
Pyroptosis and Its Role in SARS-CoV-2 Infection


Abstract
:1. Introduction
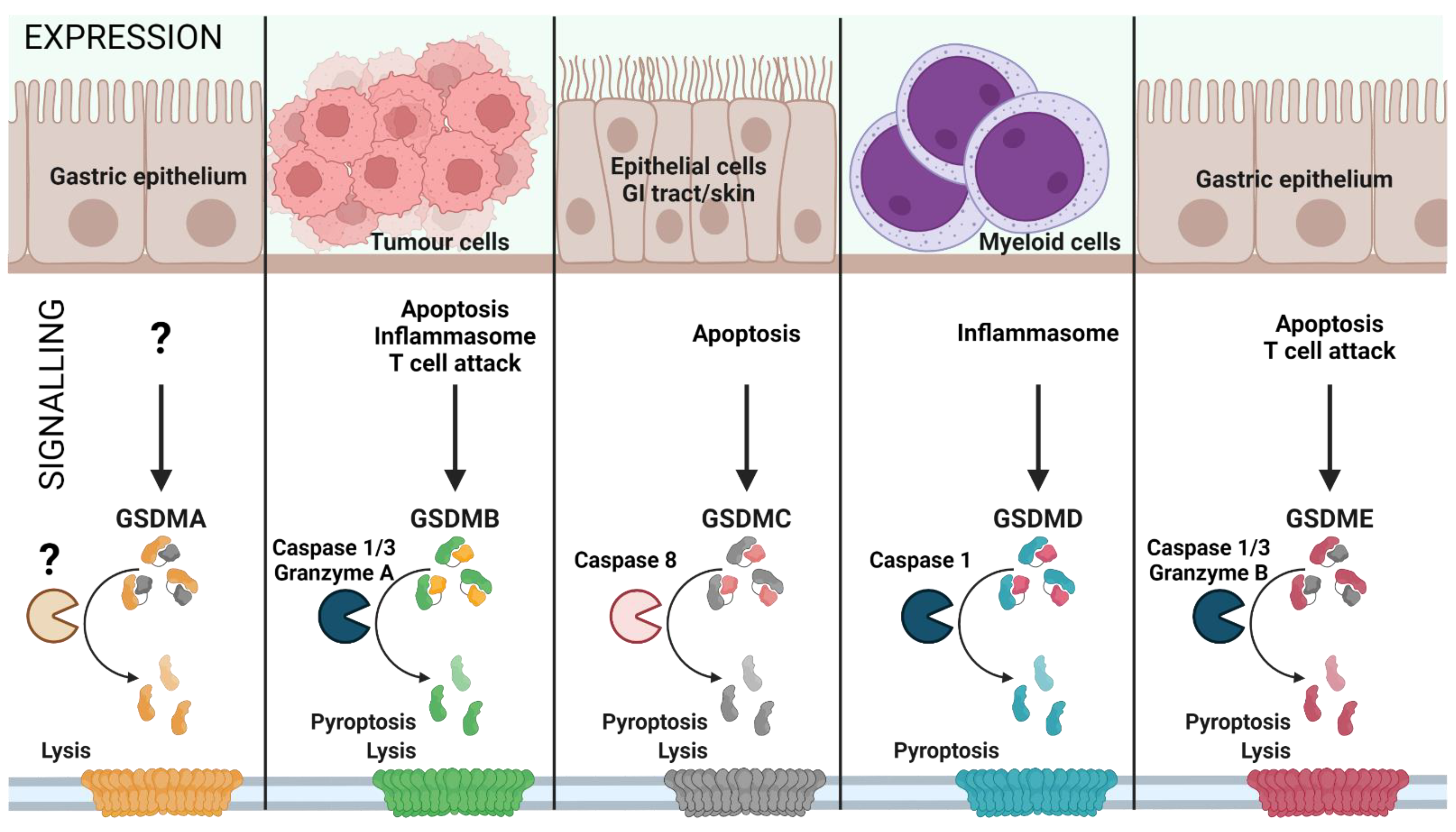
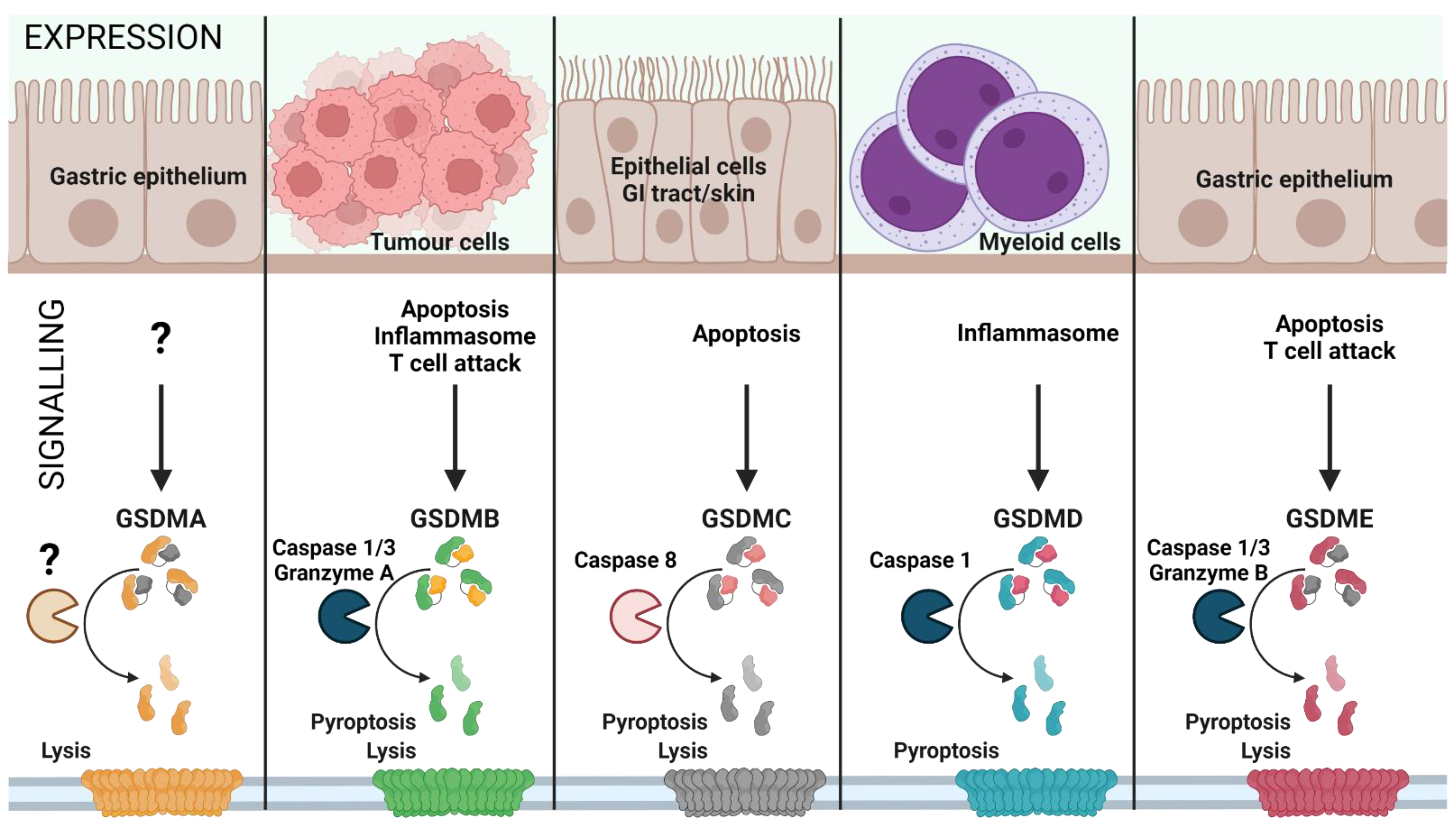
2. The GSDM Family
3. GSDMD Pore Formation
4. GSDMD Post-Translational Modifications (PTM)
5. Pyroptosis in Non-Infectious Diseases
6. GSDMs as Therapeutic Targets
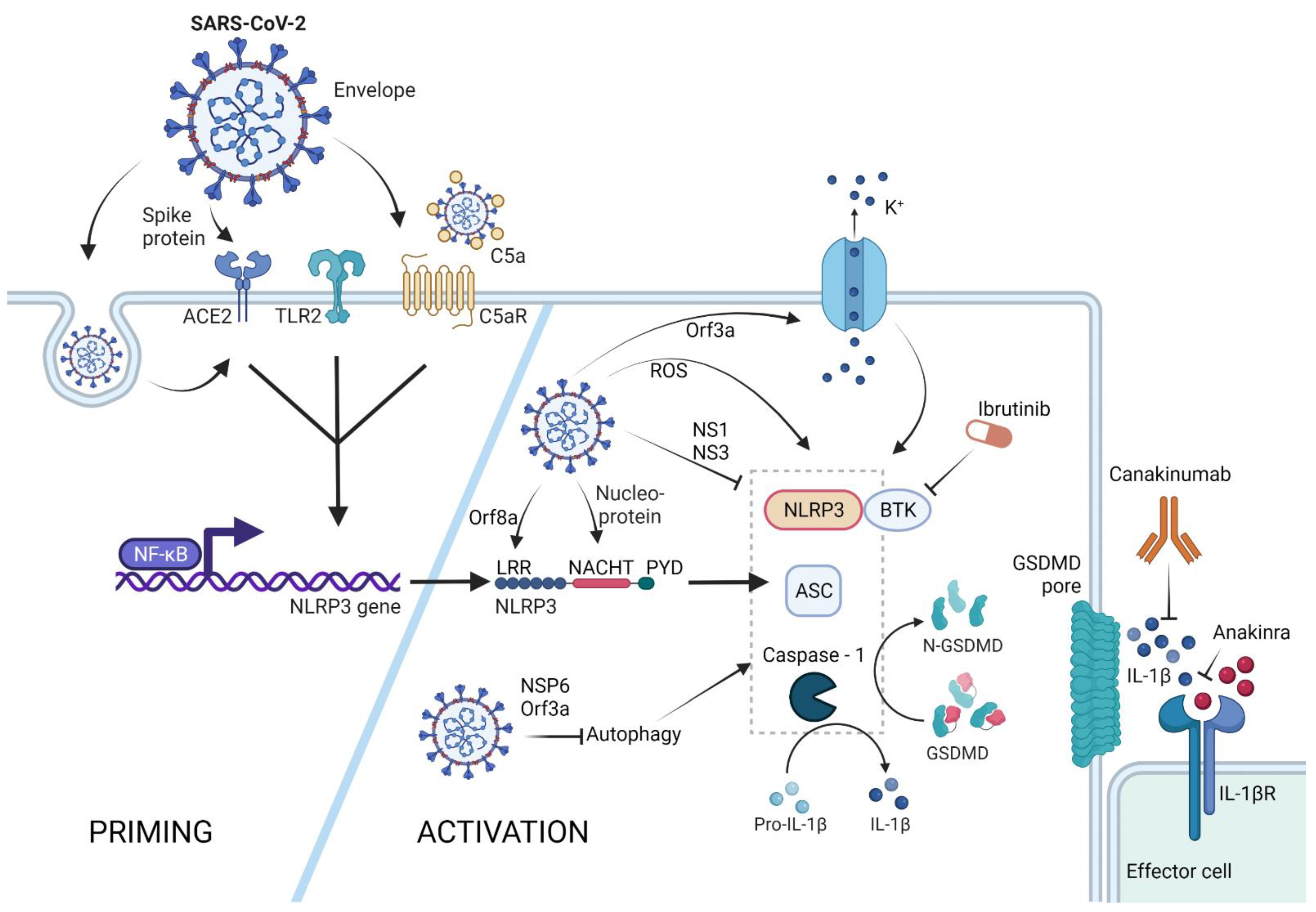
7. SARS-CoV-2 Triggered NLRP3-Mediated Pyroptosis
8. Summary and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Zychlinsky, A.; Prevost, M.C.; Sansonetti, P.J. Shigella flexneri induces apoptosis in infected macrophages. Nature 1992, 358, 167–169. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.M.; Kaniga, K.; Galán, J.E. Salmonella spp. are cytotoxic for cultured macrophages. Mol. Microbiol. 1996, 21, 1101–1115. [Google Scholar] [CrossRef] [PubMed]
- Hilbi, H.; Chen, Y.; Thirumalai, K.; Zychlinsky, A. The interleukin 1β-converting enzyme, caspase 1, is activated during Shigella flexneri-induced apoptosis in human monocyte-derived macrophages. Infect. Immun. 1997, 65, 5165–5170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, S.L.; Cookson, B.T. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell. Microbiol. 2006, 8, 1812–1825. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Fink, S.L.; Cookson, B.T. Pyroptosis and host cell death responses during Salmonella infection. Cell. Microbiol. 2007, 9, 2562–2570. [Google Scholar] [CrossRef]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116. [Google Scholar] [CrossRef]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef] [PubMed]
- He, W.-T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.-H.; Zhong, C.-Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Russo, A.J.; Vasudevan, S.O.; Méndez-Huergo, S.P.; Kumari, P.; Menoret, A.; Duduskar, S.; Wang, C.; Sáez, J.M.P.; Fettis, M.M.; Li, C.; et al. Intracellular immune sensing promotes inflammation via gasdermin D–driven release of a lectin alarmin. Nat. Immunol. 2021, 22, 154–165. [Google Scholar] [CrossRef]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.-G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.N.R.; Bittner, Z.A.; Shankar, S.; Liu, X.; Chang, T.-H.; Jin, T.; Tapia-Abellán, A. Recent insights into the regulatory networks of NLRP3 inflammasome activation. J. Cell Sci. 2020, 133, jcs248344. [Google Scholar] [CrossRef] [PubMed]
- Taabazuing, C.Y.; Okondo, M.; Bachovchin, D.A. Pyroptosis and Apoptosis Pathways Engage in Bidirectional Crosstalk in Monocytes and Macrophages. Cell Chem. Biol. 2017, 24, 507–514.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, L.; Chen, M.; Chen, X.; Zhao, C.; Fang, Z.; Wang, H.; Dai, H. Chemotherapy-induced pyroptosis is mediated by BAK/BAX-caspase-3-GSDME pathway and inhibited by 2-bromopalmitate. Cell Death Dis. 2020, 11, 281. [Google Scholar] [PubMed] [Green Version]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef]
- Saeki, N.; Kim, D.H.; Usui, K.; Aoyagi, K.; Tatsuta, T.; Aoki, K.; Yanagihara, K.; Tamura, M.; Mizushima, H.; Sakamoto, H.; et al. GASDERMIN, suppressed frequently in gastric cancer, is a target of LMO1 in TGF-β-dependent apoptotic signalling. Oncogene 2007, 26, 6488–6498. [Google Scholar] [CrossRef] [Green Version]
- Lunny, D.P.; Weed, E.; Nolan, P.M.; Marquardt, A.; Augustin, M.; Porter, R.M. Mutations in Gasdermin 3 Cause Aberrant Differentiation of the Hair Follicle and Sebaceous Gland. J. Investig. Dermatol. 2005, 124, 615–621. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.; Zhao, R.; Xia, W.; Chang, C.-W.; You, Y.; Hsu, J.-M.; Nie, L.; Chen, Y.; Wang, Y.-C.; Liu, C.; et al. Author Correction: PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis (Nature Cell Biology, (2020), 22, 10, (1264–1275), 10.1038/s41556-020-0575-z). Nat. Cell Biol. 2020, 22, 1396. [Google Scholar] [CrossRef]
- Delmaghani, S.; del Castillo, F.; Michel, V.; Leibovici, M.; Aghaie, A.; Ron, U.; van Laer, L.; Ben-Tal, N.; van Camp, G.; Weil, D.; et al. Mutations in the gene encoding pejvakin, a newly identified protein of the afferent auditory pathway, cause DFNB59 auditory neuropathy. Nat. Genet. 2006, 38, 770–778. [Google Scholar] [CrossRef]
- Volchuk, A.; Ye, A.; Chi, L.; Steinberg, B.E.; Goldenberg, N.M. Indirect regulation of HMGB1 release by gasdermin D. Nat. Commun. 2020, 11, 4561. [Google Scholar] [CrossRef] [PubMed]
- Heilig, R.; Dick, M.S.; Sborgi, L.; Meunier, E.; Hiller, S.; Broz, P. The Gasdermin-D pore acts as a conduit for IL-1β secretion in mice. Eur. J. Immunol. 2018, 48, 584–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiPeso, L.; Ji, D.; Vance, R.E.; Price, J.V. Cell death and cell lysis are separable events during pyroptosis. Cell Death Discov. 2017, 3, 17070. [Google Scholar] [CrossRef] [PubMed]
- Flores-Romero, H.; Ros, U.; Garcia-Saez, A.J. Pore formation in regulated cell death. EMBO J. 2020, 39, e105753. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, C.; Yang, J.; Zhou, B.; Yang, R.; Ramachandran, R.; Abbott, D.W.; Xiao, T.S. Crystal Structures of the Full-Length Murine and Human Gasdermin D Reveal Mechanisms of Autoinhibition, Lipid Binding, and Oligomerization. Immunity 2019, 51, 43–49. [Google Scholar] [CrossRef]
- Broz, P.; Pelegrín, P.; Shao, F. The gasdermins, a protein family executing cell death and inflammation. Nat. Rev. Immunol. 2020, 20, 143–157. [Google Scholar] [CrossRef]
- Chen, X.; He, W.-T.; Hu, L.; Li, J.; Fang, J.; Wang, X.; Xu, X.; Wang, Z.; Huang, K.; Han, J. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res. 2016, 26, 1007–1020. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef] [Green Version]
- Ruan, J.; Xia, S.; Liu, X.; Lieberman, J.; Wu, H. Cryo-EM structure of the gasdermin A3 membrane pore. Nature 2018, 557, 62–67. [Google Scholar] [CrossRef]
- Xia, S.; Zhang, Z.; Magupalli, V.G.; Pablo, J.L.; Dong, Y.; Vora, S.M.; Wang, L.; Fu, T.-M.; Jacobson, M.P.; Greka, A.; et al. Gasdermin D pore structure reveals preferential release of mature interleukin-1. Nature 2021, 593, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Evavold, C.L.; Hafner-Bratkovic, I.; Devant, P.; D’Andrea, J.M.; Ngwa, E.M.; Borsic, E.; Doench, J.G.; LaFleur, M.W.; Sharpe, A.H.; Thiagarajah, J.R.; et al. Control of gasdermin D oligomerization and pyroptosis by the Ragulator-Rag-mTORC1 pathway. Cell 2021, 184, 4495–4511. [Google Scholar] [CrossRef] [PubMed]
- Magupalli, V.G.; Fontana, P.; Wu, H. Ragulator-Rag and ROS TORment gasdermin D pore formation. Trends Immunol. 2021, 42, 948–950. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shi, P.; Chen, Q.; Huang, Z.; Zou, D.; Zhang, J.; Gao, X.; Lin, Z. Mitochondrial ROS promote macrophage pyroptosis by inducing GSDMD oxidation. J. Mol. Cell Biol. 2019, 11, 1069–1082. [Google Scholar] [CrossRef] [Green Version]
- Sborgi, L.; Rühl, S.; Mulvihill, E.; Pipercevic, J.; Heilig, R.; Stahlberg, H.; Farady, C.J.; Muller, D.J.; Broz, P.; Hiller, S. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 2016, 35, 1766–1778. [Google Scholar] [CrossRef]
- Rühl, S.; Broz, P. Regulation of Lytic and Non-Lytic Functions of Gasdermin Pores. J. Mol. Biol. 2022, 434, 167246. [Google Scholar] [CrossRef]
- De Vasconcelos, N.M.; van Opdenbosch, N.; van Gorp, H.; Parthoens, E.; Lamkanfi, M. Single-cell analysis of pyroptosis dynamics reveals conserved GSDMD-mediated subcellular events that precede plasma membrane rupture. Cell Death Differ. 2019, 26, 146–161. [Google Scholar] [CrossRef] [Green Version]
- Russo, H.M.; Rathkey, J.; Boyd-Tressler, A.; Katsnelson, M.A.; Abbott, D.W.; Dubyak, G.R. Active Caspase-1 Induces Plasma Membrane Pores That Precede Pyroptotic Lysis and Are Blocked by Lanthanides. J. Immunol. 2016, 197, 1353–1367. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Cheng, X.; Tang, Y.; Qiu, X.; Wang, Y.; Kang, H.; Wu, J.; Wang, Z.; Liu, Y.; Chen, F.; et al. Bacterial Endotoxin Activates the Coagulation Cascade through Gasdermin D-Dependent Phosphatidylserine Exposure. Immunity 2019, 51, 983–996. [Google Scholar] [CrossRef]
- Rühl, S.; Shkarina, K.; Demarco, B.; Heilig, R.; Santos, J.C.; Broz, P. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 2018, 362, 956–960. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, F.; Heit, A.; Dreher, S.; Eisenächer, K.; Mages, J.; Haas, T.; Krug, A.; Janssen, K.-P.; Kirschning, C.J.; Wagner, H. Mammalian target of rapamycin (mTOR) orchestrates the defense program of innate immune cells. Eur. J. Immunol. 2008, 38, 2981–2992. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zheng, J.; Hu, Q.; Zhao, C.; Chen, Q.; Shi, P.; Chen, Q.; Zou, Y.; Zou, D.; Liu, Q.; et al. Magnesium protects against sepsis by blocking gasdermin D N-terminal-induced pyroptosis. Cell Death Differ. 2020, 27, 466–481. [Google Scholar] [CrossRef] [PubMed]
- Loomis, W.P.; Hartigh, A.B.D.; Cookson, B.T.; Fink, S.L. Diverse small molecules prevent macrophage lysis during pyroptosis. Cell Death Dis. 2019, 10, 326. [Google Scholar] [CrossRef]
- Monteleone, M.; Stow, J.; Schroder, K. Mechanisms of unconventional secretion of IL-1 family cytokines. Cytokine 2015, 74, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Zanoni, I.; Tan, Y.; Di Gioia, M.; Broggi, A.; Ruan, J.; Shi, J.; Donado, C.A.; Shao, F.; Wu, H.; Springstead, J.R.; et al. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science 2016, 352, 1232–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kayagaki, N.; Kornfeld, O.S.; Lee, B.L.; Stowe, I.B.; O’Rourke, K.; Li, Q.; Sandoval, W.; Yan, D.; Kang, J.; Xu, M.; et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature 2021, 591, 131–136. [Google Scholar] [CrossRef]
- Fischer, F.A.; Chen, K.W.; Bezbradica, J.S. Posttranslational and Therapeutic Control of Gasdermin-Mediated Pyroptosis and Inflammation. Front. Immunol. 2021, 12, 661162. [Google Scholar] [CrossRef]
- Bambouskova, M.; Potuckova, L.; Paulenda, T.; Kerndl, M.; Mogilenko, D.A.; Lizotte, K.; Swain, A.; Hayes, S.; Sheldon, R.D.; Kim, H.; et al. Itaconate confers tolerance to late NLRP3 inflammasome activation. Cell Rep. 2021, 34, 108756. [Google Scholar] [CrossRef]
- Hoyle, C.; Green, J.P.; Allan, S.M.; Brough, D.; Lemarchand, E. Itaconate and fumarate derivatives exert a dual inhibitory effect on canonical NLRP3 activation in macrophages and microglia. bioRxiv 2021, in press. [Google Scholar] [CrossRef]
- Humphries, F.; Shmuel-Galia, L.; Ketelut-Carneiro, N.; Li, S.; Wang, B.; Nemmara, V.V.; Wilson, R.; Jiang, Z.; Khalighinejad, F.; Muneeruddin, K.; et al. Succination inactivates gasdermin D and blocks pyroptosis. Science 2020, 369, 1633–1637. [Google Scholar] [CrossRef]
- Rogers, C.; Erkes, D.A.; Nardone, A.; Aplin, A.E.; Fernandes-Alnemri, T.; Alnemri, E.S. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat. Commun. 2019, 10, 1689. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Jiang, M.; Chu, Y.; Wang, W.; Chen, D.; Li, X.; Zhang, Z.; Zhang, D.; Fan, D.; Nie, Y.; et al. Gasdermin D plays a key role as a pyroptosis executor of non-alcoholic steatohepatitis in humans and mice. J. Hepatol. 2018, 68, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Gordon, R.; Albornoz, E.A.; Christie, D.C.; Langley, M.R.; Kumar, V.; Mantovani, S.; Robertson, A.A.B.; Butler, M.S.; Rowe, D.B.; O’Neill, L.A.; et al. Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Sci. Transl. Med. 2018, 10, eaah4066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, F.; Lan, Z.; Xin, Z.; He, C.; Guo, Z.; Xia, X.; Hu, T. Emerging insights into molecular mechanisms underlying pyroptosis and functions of inflammasomes in diseases. J. Cell. Physiol. 2020, 235, 3207–3221. [Google Scholar] [CrossRef]
- Li, S.; Wu, Y.; Yang, D.; Wu, C.; Ma, C.; Liu, X.; Moynagh, P.N.; Wang, B.; Hu, G.; Yang, S. Gasdermin D in peripheral myeloid cells drives neuroinflammation in experimental autoimmune encephalomyelitis. J. Exp. Med. 2019, 216, 2562–2581. [Google Scholar] [CrossRef] [Green Version]
- McKenzie, B.; Dixit, V.M.; Power, C. Fiery Cell Death: Pyroptosis in the Central Nervous System. Trends Neurosci. 2020, 43, 55–73. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Y.; Xia, S.; Kong, Q.; Li, S.; Liu, X.; Junqueira, C.; Meza-Sosa, K.F.; Mok, T.M.Y.; Ansara, J.; et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 2020, 579, 415–420. [Google Scholar] [CrossRef]
- Man, S.M.; Karki, R.; Kanneganti, T.-D. AIM2 inflammasome in infection, cancer, and autoimmunity: Role in DNA sensing, inflammation, and innate immunity. Eur. J. Immunol. 2016, 46, 269–280. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Fang, Y.; Chen, X.; Wang, Z.; Liang, X.; Zhang, T.; Liu, M.; Zhou, N.; Lv, J.; Tang, K.; et al. Gasdermin E–mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci. Immunol. 2020, 5, eaax7969. [Google Scholar] [CrossRef]
- Xu, Y.-J.; Zheng, L.; Hu, Y.-W.; Wang, Q. Pyroptosis and its relationship to atherosclerosis. Clin. Chim. Acta 2018, 476, 28–37. [Google Scholar] [CrossRef]
- Zhaolin, Z.; Guohua, L.; Shiyuan, W.; Zuo, W. Role of pyroptosis in cardiovascular disease. Cell Prolif. 2019, 52, e12563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, W.; Li, Y.; Wu, M.; Jin, Q.; Wang, Q.; Li, S.; Huang, S.; Zhang, A.; Zhang, Y.; Jia, Z. Gasdermin E deficiency attenuates acute kidney injury by inhibiting pyroptosis and inflammation. Cell Death Dis. 2021, 12, 139. [Google Scholar] [CrossRef] [PubMed]
- Miao, N.; Yin, F.; Xie, H.; Wang, Y.; Xu, Y.; Shen, Y.; Xu, D.; Yin, J.; Wang, B.; Zhou, Z.; et al. The cleavage of gasdermin D by caspase-11 promotes tubular epithelial cell pyroptosis and urinary IL-18 excretion in acute kidney injury. Kidney Int. 2019, 96, 1105–1120. [Google Scholar] [CrossRef] [PubMed]
- Goldbach-Mansky, R. Current Status of Understanding the Pathogenesis and Management of Patients With NOMID/CINCA. Curr. Rheumatol. Rep. 2011, 13, 123–131. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Lopez, N.; Romero, R.; Xu, Y.; Plazyo, O.; Unkel, R.; Leng, Y.; Than, N.G.; Chaiworapongsa, T.; Panaitescu, B.; Dong, Z.; et al. A Role for the Inflammasome in Spontaneous Preterm Labor with Acute Histologic Chorioamnionitis. Reprod. Sci. 2017, 24, 1382–1401. [Google Scholar] [CrossRef]
- Cheng, S.-B.; Nakashima, A.; Huber, W.J.; Davis, S.; Banerjee, S.; Huang, Z.; Saito, S.; Sadovsky, Y.; Sharma, S. Pyroptosis is a critical inflammatory pathway in the placenta from early onset preeclampsia and in human trophoblasts exposed to hypoxia and endoplasmic reticulum stressors. Cell Death Dis. 2019, 10, 927. [Google Scholar] [CrossRef]
- Herster, F.; Bittner, Z.; Archer, N.; Dickhöfer, S.; Eisel, D.; Eigenbrod, T.; Knorpp, T.; Schneiderhan-Marra, N.; Löffler, M.W.; Kalbacher, H.; et al. Neutrophil extracellular trap-associated RNA and LL37 enable self-amplifying inflammation in psoriasis. Nat. Commun. 2020, 11, 105. [Google Scholar] [CrossRef] [Green Version]
- Sollberger, G.; Choidas, A.; Burn, G.L.; Habenberger, P.; Di Lucrezia, R.; Kordes, S.; Menninger, S.; Eickhoff, J.; Nussbaumer, P.; Klebl, B.; et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6689. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; He, H.; Wang, K.; Shi, X.; Wang, Y.; Su, Y.; Wang, Y.; Li, D.; Liu, W.; Zhang, Y.; et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science 2020, 368, 1833. [Google Scholar] [CrossRef]
- Rathkey, J.K.; Zhao, J.; Liu, Z.; Chen, Y.; Yang, J.; Kondolf, H.C.; Benson, B.L.; Chirieleison, S.M.; Huang, A.Y.; Dubyak, G.R.; et al. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci. Immunol. 2018, 3, eaat2738. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.J.; Liu, X.; Xia, S.; Zhang, Z.; Zhang, Y.; Zhao, J.; Ruan, J.; Luo, X.; Lou, X.; Bai, Y.; et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat. Immunol. 2020, 21, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Martín-Sánchez, F.; Diamond, C.; Zeitler, M.; Gomez, A.I.; Baroja-Mazo, A.; Bagnall, J.; Spiller, D.; White, M.; Daniels, M.; Mortellaro, A.; et al. Inflammasome-dependent IL-1β release depends upon membrane permeabilisation. Cell Death Differ. 2016, 23, 1219–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juliana, C.; Fernandes-Alnemri, T.; Wu, J.; Datta, P.; Solorzano, L.; Yu, J.-W.; Meng, R.; Quong, A.A.; Latz, E.; Scott, C.P.; et al. Anti-inflammatory Compounds Parthenolide and Bay 11-7082 Are Direct Inhibitors of the Inflammasome. J. Biol. Chem. 2010, 285, 9792–9802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, S.; Exline, M.; Habyarimana, F.; Gavrilin, M.; Baker, P.; Masters, S.L.; Wewers, M.D.; Sarkar, A. Microparticulate Caspase 1 Regulates Gasdermin D and Pulmonary Vascular Endothelial Cell Injury. Am. J. Respir. Cell Mol. Biol. 2018, 59, 56–64. [Google Scholar] [CrossRef]
- Diaz, G.M.; Hupperts, R.; Fraussen, J.; Somers, V. Dimethyl fumarate treatment in multiple sclerosis: Recent advances in clinical and immunological studies. Autoimmun. Rev. 2018, 17, 1240–1250. [Google Scholar] [CrossRef]
- Molina-Crespo, A.; Cadete, A.; Sarrio, D.; Gamez-Chiachio, M.; Martinez, L.; Chao, K.; Olivera, A.; Gonella, A.; Diaz, E.; Palacio, H.; et al. Intracellular delivery of an antibody targeting gasdermin-b reduces her2 breast cancer aggressiveness. Clin. Cancer Res. 2019, 25, 4846–4858. [Google Scholar] [CrossRef] [Green Version]
- Landi, L.; Ravaglia, C.; Russo, E.; Cataleta, P.; Fusari, M.; Boschi, A.; Giannarelli, D.; Facondini, F.; Valentini, I.; Panzini, I.; et al. Blockage of interleukin-1β with canakinumab in patients with Covid-19. Sci. Rep. 2020, 10, 21775. [Google Scholar] [CrossRef]
- Cavalli, G.; de Luca, G.; Campochiaro, C.; Della-Torre, E.; Ripa, M.; Canetti, D.; Oltolini, C.; Castiglioni, B.; Din, C.T.; Boffini, N.; et al. Interleukin-1 blockade with high-dose anakinra in patients with COVID-19, acute respiratory distress syndrome, and hyperinflammation: A retrospective cohort study. Lancet Rheumatol. 2020, 2, e325–e331. [Google Scholar] [CrossRef]
- Hergueta-Redondo, M.; Sarrio, D.; Molina-Crespo, A.; Vicario, R.; Morales, C.B.; Martínez, L.; Rojo-Sebastián, A.; Serra-Musach, J.; Mota, A.; Martínez-Ramírez, A.; et al. Gasdermin B expression predicts poor clinical outcome in HER2-positive breast cancer. Oncotarget 2016, 7, 56295–56308. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.F.-W.; Kok, K.-H.; Zhu, Z.; Chu, H.; To, K.K.-W.; Yuan, S.; Yuen, K.-Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef] [Green Version]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Toldo, S.; Bussani, R.; Nuzzi, V.; Bonaventura, A.; Mauro, A.G.; Cannatà, A.; Pillappa, R.; Sinagra, G.; Nana-Sinkam, P.; Sime, P.; et al. Inflammasome formation in the lungs of patients with fatal COVID-19. Agents Actions 2021, 70, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, T.S.; de Sa, K.S.G.; Ishimoto, A.Y.; Becerra, A.; Oliveira, S.; Almeida, L.; Goncalves, A.V.; Perucello, D.B.; Andrade, W.A.; Castro, R.; et al. Inflammasomes are activated in response to SARS-cov-2 infection and are associated with COVID-19 severity in patients. J. Exp. Med. 2020, 218, e20201707. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Xie, X.; Feng, X.-L.; Xu, L.; Han, J.-B.; Yu, D.; Zou, Q.-C.; Liu, Q.; Li, X.; Ma, G.; et al. Specific inhibition of the NLRP3 inflammasome suppresses immune overactivation and alleviates COVID-19 like pathology in mice. eBioMedicine 2021, 75, 103803. [Google Scholar] [CrossRef]
- Coll, R.C.; Hill, J.R.; Day, C.J.; Zamoshnikova, A.; Boucher, D.; Massey, N.L.; Chitty, J.; Fraser, J.A.; Jennings, M.P.; Robertson, A.A.B.; et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat. Chem. Biol. 2019, 15, 556–559. [Google Scholar] [CrossRef]
- Ferreira, A.C.; Soares, V.C.; de Azevedo-Quintanilha, I.G.; Dias, S.d.S.G.; Fintelman-Rodrigues, N.; Sacramento, C.Q.; Mattos, M.; de Freitas, C.S.; Temerozo, J.R.; Teixeira, L.; et al. Correction: SARS-CoV-2 engages inflammasome and pyroptosis in human primary monocytes. Cell Death Discov. 2021, 7, 43. [Google Scholar] [CrossRef]
- Grein, J.; Ohmagari, N.; Shin, D.; Diaz, G.; Asperges, E.; Castagna, A.; Feldt, T.; Green, G.; Green, M.L.; Lescure, F.X.; et al. Compassionate Use of Remdesivir for Patients with Severe Covid-19. N. Engl. J. Med. 2020, 382, 2327–2336. [Google Scholar] [CrossRef]
- Pontali, E.; Volpi, S.; Antonucci, G.; Castellaneta, M.; Buzzi, D.; Tricerri, F.; Angelelli, A.; Caorsi, R.; Feasi, M.; Calautti, F.; et al. Safety and efficacy of early high-dose IV anakinra in severe COVID-19 lung disease. J. Allergy Clin. Immunol. 2020, 146, 213–215. [Google Scholar] [CrossRef]
- Pasin, L.; Cavalli, G.; Navalesi, P.; Sella, N.; Landoni, G.; Yavorovskiy, A.G.; Likhvantsev, V.V.; Zangrillo, A.; Dagna, L.; Monti, G. Anakinra for patients with COVID-19: A meta-analysis of non-randomized cohort studies. Eur. J. Intern. Med. 2021, 86, 34–40. [Google Scholar] [CrossRef]
- Kyriazopoulou, E.; Poulakou, G.; Milionis, H.; Metallidis, S.; Adamis, G.; Tsiakos, K.; Fragkou, A.; Rapti, A.; Damoulari, C.; Fantoni, M.; et al. Early treatment of COVID-19 with anakinra guided by soluble urokinase plasminogen receptor plasma levels: A double-blind, randomized controlled phase 3 trial. Nat. Med. 2021, 27, 1752–1760. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, H.; Yao, X.; Zhang, D.; Zhou, Y.; Fu, B.; Wang, W.; Li, H.; Wang, Z.; Hu, Z.; et al. Pyroptotic macrophages stimulate the SARS-CoV-2-associated cytokine storm. Cell. Mol. Immunol. 2021, 18, 1305–1307. [Google Scholar] [CrossRef] [PubMed]
- Siu, K.-L.; Yuen, K.-S.; Castano-Rodriguez, C.; Ye, Z.-W.; Yeung, M.-L.; Fung, S.-Y.; Yuan, S.; Chan, C.-P.; Yuen, K.-Y.; Enjuanes, L.; et al. Severe acute respiratory syndrome Coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019, 33, 8865–8877. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.-Y.; Moriyama, M.; Chang, M.-F.; Ichinohe, T. Severe Acute Respiratory Syndrome Coronavirus Viroporin 3a Activates the NLRP3 Inflammasome. Front. Microbiol. 2019, 10, 50. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Chitre, S.A.; Akinyemi, I.A.; Loeb, J.C.; Lednicky, J.A.; McIntosh, M.T.; Bhaduri-McIntosh, S. SARS-CoV-2 viroporin triggers the NLRP3 inflammatory pathway. bioRxiv 2020, in press. [Google Scholar]
- Shi, C.-S.; Nabar, N.R.; Huang, N.-N.; Kehrl, J.H. SARS-Coronavirus Open Reading Frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov. 2019, 5, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, P.; Shen, M.; Yu, Z.; Ge, W.; Chen, K.; Tian, M.; Xiao, F.; Wang, Z.; Wang, J.; Jia, Y.; et al. SARS-CoV-2 N protein promotes NLRP3 inflammasome activation to induce hyperinflammation. Nat. Commun. 2021, 12, 4664. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Liu, Y.; Huang, Z.; Xu, W.; Hu, W.; Yi, L.; Liu, Z.; Chan, H.; Zeng, J.; Liu, X.; et al. SARS-CoV-2 non-structural protein 6 triggers NLRP3-dependent pyroptosis by targeting ATP6AP1. Cell Death Differ. 2022, in press. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Karki, R.; Williams, E.P.; Yang, D.; Fitzpatrick, E.; Vogel, P.; Jonsson, C.B.; Kanneganti, T.-D. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat. Immunol. 2021, 22, 829–838. [Google Scholar] [CrossRef]
- Cohen, M.E.; Eichel, R.; Steiner-Birmanns, B.; Janah, A.; Ioshpa, M.; Bar-Shalom, R.; Paul, J.J.; Gaber, H.; Skrahina, V.; Bornstein, N.M.; et al. A case of probable Parkinson’s disease after SARS-CoV-2 infection. Lancet Neurol. 2020, 19, 804–805. [Google Scholar] [CrossRef]
- Albornoz, E.; Amarilla, A.A.; Modhiran, N.; Parker, S.; Li, X.X.; Wijesundara, D.K.; Zamora, A.P.; McMillan, C.L.D.; Liang, B.; Peng, N.Y.G.; et al. SARS-CoV-2 drives NLRP3 inflammasome activation in human microglia through spike-ACE2 receptor interaction. bioRxiv 2022, in press. [Google Scholar]
- Theobald, S.J.; Simonis, A.; Georgomanolis, T.; Kreer, C.; Zehner, M.; Eisfeld, H.S.; Albert, M.-C.; Chhen, J.; Motameny, S.; Erger, F.; et al. Long-lived macrophage reprogramming drives spike protein-mediated inflammasome activation in COVID-19. EMBO Mol. Med. 2021, 13, e14150. [Google Scholar] [CrossRef] [PubMed]
- Junqueira, C.; Crespo, A.; Ranjbar, S.; Ingber, J.; Parry, B.; Ravid, S.; de Lacerda, L.B.; Lewandrowski, M.; Clark, S.; Ho, F.; et al. SARS-CoV-2 infects blood monocytes to activate NLRP3 and AIM2 inflammasomes, pyroptosis and cytokine release. medRxiv 2021, in press. [Google Scholar]
- Yu, J.; Yuan, X.; Chen, H.; Chaturvedi, S.; Braunstein, E.M.; Brodsky, R.A. Direct activation of the alternative complement pathway by SARS-CoV-2 spike proteins is blocked by factor D Inhibition. Blood 2020, 136, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Bournazos, S.; Gupta, A.; Ravetch, J.V. The role of IgG Fc receptors in antibody-dependent enhancement. Nat. Rev. Immunol. 2020, 20, 633–643. [Google Scholar] [CrossRef]
- Kim, N.E.; Kim, D.K.; Song, Y.J. Sars-cov-2 nonstructural proteins 1 and 13 suppress caspase-1 and the NLRP3 inflammasome activation. Microorganisms 2021, 9, 494. [Google Scholar] [CrossRef]
- Maggiore, U. Colchicine Counteracting Inflammation in COVID-19 Pneumonia (ColCOVID-19). 2020. Available online: https://clinicaltrials.gov/show/NCT04322565 (accessed on 13 April 2022).
- Mehta, K.G.; Patel, T.; Chavda, P.D.; Patel, P. Efficacy and safety of colchicine in COVID-19: A meta-analysis of randomised controlled trials. RMD Open 2021, 7, e001746. [Google Scholar] [CrossRef]
- Treon, S.P.; Castillo, J.J.; Skarbnik, A.P.; Soumerai, J.D.; Ghobrial, I.M.; Guerrera, M.L.; Meid, K.E.; Yang, G. The BTK inhibitor ibrutinib may protect against pulmonary injury in COVID-19–infected patients. Blood 2020, 135, 1912–1915. [Google Scholar] [CrossRef] [Green Version]
- Roschewski, M.; Lionakis, M.S.; Sharman, J.P.; Roswarski, J.; Goy, A.; Monticelli, M.A.; Roshon, M.; Wrzesinski, S.H.; Desai, J.V.; Zarakas, M.A.; et al. Inhibition of Bruton tyrosine kinase in patients with severe COVID-19. Sci. Immunol. 2020, 5, eabd0110. [Google Scholar] [CrossRef]
- Zheng, M.; Williams, E.P.; Malireddi, R.K.S.; Karki, R.; Banoth, B.; Burton, A.; Webby, R.; Channappanavar, R.; Jonsson, C.B.; Kanneganti, T.-D. Impaired NLRP3 inflammasome activation/pyroptosis leads to robust inflammatory cell death via caspase-8/RIPK3 during coronavirus infection. J. Biol. Chem. 2020, 295, 14040–14052. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Target | Agent | Mode of Action | Citation |
|---|---|---|---|
| NF-κB | Bay 11–7082 | Upstream inhibition of NLRP3 priming | [73] |
| Caspase 1 | z-VAD-fmk | Inhibition of GSDMs and pro-IL-1β cleavage | [74] |
| GSDMB | Anti-GSDMB antibody | Direct binding of GSDMB, thereby reducing tumour growth and distant metastasis in HER2 positive cancer | [76] |
| GSDMD | Disulfiram | Inhibits assembly of NT-GSDMD by binding at Cys191 | [71] |
| Dimethylfumarate (DMF) | DMF inhibits GSDMD and GSDME at Cys45 and Cys191 and reduces demyelination in MS | [75] | |
| Necrosulfonamide (NSA) | Inhibits formation of NT-GSDMD by binding at Cys191 | [70] | |
| Punicalagin | Probably inhibits GSDMD insertion into cell membrane | [72] | |
| GSDME | 2-Bromopalmitate (2-BP) | Counteracts the CT-GSDME palmitoylation; prevents chemotherapy-induced GSDME-mediated pyroptosis | [15] |
| IL-1β | Canakinumab | Binds and neutralizes IL-1β | [77] |
| IL-1βR | Anakinra | IL-1βR antagonist | [78] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bittner, Z.A.; Schrader, M.; George, S.E.; Amann, R. Pyroptosis and Its Role in SARS-CoV-2 Infection. Cells 2022, 11, 1717. https://doi.org/10.3390/cells11101717
Bittner ZA, Schrader M, George SE, Amann R. Pyroptosis and Its Role in SARS-CoV-2 Infection. Cells. 2022; 11(10):1717. https://doi.org/10.3390/cells11101717
Chicago/Turabian StyleBittner, Zsofia Agnes, Markus Schrader, Shilpa Elizabeth George, and Ralf Amann. 2022. "Pyroptosis and Its Role in SARS-CoV-2 Infection" Cells 11, no. 10: 1717. https://doi.org/10.3390/cells11101717
APA StyleBittner, Z. A., Schrader, M., George, S. E., & Amann, R. (2022). Pyroptosis and Its Role in SARS-CoV-2 Infection. Cells, 11(10), 1717. https://doi.org/10.3390/cells11101717