa, H.; Kamoi, H.; Shiraishi, S.; Hirata, K.; Yoshikawa, J. Increased Levels of Vascular Endothelial Growth Factor in Induced Sputum in Asthmatic Patients. Clin. Exp. Allergy 2003, 33, 595–599. [Google Scholar] [CrossRef]
- Elias, J.A.; Kang, M.J.; Crothers, K.; Homer, R.; Lee, C.G. State of the Art. Mechanistic Heterogeneity in Chronic Obstructive Pulmonary Disease: Insights from Transgenic Mice. Proc. Am. Thorac. Soc. 2006, 3, 494–498. [Google Scholar] [CrossRef]
- Saharinen, P.; Eklund, L.; Alitalo, K. Therapeutic Targeting of the Angiopoietin-Tie Pathway. Nat. Rev. Drug Discov. 2017, 16, 635–661. [Google Scholar] [CrossRef]
- Lee, K.S.; Kim, S.R.; Park, H.S.; Jin, G.Y.; Lee, Y.C. Cysteinyl Leukotriene Receptor Antagonist Regulates Vascular Permeability by Reducing Vascular Endothelial Growth Factor Expression. J. Allergy Clin. Immunol. 2004, 114, 1093–1099. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poto, R.; Loffredo, S.; Palestra, F.; Marone, G.; Patella, V.; Varricchi, G. Angiogenesis, Lymphangiogenesis, and Inflammation in Chronic Obstructive Pulmonary Disease (COPD): Few Certainties and Many Outstanding Questions. Cells 2022, 11, 1720. https://doi.org/10.3390/cells11101720
Poto R, Loffredo S, Palestra F, Marone G, Patella V, Varricchi G. Angiogenesis, Lymphangiogenesis, and Inflammation in Chronic Obstructive Pulmonary Disease (COPD): Few Certainties and Many Outstanding Questions. Cells. 2022; 11(10):1720. https://doi.org/10.3390/cells11101720
Chicago/Turabian StylePoto, Remo, Stefania Loffredo, Francesco Palestra, Gianni Marone, Vincenzo Patella, and Gilda Varricchi. 2022. "Angiogenesis, Lymphangiogenesis, and Inflammation in Chronic Obstructive Pulmonary Disease (COPD): Few Certainties and Many Outstanding Questions" Cells 11, no. 10: 1720. https://doi.org/10.3390/cells11101720
APA StylePoto, R., Loffredo, S., Palestra, F., Marone, G., Patella, V., & Varricchi, G. (2022). Angiogenesis, Lymphangiogenesis, and Inflammation in Chronic Obstructive Pulmonary Disease (COPD): Few Certainties and Many Outstanding Questions. Cells, 11(10), 1720. https://doi.org/10.3390/cells11101720