
Fibrotic Signaling in Cardiac Fibroblasts and Vascular Smooth Muscle Cells: The Dual Roles of Fibrosis in HFpEF and CAD

 , and
, and 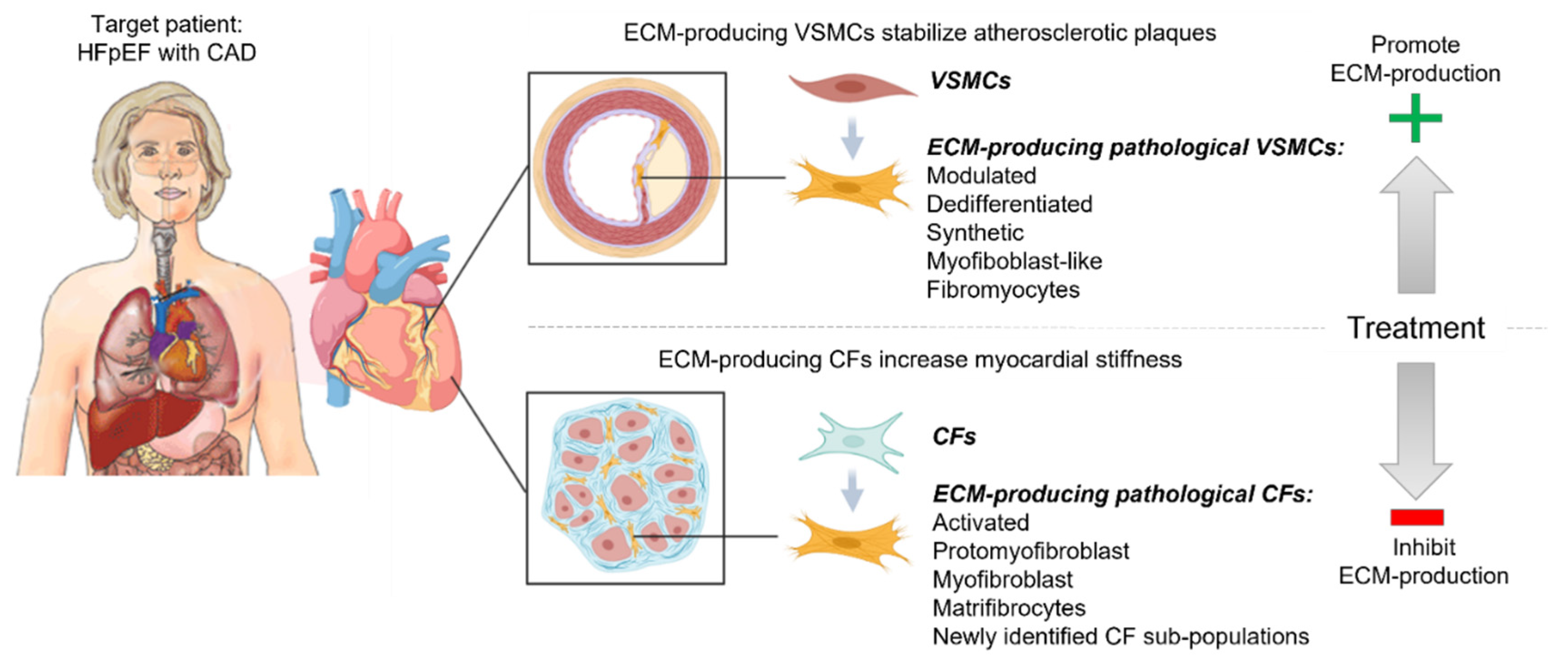{kind=link}
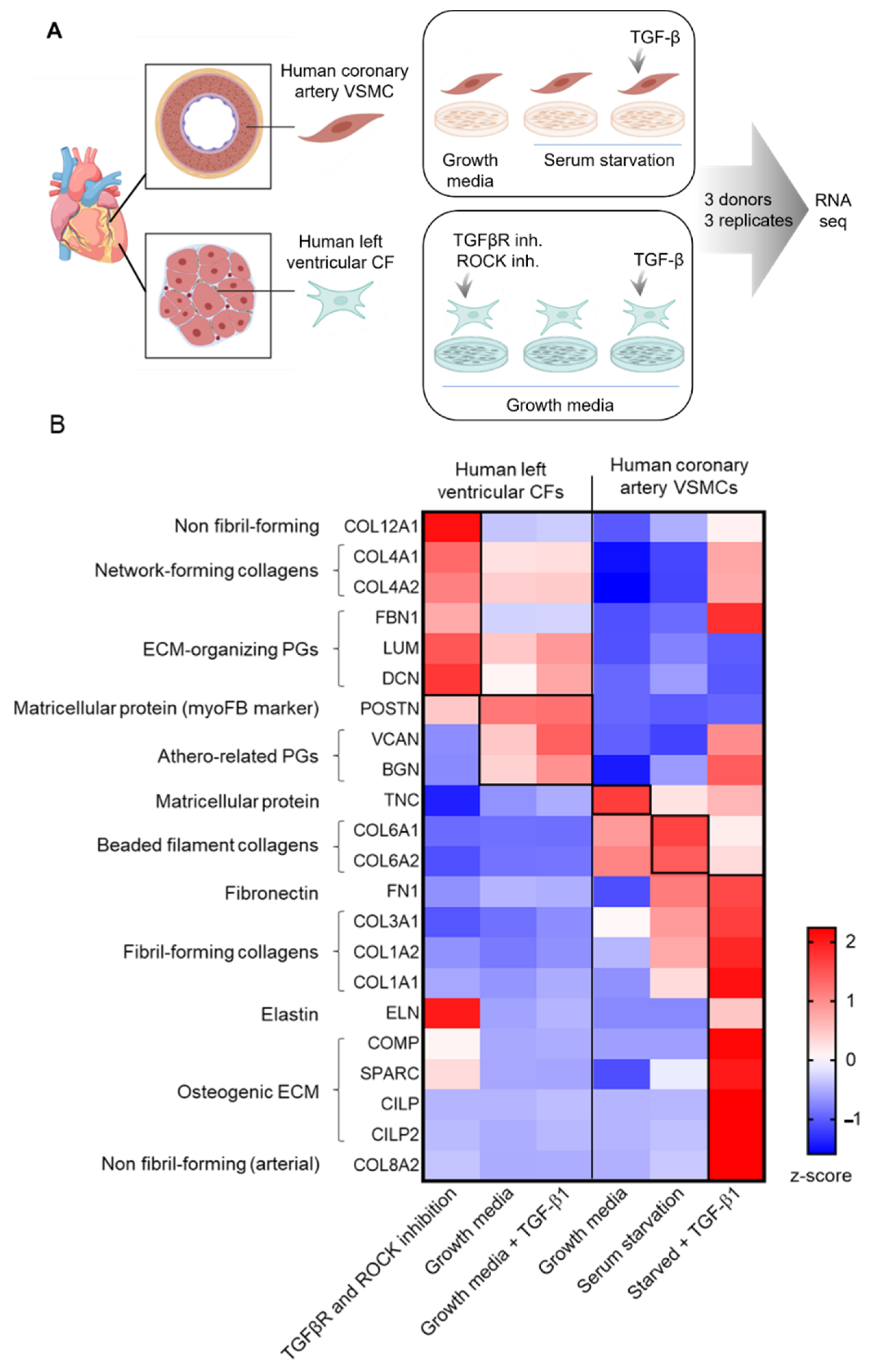{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Dual Roles of Fibrotic Remodeling in Cardiovascular Disease
1.2. Cardiac Fibroblasts Regulate Fibrotic Remodeling of the Myocardium
1.3. Vascular Smooth Muscle Cells Regulate Fibrotic Remodeling in Coronary Atherosclerotic Plaques
2. Studying CFs and VSMCs In Vitro

3. Signaling Pathways That Drive ECM Remodeling in CFs and VSMCs
3.1. TGF-β Signaling
3.2. Renin-Angiotensin-Aldosterone System (RAAS)
3.3. Wnt/β-Catenin Signaling
3.4. PDGF Signaling
3.5. Interleukins
3.5.1. IL-1
3.5.2. IL-6
3.5.3. IL-11
3.5.4. IL-17
3.5.5. IL-18
3.6. Mechanical Signaling
4. TGF-β and Angiotensin II Signaling Pathways—Center of Stage for ECM Remodeling in Both CFs and VSMCs
5. Clinical Studies of TGF-β or RAAS Inhibition: Effects on Fibrotic Remodeling
5.1. Anti-TGF-β Treatment
5.2. Pirfenidone
5.3. RAAS Inhibitors
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gao, X.M.; White, D.A.; Dart, A.M.; Du, X.J. Post-infarct cardiac rupture: Recent insights on pathogenesis and therapeutic interventions. Pharm. Ther. 2012, 134, 156–179. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef] [PubMed]
- Owan, T.E.; Hodge, D.O.; Herges, R.M.; Jacobsen, S.J.; Roger, V.L.; Redfield, M.M. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N. Engl. J. Med. 2006, 355, 251–259. [Google Scholar] [CrossRef]
- Schiattarella, G.G.; Rodolico, D.; Hill, J.A. Metabolic inflammation in heart failure with preserved ejection fraction. Cardiovasc. Res. 2021, 117, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Van Heerebeek, L.; Franssen, C.P.; Hamdani, N.; Verheugt, F.W.; Somsen, G.A.; Paulus, W.J. Molecular and cellular basis for diastolic dysfunction. Curr. Heart Fail. Rep. 2012, 9, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.Y.; Ghosn, M.G.; Khan, M.A.; Gramze, N.L.; Brunner, G.; Nabi, F.; Nambi, V.; Nagueh, S.F.; Nguyen, D.T.; Graviss, E.A.; et al. Myocardial Extracellular Volume Fraction Adds Prognostic Information Beyond Myocardial Replacement Fibrosis. Circ. Cardiovasc. Imaging 2019, 12, e009535. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. [Google Scholar] [CrossRef]
- Pakshir, P.; Noskovicova, N.; Lodyga, M.; Son, D.O.; Schuster, R.; Goodwin, A.; Karvonen, H.; Hinz, B. The myofibroblast at a glance. J. Cell Sci. 2020, 133, jcs227900. [Google Scholar] [CrossRef]
- Rush, C.J.; Berry, C.; Oldroyd, K.G.; Rocchiccioli, J.P.; Lindsay, M.M.; Touyz, R.M.; Murphy, C.L.; Ford, T.J.; Sidik, N.; McEntegart, M.B.; et al. Prevalence of Coronary Artery Disease and Coronary Microvascular Dysfunction in Patients With Heart Failure With Preserved Ejection Fraction. JAMA Cardiol. 2021, 6, 1130–1143. [Google Scholar] [CrossRef]
- Vergallo, R.; Crea, F. Atherosclerotic Plaque Healing. N. Engl. J. Med. 2020, 383, 846–857. [Google Scholar] [CrossRef]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgozoglu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef]
- Porter, K.E.; Turner, N.A. Cardiac fibroblasts: At the heart of myocardial remodeling. Pharmacol. Ther. 2009, 123, 255–278. [Google Scholar] [CrossRef]
- Cowling, R.T.; Yeo, S.J.; Kim, I.J.; Park, J.I.; Gu, Y.; Dalton, N.D.; Peterson, K.L.; Greenberg, B.H. Discoidin domain receptor 2 germline gene deletion leads to altered heart structure and function in the mouse. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H773–H781. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’Antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.A.; et al. Revisiting Cardiac Cellular Composition. Circ. Res. 2016, 118, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Tallquist, M.D. Cardiac Fibroblast Diversity. Annu. Rev. Physiol. 2020, 82, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Litvinukova, M.; Talavera-Lopez, C.; Maatz, H.; Reichart, D.; Worth, C.L.; Lindberg, E.L.; Kanda, M.; Polanski, K.; Heinig, M.; Lee, M.; et al. Cells of the adult human heart. Nature 2020, 588, 466–472. [Google Scholar] [CrossRef]
- Acharya, A.; Baek, S.T.; Huang, G.; Eskiocak, B.; Goetsch, S.; Sung, C.Y.; Banfi, S.; Sauer, M.F.; Olsen, G.S.; Duffield, J.S.; et al. The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development 2012, 139, 2139–2149. [Google Scholar] [CrossRef]
- Nurnberg, S.T.; Cheng, K.; Raiesdana, A.; Kundu, R.; Miller, C.L.; Kim, J.B.; Arora, K.; Carcamo-Oribe, I.; Xiong, Y.; Tellakula, N.; et al. Coronary Artery Disease Associated Transcription Factor TCF21 Regulates Smooth Muscle Precursor Cells that Contribute to the Fibrous Cap. Genom. Data 2015, 5, 36–37. [Google Scholar] [CrossRef]
- Kanisicak, O.; Khalil, H.; Ivey, M.J.; Karch, J.; Maliken, B.D.; Correll, R.N.; Brody, M.J.; SC, J.L.; Aronow, B.J.; Tallquist, M.D.; et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 2016, 7, 1–14. [Google Scholar] [CrossRef]
- Majno, G.; Gabbiani, G.; Hirschel, B.J.; Ryan, G.B.; Statkov, P.R. Contraction of granulation tissue in vitro: Similarity to smooth muscle. Science 1971, 173, 548–550. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, C.; Liu, Q.; Wang, L.; Bao, A.X.; Jung, J.P.; Francis, J.; Molkentin, J.D.; Fu, X. Loss of Acta2 in cardiac fibroblasts does not affect myofibroblast differentiation or cardiac repair after myocardial infarction. BioRxiv 2021. version posted 21 May 2021. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Moore-Morris, T.; Guimaraes-Camboa, N.; Banerjee, I.; Zambon, A.C.; Kisseleva, T.; Velayoudon, A.; Stallcup, W.B.; Gu, Y.; Dalton, N.D.; Cedenilla, M.; et al. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J. Clin. Investig. 2014, 124, 2921–2934. [Google Scholar] [CrossRef] [PubMed]
- Ivey, M.J.; Kuwabara, J.T.; Pai, J.T.; Moore, R.E.; Sun, Z.; Tallquist, M.D. Resident fibroblast expansion during cardiac growth and remodeling. J. Mol. Cell Cardiol. 2018, 114, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Kramann, R.; Schneider, R.K.; DiRocco, D.P.; Machado, F.; Fleig, S.; Bondzie, P.A.; Henderson, J.M.; Ebert, B.L.; Humphreys, B.D. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell 2015, 16, 51–66. [Google Scholar] [CrossRef]
- Kuppe, C.; Ramirez Flores, R.O.; Li, Z.; Hannani, M.; Tanevski, J.; Halder, M.; Cheng, M.; Ziegler, S.; Zhang, X.; Preisker, F.; et al. Spatial multi-omic map of human myocardial infarction. BioRxiv 2020. version posted 10 December 2020. [Google Scholar] [CrossRef]
- Farbehi, N.; Patrick, R.; Dorison, A.; Xaymardan, M.; Janbandhu, V.; Wystub-Lis, K.; Ho, J.W.; Nordon, R.E.; Harvey, R.P. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. Elife 2019, 8, e43882. [Google Scholar] [CrossRef]
- Skelly, D.A.; Squiers, G.T.; McLellan, M.A.; Bolisetty, M.T.; Robson, P.; Rosenthal, N.A.; Pinto, A.R. Single-Cell Transcriptional Profiling Reveals Cellular Diversity and Intercommunication in the Mouse Heart. Cell Rep. 2018, 22, 600–610. [Google Scholar] [CrossRef]
- McLellan, M.A.; Skelly, D.A.; Dona, M.S.I.; Squiers, G.T.; Farrugia, G.E.; Gaynor, T.L.; Cohen, C.D.; Pandey, R.; Diep, H.; Vinh, A.; et al. High-Resolution Transcriptomic Profiling of the Heart During Chronic Stress Reveals Cellular Drivers of Cardiac Fibrosis and Hypertrophy. Circulation 2020, 142, 1448–1463. [Google Scholar] [CrossRef]
- Tucker, N.R.; Chaffin, M.; Fleming, S.J.; Hall, A.W.; Parsons, V.A.; Bedi, K.C., Jr.; Akkad, A.D.; Herndon, C.N.; Arduini, A.; Papangeli, I.; et al. Transcriptional and Cellular Diversity of the Human Heart. Circulation 2020, 142, 466–482. [Google Scholar] [CrossRef] [PubMed]
- Koenig, A.L.; Shchukina, I.; Amrute, J.; Andhey, P.S.; Zaitsev, K.; Lai, L.; Bajpai, G.; Bredemeyer, A.; Smith, G.; Jones, C.; et al. Single-cell transcriptomics reveals cell-type-specific diversification in human heart failure. Nat. Cardiovasc. Res. 2022, 1, 263–280. [Google Scholar] [CrossRef]
- Krstevski, C.; Cohen, C.D.; Dona, M.S.I.; Pinto, A.R. New perspectives of the cardiac cellular landscape: Mapping cellular mediators of cardiac fibrosis using single-cell transcriptomics. Biochem. Soc. Trans. 2020, 48, 2483–2493. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Khalil, H.; Kanisicak, O.; Boyer, J.G.; Vagnozzi, R.J.; Maliken, B.D.; Sargent, M.A.; Prasad, V.; Valiente-Alandi, I.; Blaxall, B.C.; et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J. Clin. Investig. 2018, 128, 2127–2143. [Google Scholar] [CrossRef]
- Pillai, I.C.L.; Li, S.; Romay, M.; Lam, L.; Lu, Y.; Huang, J.; Dillard, N.; Zemanova, M.; Rubbi, L.; Wang, Y.; et al. Cardiac Fibroblasts Adopt Osteogenic Fates and Can Be Targeted to Attenuate Pathological Heart Calcification. Cell Stem Cell 2017, 20, 218–232.e215. [Google Scholar] [CrossRef]
- Linna-Kuosmanen, S.; Schmauch, E.; Galani, K.; Boix, C.A.; Hou, L.; Örd, T.; Toropainen, A.; Stolze, L.K.; Meibalan, E.; Mantero, J.C.; et al. Single-cell dissection of live human hearts in ischemic heart disease and heart failure reveals cell-type-specific driver genes and pathways. BioRxiv 2021. version posted 24 June 2021. [Google Scholar] [CrossRef]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef]
- Kurose, H. Cardiac Fibrosis and Fibroblasts. Cells 2021, 10, 1716. [Google Scholar] [CrossRef]
- Herring, B.P.; Hoggatt, A.M.; Burlak, C.; Offermanns, S. Previously differentiated medial vascular smooth muscle cells contribute to neointima formation following vascular injury. Vasc. Cell 2014, 6, 1–14. [Google Scholar] [CrossRef]
- Grootaert, M.O.J.; Bennett, M.R. Vascular smooth muscle cells in atherosclerosis: Time for a re-assessment. Cardiovasc. Res. 2021, 117, 2326–2339. [Google Scholar] [CrossRef]
- Basatemur, G.L.; Jorgensen, H.F.; Clarke, M.C.H.; Bennett, M.R.; Mallat, Z. Vascular smooth muscle cells in atherosclerosis. Nat. Rev. Cardiol. 2019, 16, 727–744. [Google Scholar] [CrossRef] [PubMed]
- Chappell, J.; Harman, J.L.; Narasimhan, V.M.; Yu, H.; Foote, K.; Simons, B.D.; Bennett, M.R.; Jorgensen, H.F. Extensive Proliferation of a Subset of Differentiated, yet Plastic, Medial Vascular Smooth Muscle Cells Contributes to Neointimal Formation in Mouse Injury and Atherosclerosis Models. Circ. Res. 2016, 119, 1313–1323. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, K.; Lund, M.B.; Shim, J.; Gunnersen, S.; Fuchtbauer, E.M.; Kjolby, M.; Carramolino, L.; Bentzon, J.F. Diverse cellular architecture of atherosclerotic plaque derives from clonal expansion of a few medial SMCs. JCI Insight 2017, 2, e95890. [Google Scholar] [CrossRef] [PubMed]
- Shankman, L.S.; Gomez, D.; Cherepanova, O.A.; Salmon, M.; Alencar, G.F.; Haskins, R.M.; Swiatlowska, P.; Newman, A.A.; Greene, E.S.; Straub, A.C.; et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat. Med. 2015, 21, 628–637. [Google Scholar] [CrossRef]
- Wirka, R.C.; Wagh, D.; Paik, D.T.; Pjanic, M.; Nguyen, T.; Miller, C.L.; Kundu, R.; Nagao, M.; Coller, J.; Koyano, T.K.; et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat. Med. 2019, 25, 1280–1289. [Google Scholar] [CrossRef]
- Herum, K.M.; Romaine, A.; Wang, A.; Melleby, A.O.; Strand, M.E.; Pacheco, J.; Braathen, B.; Duner, P.; Tonnessen, T.; Lunde, I.G.; et al. Syndecan-4 Protects the Heart From the Profibrotic Effects of Thrombin-Cleaved Osteopontin. J. Am. Heart Assoc. 2020, 9, e013518. [Google Scholar] [CrossRef]
- Snider, P.; Hinton, R.B.; Moreno-Rodriguez, R.A.; Wang, J.; Rogers, R.; Lindsley, A.; Li, F.; Ingram, D.A.; Menick, D.; Field, L.; et al. Periostin is required for maturation and extracellular matrix stabilization of noncardiomyocyte lineages of the heart. Circ. Res. 2008, 102, 752–760. [Google Scholar] [CrossRef]
- Alsaigh, T.; Evans, D.; Frankel, D.; Torkamani, A. Decoding the transcriptome of atherosclerotic plaque at single-cell resolution. BioRxiv 2020. version posted 4 March 2020. [Google Scholar] [CrossRef]
- Gilles, G.; McCulloch, A.D.; Brakebusch, C.H.; Herum, K.M. Maintaining resting cardiac fibroblasts in vitro by disrupting mechanotransduction. PLoS ONE 2020, 15, e0241390. [Google Scholar] [CrossRef]
- Jensen, L.F.; Bentzon, J.F.; Albarran-Juarez, J. The Phenotypic Responses of Vascular Smooth Muscle Cells Exposed to Mechanical Cues. Cells 2021, 10, 2209. [Google Scholar] [CrossRef]
- van Putten, S.; Shafieyan, Y.; Hinz, B. Mechanical control of cardiac myofibroblasts. J. Mol. Cell Cardiol. 2016, 93, 133–142. [Google Scholar] [CrossRef]
- Han, M.; Wen, J.K.; Zheng, B.; Cheng, Y.; Zhang, C. Serum deprivation results in redifferentiation of human umbilical vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 2006, 291, C50–C58. [Google Scholar] [CrossRef] [PubMed]
- Hemmer, J.D.; Dean, D.; Vertegel, A.; Langan, E., 3rd; LaBerge, M. Effects of serum deprivation on the mechanical properties of adherent vascular smooth muscle cells. Proc. Inst. Mech. Eng. H 2008, 222, 761–772. [Google Scholar] [CrossRef]
- Vaes, R.D.W.; van den Berk, L.; Boonen, B.; van Dijk, D.P.J.; Olde Damink, S.W.M.; Rensen, S.S. A novel human cell culture model to study visceral smooth muscle phenotypic modulation in health and disease. Am. J. Physiol. Cell Physiol. 2018, 315, C598–C607. [Google Scholar] [CrossRef] [PubMed]
- Christensen, G.; Herum, K.M.; Lunde, I.G. Sweet, yet underappreciated: Proteoglycans and extracellular matrix remodeling in heart disease. Matrix Biol. 2019, 75–76, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Khalil, M.F.; Wagner, W.D.; Goldberg, I.J. Molecular interactions leading to lipoprotein retention and the initiation of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2211–2218. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Zheng, Y.; Gardner, S.E.; Clarke, M.C. Cell death, damage-associated molecular patterns, and sterile inflammation in cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2781–2786. [Google Scholar] [CrossRef]
- Paulus, W.J.; Tschope, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef]
- Paulus, W.J.; Zile, M.R. From Systemic Inflammation to Myocardial Fibrosis: The Heart Failure With Preserved Ejection Fraction Paradigm Revisited. Circ. Res. 2021, 128, 1451–1467. [Google Scholar] [CrossRef] [PubMed]
- Herum, K.M.; Choppe, J.; Kumar, A.; Engler, A.J.; McCulloch, A.D. Mechanical regulation of cardiac fibroblast profibrotic phenotypes. Mol. Biol. Cell 2017, 28, 1871–1882. [Google Scholar] [CrossRef]
- Herum, K.M.; Lunde, I.G.; McCulloch, A.D.; Christensen, G. The Soft- and Hard-Heartedness of Cardiac Fibroblasts: Mechanotransduction Signaling Pathways in Fibrosis of the Heart. J. Clin. Med. 2017, 6, 53. [Google Scholar] [CrossRef] [PubMed]
- Zacchigna, L.; Vecchione, C.; Notte, A.; Cordenonsi, M.; Dupont, S.; Maretto, S.; Cifelli, G.; Ferrari, A.; Maffei, A.; Fabbro, C.; et al. Emilin1 links TGF-beta maturation to blood pressure homeostasis. Cell 2006, 124, 929–942. [Google Scholar] [CrossRef]
- Guo, X.; Wang, X.F. Signaling cross-talk between TGF-beta/BMP and other pathways. Cell Res. 2009, 19, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, F.; Shabaninejad, Z.; Vakili, S.; Derakhshan, M.; Movahedpour, A.; Dabiri, H.; Ghasemi, Y.; Mahjoubin-Tehran, M.; Nikoozadeh, A.; Savardashtaki, A.; et al. TGF-beta and WNT signaling pathways in cardiac fibrosis: Non-coding RNAs come into focus. Cell Commun. Signal. 2020, 18, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Transforming growth factor-beta in myocardial disease. Nat. Rev. Cardiol. 2022, 1–21. [Google Scholar] [CrossRef]
- Grainger, D.J. TGF-beta and atherosclerosis in man. Cardiovasc. Res. 2007, 74, 213–222. [Google Scholar] [CrossRef]
- Sweeney, M.; Corden, B.; Cook, S.A. Targeting cardiac fibrosis in heart failure with preserved ejection fraction: Mirage or miracle? EMBO Mol. Med. 2020, 12, e10865. [Google Scholar] [CrossRef]
- Huang, S.; Chen, B.; Humeres, C.; Alex, L.; Hanna, A.; Frangogiannis, N.G. The role of Smad2 and Smad3 in regulating homeostatic functions of fibroblasts in vitro and in adult mice. Biochim. Biophys. Acta. Mol. Cell Res. 2020, 1867, 118703. [Google Scholar] [CrossRef]
- Huang, S.; Chen, B.; Su, Y.; Alex, L.; Humeres, C.; Shinde, A.V.; Conway, S.J.; Frangogiannis, N.G. Distinct roles of myofibroblast-specific Smad2 and Smad3 signaling in repair and remodeling of the infarcted heart. J. Mol. Cell Cardiol. 2019, 132, 84–97. [Google Scholar] [CrossRef]
- Khalil, H.; Kanisicak, O.; Prasad, V.; Correll, R.N.; Fu, X.; Schips, T.; Vagnozzi, R.J.; Liu, R.; Huynh, T.; Lee, S.J.; et al. Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 2017, 127, 3770–3783. [Google Scholar] [CrossRef]
- Zhao, J.; Shi, W.; Wang, Y.L.; Chen, H.; Bringas, P., Jr.; Datto, M.B.; Frederick, J.P.; Wang, X.F.; Warburton, D. Smad3 deficiency attenuates bleomycin-induced pulmonary fibrosis in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L585–L593. [Google Scholar] [CrossRef] [PubMed]
- Flanders, K.C.; Sullivan, C.D.; Fujii, M.; Sowers, A.; Anzano, M.A.; Arabshahi, A.; Major, C.; Deng, C.; Russo, A.; Mitchell, J.B.; et al. Mice lacking Smad3 are protected against cutaneous injury induced by ionizing radiation. Am. J. Pathol. 2002, 160, 1057–1068. [Google Scholar] [CrossRef]
- Sato, M.; Muragaki, Y.; Saika, S.; Roberts, A.B.; Ooshima, A. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J. Clin. Investig. 2003, 112, 1486–1494. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Xiao, H.; Luo, H.; Chen, Y.; Zhang, Y.; Tao, L.; Jiang, Y.; Chen, Y.; Shen, X. Inhibitory effects of oxymatrine on TGFbeta1induced proliferation and abnormal differentiation in rat cardiac fibroblasts via the p38MAPK and ERK1/2 signaling pathways. Mol. Med. Rep. 2017, 16, 5354–5362. [Google Scholar] [CrossRef] [PubMed]
- Villarreal, F.J.; Dillmann, W.H. Cardiac hypertrophy-induced changes in mRNA levels for TGF-beta 1, fibronectin, and collagen. Am. J. Physiol. 1992, 262, H1861–H1866. [Google Scholar] [CrossRef]
- Small, E.M.; Thatcher, J.E.; Sutherland, L.B.; Kinoshita, H.; Gerard, R.D.; Richardson, J.A.; Dimaio, J.M.; Sadek, H.; Kuwahara, K.; Olson, E.N. Myocardin-related transcription factor-a controls myofibroblast activation and fibrosis in response to myocardial infarction. Circ. Res. 2010, 107, 294–304. [Google Scholar] [CrossRef]
- Santos, G.L.; Hartmann, S.; Zimmermann, W.H.; Ridley, A.; Lutz, S. Inhibition of Rho-associated kinases suppresses cardiac myofibroblast function in engineered connective and heart muscle tissues. J. Mol. Cell. Cardiol. 2019, 134, 13–28. [Google Scholar] [CrossRef]
- Akhmetshina, A.; Dees, C.; Pileckyte, M.; Szucs, G.; Spriewald, B.M.; Zwerina, J.; Distler, O.; Schett, G.; Distler, J.H. Rho-associated kinases are crucial for myofibroblast differentiation and production of extracellular matrix in scleroderma fibroblasts. Arthritis Rheum. 2008, 58, 2553–2564. [Google Scholar] [CrossRef]
- Small, E.M. The actin-MRTF-SRF gene regulatory axis and myofibroblast differentiation. J. Cardiovasc. Transl. Res. 2012, 5, 794–804. [Google Scholar] [CrossRef] [PubMed]
- Olson, E.N.; Nordheim, A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat. Rev. Mol. Cell Biol. 2010, 11, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Sladojevic, N.; Blair, J.E.; Liao, J.K. Targeting Rho-associated coiled-coil forming protein kinase (ROCK) in cardiovascular fibrosis and stiffening. Expert Opin. Ther. Targets 2020, 24, 47–62. [Google Scholar] [CrossRef]
- Mack, C.P. Signaling mechanisms that regulate smooth muscle cell differentiation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1495–1505. [Google Scholar] [CrossRef] [PubMed]
- Bobik, A. Transforming growth factor-betas and vascular disorders. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1712–1720. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.Y.; Qin, L.; Li, G.; Malagon-Lopez, J.; Wang, Z.; Bergaya, S.; Gujja, S.; Caulk, A.W.; Murtada, S.I.; Zhang, X.; et al. Smooth Muscle Cell Reprogramming in Aortic Aneurysms. Cell Stem Cell 2020, 26, 542–557. [Google Scholar] [CrossRef] [PubMed]
- Kawai-Kowase, K.; Ohshima, T.; Matsui, H.; Tanaka, T.; Shimizu, T.; Iso, T.; Arai, M.; Owens, G.K.; Kurabayashi, M. PIAS1 mediates TGFbeta-induced SM alpha-actin gene expression through inhibition of KLF4 function-expression by protein sumoylation. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 99–106. [Google Scholar] [CrossRef]
- Liu, Y.; Sinha, S.; McDonald, O.G.; Shang, Y.; Hoofnagle, M.H.; Owens, G.K. Kruppel-like factor 4 abrogates myocardin-induced activation of smooth muscle gene expression. J. Biol. Chem. 2005, 280, 9719–9727. [Google Scholar] [CrossRef]
- Hocevar, B.A.; Prunier, C.; Howe, P.H. Disabled-2 (Dab2) mediates transforming growth factor beta (TGFbeta)-stimulated fibronectin synthesis through TGFbeta-activated kinase 1 and activation of the JNK pathway. J. Biol. Chem. 2005, 280, 25920–25927. [Google Scholar] [CrossRef]
- Li, H.X.; Han, M.; Bernier, M.; Zheng, B.; Sun, S.G.; Su, M.; Zhang, R.; Fu, J.R.; Wen, J.K. Kruppel-like factor 4 promotes differentiation by transforming growth factor-beta receptor-mediated Smad and p38 MAPK signaling in vascular smooth muscle cells. J. Biol. Chem. 2010, 285, 17846–17856. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Liu, Y.; Wang, N.; Qi, Y.; Du, J. Kruppel-like factor 4 transcriptionally regulates TGF-beta1 and contributes to cardiac myofibroblast differentiation. PLoS ONE 2013, 8, e63424. [Google Scholar] [CrossRef]
- Zheng, B.; Han, M.; Wen, J.K. Role of Kruppel-like factor 4 in phenotypic switching and proliferation of vascular smooth muscle cells. IUBMB Life 2010, 62, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, M.W.; Watanabe, M.; Lebedeva, M.A.; Depina, A.S.; Hanai, J.; Mammoto, T.; Frederick, J.P.; Wang, X.F.; Sukhatme, V.P.; Jain, M.K. Transforming growth factor-beta1 inhibition of vascular smooth muscle cell activation is mediated via Smad3. J. Biol. Chem. 2004, 279, 16388–16393. [Google Scholar] [CrossRef] [PubMed]
- Vander Ark, A.; Cao, J.; Li, X. TGF-beta receptors: In and beyond TGF-beta signaling. Cell Signal. 2018, 52, 112–120. [Google Scholar] [CrossRef]
- Liu, C.; Li, J.; Xiang, X.; Guo, L.; Tu, K.; Liu, Q.; Shah, V.H.; Kang, N. PDGF receptor-alpha promotes TGF-beta signaling in hepatic stellate cells via transcriptional and posttranscriptional regulation of TGF-beta receptors. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G749–G759. [Google Scholar] [CrossRef]
- McCaffrey, T.A.; Consigli, S.; Du, B.; Falcone, D.J.; Sanborn, T.A.; Spokojny, A.M.; Bush, H.L., Jr. Decreased type II/type I TGF-beta receptor ratio in cells derived from human atherosclerotic lesions. Conversion from an antiproliferative to profibrotic response to TGF-beta1. J. Clin. Investig. 1995, 96, 2667–2675. [Google Scholar] [CrossRef]
- Mallat, Z.; Gojova, A.; Marchiol-Fournigault, C.; Esposito, B.; Kamate, C.; Merval, R.; Fradelizi, D.; Tedgui, A. Inhibition of transforming growth factor-beta signaling accelerates atherosclerosis and induces an unstable plaque phenotype in mice. Circ. Res. 2001, 89, 930–934. [Google Scholar] [CrossRef]
- Cipollone, F.; Fazia, M.; Mincione, G.; Iezzi, A.; Pini, B.; Cuccurullo, C.; Ucchino, S.; Spigonardo, F.; Di Nisio, M.; Cuccurullo, F.; et al. Increased expression of transforming growth factor-beta1 as a stabilizing factor in human atherosclerotic plaques. Stroke 2004, 35, 2253–2257. [Google Scholar] [CrossRef]
- Kalinina, N.; Agrotis, A.; Antropova, Y.; Ilyinskaya, O.; Smirnov, V.; Tararak, E.; Bobik, A. Smad expression in human atherosclerotic lesions: Evidence for impaired TGF-beta/Smad signaling in smooth muscle cells of fibrofatty lesions. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1391–1396. [Google Scholar] [CrossRef]
- Mehta, P.K.; Griendling, K.K. Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 2007, 292, C82–C97. [Google Scholar] [CrossRef]
- Santos, R.A.S.; Oudit, G.Y.; Verano-Braga, T.; Canta, G.; Steckelings, U.M.; Bader, M. The renin-angiotensin system: Going beyond the classical paradigms. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H958–H970. [Google Scholar] [CrossRef]
- Schorb, W.; Booz, G.W.; Dostal, D.E.; Conrad, K.M.; Chang, K.C.; Baker, K.M. Angiotensin II is mitogenic in neonatal rat cardiac fibroblasts. Circ. Res. 1993, 72, 1245–1254. [Google Scholar] [CrossRef] [PubMed]
- Brilla, C.G.; Zhou, G.; Matsubara, L.; Weber, K.T. Collagen metabolism in cultured adult rat cardiac fibroblasts: Response to angiotensin II and aldosterone. J. Mol. Cell. Cardiol. 1994, 26, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Siddesha, J.M.; Valente, A.J.; Sakamuri, S.S.; Yoshida, T.; Gardner, J.D.; Somanna, N.; Takahashi, C.; Noda, M.; Chandrasekar, B. Angiotensin II stimulates cardiac fibroblast migration via the differential regulation of matrixins and RECK. J. Mol. Cell. Cardiol. 2013, 65, 9–18. [Google Scholar] [CrossRef]
- Houser, S.R.; Margulies, K.B.; Murphy, A.M.; Spinale, F.G.; Francis, G.S.; Prabhu, S.D.; Rockman, H.A.; Kass, D.A.; Molkentin, J.D.; Sussman, M.A.; et al. Animal models of heart failure: A scientific statement from the American Heart Association. Circ. Res. 2012, 111, 131–150. [Google Scholar] [CrossRef] [PubMed]
- van Kesteren, C.A.; van Heugten, H.A.; Lamers, J.M.; Saxena, P.R.; Schalekamp, M.A.; Danser, A.H. Angiotensin II-mediated growth and antigrowth effects in cultured neonatal rat cardiac myocytes and fibroblasts. J. Mol. Cell. Cardiol. 1997, 29, 2147–2157. [Google Scholar] [CrossRef]
- Sharma, H.S.; van Heugten, H.A.; Goedbloed, M.A.; Verdouw, P.D.; Lamers, J.M. Angiotensin II induced expression of transcription factors precedes increase in transforming growth factor-beta 1 mRNA in neonatal cardiac fibroblasts. Biochem. Biophys. Res. Commun. 1994, 205, 105–112. [Google Scholar] [CrossRef]
- Bouzegrhane, F.; Thibault, G. Is angiotensin II a proliferative factor of cardiac fibroblasts? Cardiovasc. Res. 2002, 53, 304–312. [Google Scholar] [CrossRef]
- Moriguchi, Y.; Matsubara, H.; Mori, Y.; Murasawa, S.; Masaki, H.; Maruyama, K.; Tsutsumi, Y.; Shibasaki, Y.; Tanaka, Y.; Nakajima, T.; et al. Angiotensin II-induced transactivation of epidermal growth factor receptor regulates fibronectin and transforming growth factor-beta synthesis via transcriptional and posttranscriptional mechanisms. Circ. Res. 1999, 84, 1073–1084. [Google Scholar] [CrossRef]
- Rosenkranz, S. TGF-beta1 and angiotensin networking in cardiac remodeling. Cardiovasc. Res. 2004, 63, 423–432. [Google Scholar] [CrossRef]
- Campbell, S.E.; Katwa, L.C. Angiotensin II stimulated expression of transforming growth factor-beta1 in cardiac fibroblasts and myofibroblasts. J. Mol. Cell. Cardiol. 1997, 29, 1947–1958. [Google Scholar] [CrossRef] [PubMed]
- Gurantz, D.; Cowling, R.T.; Varki, N.; Frikovsky, E.; Moore, C.D.; Greenberg, B.H. IL-1beta and TNF-alpha upregulate angiotensin II type 1 (AT1) receptors on cardiac fibroblasts and are associated with increased AT1 density in the post-MI heart. J. Mol. Cell. Cardiol. 2005, 38, 505–515. [Google Scholar] [CrossRef]
- Griendling, K.K.; Ushio-Fukai, M.; Lassegue, B.; Alexander, R.W. Angiotensin II signaling in vascular smooth muscle. New concepts. Hypertension 1997, 29, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Ott, K.M.; Kagiyama, S.; Phillips, M.I. The multiple actions of angiotensin II in atherosclerosis. Regul. Pept. 2000, 93, 65–77. [Google Scholar] [CrossRef]
- Geisterfer, A.A.; Peach, M.J.; Owens, G.K. Angiotensin II induces hypertrophy, not hyperplasia, of cultured rat aortic smooth muscle cells. Circ. Res. 1988, 62, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, M.A.; Myers, P.R. Nitric oxide modulates basal and endothelin-induced coronary artery vascular smooth muscle cell proliferation and collagen levels. J. Mol. Cell. Cardiol. 1997, 29, 1779–1789. [Google Scholar] [CrossRef]
- Xi, X.P.; Graf, K.; Goetze, S.; Fleck, E.; Hsueh, W.A.; Law, R.E. Central role of the MAPK pathway in ang II-mediated DNA synthesis and migration in rat vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 73–82. [Google Scholar] [CrossRef]
- Zhang, F.; Ren, X.; Zhao, M.; Zhou, B.; Han, Y. Angiotensin-(1–7) abrogates angiotensin II-induced proliferation, migration and inflammation in VSMCs through inactivation of ROS-mediated PI3K/Akt and MAPK/ERK signaling pathways. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef]
- Wynne, B.M.; Chiao, C.W.; Webb, R.C. Vascular Smooth Muscle Cell Signaling Mechanisms for Contraction to Angiotensin II and Endothelin-1. J. Am. Soc. Hypertens. 2009, 3, 84–95. [Google Scholar] [CrossRef]
- Rodriguez-Vita, J.; Sanchez-Lopez, E.; Esteban, V.; Ruperez, M.; Egido, J.; Ruiz-Ortega, M. Angiotensin II activates the Smad pathway in vascular smooth muscle cells by a transforming growth factor-beta-independent mechanism. Circulation 2005, 111, 2509–2517. [Google Scholar] [CrossRef]
- Ruiz-Ortega, M.; Lorenzo, O.; Ruperez, M.; Suzuki, Y.; Egido, J. Angiotensin II activates nuclear transcription factor-kappaB in aorta of normal rats and in vascular smooth muscle cells of AT1 knockout mice. Nephrol. Dial. Transplant. 2001, 16 (Suppl. S1), 27–33. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ortega, M.; Lorenzo, O.; Ruperez, M.; Konig, S.; Wittig, B.; Egido, J. Angiotensin II activates nuclear transcription factor kappaB through AT(1) and AT(2) in vascular smooth muscle cells: Molecular mechanisms. Circ. Res. 2000, 86, 1266–1272. [Google Scholar] [CrossRef] [PubMed]
- St Paul, A.; Corbett, C.B.; Okune, R.; Autieri, M.V. Angiotensin II, Hypercholesterolemia, and Vascular Smooth Muscle Cells: A Perfect Trio for Vascular Pathology. Int. J. Mol. Sci. 2020, 21, 4525. [Google Scholar] [CrossRef]
- Schieffer, B.; Schieffer, E.; Hilfiker-Kleiner, D.; Hilfiker, A.; Kovanen, P.T.; Kaartinen, M.; Nussberger, J.; Harringer, W.; Drexler, H. Expression of angiotensin II and interleukin 6 in human coronary atherosclerotic plaques: Potential implications for inflammation and plaque instability. Circulation 2000, 101, 1372–1378. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.W.; Yang, L.X.; Wang, H.; Liu, B.; Wang, L. Angiotensin II induces matrix metalloproteinase-9 expression via a nuclear factor-kappaB-dependent pathway in vascular smooth muscle cells. Regul. Pept. 2008, 147, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Mazzolai, L.; Duchosal, M.A.; Korber, M.; Bouzourene, K.; Aubert, J.F.; Hao, H.; Vallet, V.; Brunner, H.R.; Nussberger, J.; Gabbiani, G.; et al. Endogenous angiotensin II induces atherosclerotic plaque vulnerability and elicits a Th1 response in ApoE−/− mice. Hypertension 2004, 44, 277–282. [Google Scholar] [CrossRef]
- Yoon, S.; Eom, G.H. Heart failure with preserved ejection fraction: Present status and future directions. Exp. Mol. Med. 2019, 51, 1–9. [Google Scholar] [CrossRef]
- Duan, J.; Gherghe, C.; Liu, D.; Hamlett, E.; Srikantha, L.; Rodgers, L.; Regan, J.N.; Rojas, M.; Willis, M.; Leask, A.; et al. Wnt1/betacatenin injury response activates the epicardium and cardiac fibroblasts to promote cardiac repair. EMBO J. 2012, 31, 429–442. [Google Scholar] [CrossRef]
- Weerackoon, N.; Gunawardhana, K.L.; Mani, A. Wnt Signaling Cascades and Their Role in Coronary Artery Health and Disease. J. Cell. Signal. 2021, 2, 52–62. [Google Scholar] [CrossRef]
- Clevers, H.; Nusse, R. Wnt/beta-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef]
- Zeng, X.; Tamai, K.; Doble, B.; Li, S.; Huang, H.; Habas, R.; Okamura, H.; Woodgett, J.; He, X. A dual-kinase mechanism for Wnt co-receptor phosphorylation and activation. Nature 2005, 438, 873–877. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, Y.; Semenov, M.; Han, C.; Baeg, G.H.; Tan, Y.; Zhang, Z.; Lin, X.; He, X. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 2002, 108, 837–847. [Google Scholar] [CrossRef]
- Schlessinger, K.; Hall, A.; Tolwinski, N. Wnt signaling pathways meet Rho GTPases. Genes. Dev. 2009, 23, 265–277. [Google Scholar] [CrossRef] [PubMed]
- De, A. Wnt/Ca2+ signaling pathway: A brief overview. Acta. Biochim. Biophys. Sin. (Shanghai) 2011, 43, 745–756. [Google Scholar] [CrossRef]
- Wamhoff, B.R.; Bowles, D.K.; Owens, G.K. Excitation-transcription coupling in arterial smooth muscle. Circ. Res. 2006, 98, 868–878. [Google Scholar] [CrossRef] [PubMed]
- Cruciat, C.M.; Niehrs, C. Secreted and transmembrane wnt inhibitors and activators. Cold Spring Harb. Perspect. Biol. 2013, 5, a015081. [Google Scholar] [CrossRef]
- Baetta, R.; Banfi, C. Dkk (Dickkopf) Proteins. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1330–1342. [Google Scholar] [CrossRef]
- Das, D.S.; Wadhwa, N.; Kunj, N.; Sarda, K.; Pradhan, B.S.; Majumdar, S.S. Dickkopf homolog 3 (DKK3) plays a crucial role upstream of WNT/beta-CATENIN signaling for Sertoli cell mediated regulation of spermatogenesis. PLoS ONE 2013, 8, e63603. [Google Scholar] [CrossRef]
- Blyszczuk, P.; Muller-Edenborn, B.; Valenta, T.; Osto, E.; Stellato, M.; Behnke, S.; Glatz, K.; Basler, K.; Luscher, T.F.; Distler, O.; et al. Transforming growth factor-beta-dependent Wnt secretion controls myofibroblast formation and myocardial fibrosis progression in experimental autoimmune myocarditis. Eur. Heart J. 2017, 38, 1413–1425. [Google Scholar] [CrossRef]
- Akhmetshina, A.; Palumbo, K.; Dees, C.; Bergmann, C.; Venalis, P.; Zerr, P.; Horn, A.; Kireva, T.; Beyer, C.; Zwerina, J.; et al. Activation of canonical Wnt signalling is required for TGF-beta-mediated fibrosis. Nat. Commun. 2012, 3, 1–12. [Google Scholar] [CrossRef]
- Carthy, J.M.; Garmaroudi, F.S.; Luo, Z.; McManus, B.M. Wnt3a induces myofibroblast differentiation by upregulating TGF-beta signaling through SMAD2 in a beta-catenin-dependent manner. PLoS ONE 2011, 6, e19809. [Google Scholar] [CrossRef] [PubMed]
- Shafer, S.L.; Towler, D.A. Transcriptional regulation of SM22alpha by Wnt3a: Convergence with TGFbeta(1)/Smad signaling at a novel regulatory element. J. Mol. Cell. Cardiol. 2009, 46, 621–635. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Zhu, X.H.; Zhang, X.D.; Jiang, D.S.; Bian, Z.Y.; Zhang, X.F.; Chen, K.; Wei, X.; Gao, L.; et al. Dickkopf-3 attenuates pressure overload-induced cardiac remodelling. Cardiovasc. Res. 2014, 102, 35–45. [Google Scholar] [CrossRef]
- Zhai, C.G.; Xu, Y.Y.; Tie, Y.Y.; Zhang, Y.; Chen, W.Q.; Ji, X.P.; Mao, Y.; Qiao, L.; Cheng, J.; Xu, Q.B.; et al. DKK3 overexpression attenuates cardiac hypertrophy and fibrosis in an angiotensin-perfused animal model by regulating the ADAM17/ACE2 and GSK-3beta/beta-catenin pathways. J. Mol. Cell. Cardiol. 2018, 114, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Bao, M.W.; Cai, Z.; Zhang, X.J.; Li, L.; Liu, X.; Wan, N.; Hu, G.; Wan, F.; Zhang, R.; Zhu, X.; et al. Dickkopf-3 protects against cardiac dysfunction and ventricular remodelling following myocardial infarction. Basic. Res. Cardiol. 2015, 110, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Bao, D.; Dong, W.; Liu, N.; Zhang, X.; Gao, S.; Ge, W.; Gao, X.; Zhang, L. Dkk3 prevents familial dilated cardiomyopathy development through Wnt pathway. Lab. Investig. 2016, 96, 239–248. [Google Scholar] [CrossRef]
- Rakipovski, G.; Rolin, B.; Barascuk, N.; Lund, H.E.; Bjorn Bonde, M.F.; Djordjevic, D.; Wulff-Larsen, P.G.; Petersen, M.; Kirk, R.K.; Hultman, K.; et al. A neutralizing antibody against DKK1 does not reduce plaque formation in classical murine models of atherosclerosis: Is the therapeutic potential lost in translation? Atherosclerosis 2020, 314, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Di, M.; Wang, L.; Li, M.; Zhang, Y.; Liu, X.; Zeng, R.; Wang, H.; Chen, Y.; Chen, W.; Zhang, Y.; et al. Dickkopf1 destabilizes atherosclerotic plaques and promotes plaque formation by inducing apoptosis of endothelial cells through activation of ER stress. Cell Death Dis. 2017, 8, e2917. [Google Scholar] [CrossRef]
- Baylis, R.A.; Gomez, D.; Owens, G.K. Shifting the Focus of Preclinical, Murine Atherosclerosis Studies From Prevention to Late-Stage Intervention. Circ. Res. 2017, 120, 775–777. [Google Scholar] [CrossRef]
- Lee, Y.T.; Lin, H.Y.; Chan, Y.W.; Li, K.H.; To, O.T.; Yan, B.P.; Liu, T.; Li, G.; Wong, W.T.; Keung, W.; et al. Mouse models of atherosclerosis: A historical perspective and recent advances. Lipids Health Dis. 2017, 16, 1–11. [Google Scholar] [CrossRef]
- Yu, B.; Kiechl, S.; Qi, D.; Wang, X.; Song, Y.; Weger, S.; Mayr, A.; Le Bras, A.; Karamariti, E.; Zhang, Z.; et al. A Cytokine-Like Protein Dickkopf-Related Protein 3 Is Atheroprotective. Circulation 2017, 136, 1022–1036. [Google Scholar] [CrossRef]
- Cheng, W.L.; Yang, Y.; Zhang, X.J.; Guo, J.; Gong, J.; Gong, F.H.; She, Z.G.; Huang, Z.; Xia, H.; Li, H. Dickkopf-3 Ablation Attenuates the Development of Atherosclerosis in ApoE-Deficient Mice. J. Am. Heart Assoc. 2017, 6, e004690. [Google Scholar] [CrossRef] [PubMed]
- Karamariti, E.; Zhai, C.; Yu, B.; Qiao, L.; Wang, Z.; Potter, C.M.F.; Wong, M.M.; Simpson, R.M.L.; Zhang, Z.; Wang, X.; et al. DKK3 (Dickkopf 3) Alters Atherosclerotic Plaque Phenotype Involving Vascular Progenitor and Fibroblast Differentiation Into Smooth Muscle Cells. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 425–437. [Google Scholar] [CrossRef]
- Schunk, S.J.; Speer, T.; Petrakis, I.; Fliser, D. Dickkopf 3-a novel biomarker of the ‘kidney injury continuum’. Nephrol. Dial. Transplant. 2021, 36, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Federico, G.; Meister, M.; Mathow, D.; Heine, G.H.; Moldenhauer, G.; Popovic, Z.V.; Nordstrom, V.; Kopp-Schneider, A.; Hielscher, T.; Nelson, P.J.; et al. Tubular Dickkopf-3 promotes the development of renal atrophy and fibrosis. JCI Insight 2016, 1, e84916. [Google Scholar] [CrossRef] [PubMed]
- Ponten, A.; Folestad, E.B.; Pietras, K.; Eriksson, U. Platelet-derived growth factor D induces cardiac fibrosis and proliferation of vascular smooth muscle cells in heart-specific transgenic mice. Circ. Res. 2005, 97, 1036–1045. [Google Scholar] [CrossRef] [PubMed]
- Guerit, E.; Arts, F.; Dachy, G.; Boulouadnine, B.; Demoulin, J.B. PDGF receptor mutations in human diseases. Cell Mol. Life Sci. 2021, 78, 3867–3881. [Google Scholar] [CrossRef]
- Zymek, P.; Bujak, M.; Chatila, K.; Cieslak, A.; Thakker, G.; Entman, M.L.; Frangogiannis, N.G. The role of platelet-derived growth factor signaling in healing myocardial infarcts. J. Am. Coll. Cardiol. 2006, 48, 2315–2323. [Google Scholar] [CrossRef]
- Andrae, J.; Gallini, R.; Betsholtz, C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008, 22, 1276–1312. [Google Scholar] [CrossRef]
- Lindahl, P.; Johansson, B.R.; Leveen, P.; Betsholtz, C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science 1997, 277, 242–245. [Google Scholar] [CrossRef]
- Soriano, P. Abnormal kidney development and hematological disorders in PDGF beta-receptor mutant mice. Genes Dev. 1994, 8, 1888–1896. [Google Scholar] [CrossRef]
- Ivey, M.J.; Kuwabara, J.T.; Riggsbee, K.L.; Tallquist, M.D. Platelet-derived growth factor receptor-alpha is essential for cardiac fibroblast survival. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H330–H344. [Google Scholar] [CrossRef]
- Bergsten, E.; Uutela, M.; Li, X.; Pietras, K.; Ostman, A.; Heldin, C.H.; Alitalo, K.; Eriksson, U. PDGF-D is a specific, protease-activated ligand for the PDGF beta-receptor. Nat. Cell Biol. 2001, 3, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Gladh, H.; Folestad, E.B.; Muhl, L.; Ehnman, M.; Tannenberg, P.; Lawrence, A.L.; Betsholtz, C.; Eriksson, U. Mice Lacking Platelet-Derived Growth Factor D Display a Mild Vascular Phenotype. PLoS ONE 2016, 11, e0152276. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yue, Y.; Yang, X.; Fan, T.; Mei, B.; Hou, J.; Liang, M.; Chen, G.; Wu, Z. Platelet Derived Growth Factor Alpha (PDGFRalpha) Induces the Activation of Cardiac Fibroblasts by Activating c-Kit. Med. Sci. Monit. 2017, 23, 3808–3816. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Zhao, W.; Chen, Y.; Li, V.S.; Meng, W.; Sun, Y. Platelet-derived growth factor-D promotes fibrogenesis of cardiac fibroblasts. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1719–H1726. [Google Scholar] [CrossRef]
- Ponten, A.; Li, X.; Thoren, P.; Aase, K.; Sjoblom, T.; Ostman, A.; Eriksson, U. Transgenic overexpression of platelet-derived growth factor-C in the mouse heart induces cardiac fibrosis, hypertrophy, and dilated cardiomyopathy. Am. J. Pathol. 2003, 163, 673–682. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, D.Z.; Hockemeyer, D.; McAnally, J.; Nordheim, A.; Olson, E.N. Myocardin and ternary complex factors compete for SRF to control smooth muscle gene expression. Nature 2004, 428, 185–189. [Google Scholar] [CrossRef]
- Deaton, R.A.; Gan, Q.; Owens, G.K. Sp1-dependent activation of KLF4 is required for PDGF-BB-induced phenotypic modulation of smooth muscle. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1027–H1037. [Google Scholar] [CrossRef]
- Wong, D.; Turner, A.W.; Miller, C.L. Genetic Insights Into Smooth Muscle Cell Contributions to Coronary Artery Disease. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1006–1017. [Google Scholar] [CrossRef]
- Tsaousi, A.; Mill, C.; George, S.J. The Wnt pathways in vascular disease: Lessons from vascular development. Curr. Opin. Lipidol. 2011, 22, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.P.; Kolodgie, F.D.; Farb, A.; Weber, D.K.; Malcom, G.T.; Smialek, J.; Virmani, R. Healed plaque ruptures and sudden coronary death: Evidence that subclinical rupture has a role in plaque progression. Circulation 2001, 103, 934–940. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, N.; Kurata, M.; Yamamoto, T.; Morikawa, S.; Masumoto, J. The role of interleukin-1 in general pathology. Inflamm. Regen. 2019, 39, 1–16. [Google Scholar] [CrossRef]
- Daseke, M.J., 2nd; Chalise, U.; Becirovic-Agic, M.; Salomon, J.D.; Cook, L.M.; Case, A.J.; Lindsey, M.L. Neutrophil signaling during myocardial infarction wound repair. Cell. Signal. 2021, 77, 109816. [Google Scholar] [CrossRef]
- Libby, P. Interleukin-1 Beta as a Target for Atherosclerosis Therapy: Biological Basis of CANTOS and Beyond. J. Am. Coll. Cardiol. 2017, 70, 2278–2289. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Y.Y.; Li, T.T.; Wang, J.; Jiang, Y.; Zhao, Y.; Jin, X.X.; Xue, G.L.; Yang, Y.; Zhang, X.F.; et al. Ablation of interleukin-17 alleviated cardiac interstitial fibrosis and improved cardiac function via inhibiting long non-coding RNA-AK081284 in diabetic mice. J. Mol. Cell. Cardiol. 2018, 115, 64–72. [Google Scholar] [CrossRef]
- Schafer, S.; Viswanathan, S.; Widjaja, A.A.; Lim, W.W.; Moreno-Moral, A.; DeLaughter, D.M.; Ng, B.; Patone, G.; Chow, K.; Khin, E.; et al. IL-11 is a crucial determinant of cardiovascular fibrosis. Nature 2017, 552, 110–115. [Google Scholar] [CrossRef]
- Turner, N.A. Effects of interleukin-1 on cardiac fibroblast function: Relevance to post-myocardial infarction remodelling. Vascul. Pharmacol. 2014, 60, 1–7. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Interleukin-1 in cardiac injury, repair, and remodeling: Pathophysiologic and translational concepts. Discoveries (Craiova) 2015, 3, e41. [Google Scholar] [CrossRef]
- Lugrin, J.; Parapanov, R.; Rosenblatt-Velin, N.; Rignault-Clerc, S.; Feihl, F.; Waeber, B.; Muller, O.; Vergely, C.; Zeller, M.; Tardivel, A.; et al. Cutting edge: IL-1alpha is a crucial danger signal triggering acute myocardial inflammation during myocardial infarction. J. Immunol. 2015, 194, 499–503. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, F.; Wang, Y.; Gistera, A.; Roy, J.; Paulsson-Berne, G.; Hedin, U.; Lerman, A.; Hansson, G.K.; Herrmann, J.; et al. Inflammasome-Driven Interleukin-1alpha and Interleukin-1beta Production in Atherosclerotic Plaques Relates to Hyperlipidemia and Plaque Complexity. JACC Basic Transl. Sci. 2019, 4, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Martin-Sanchez, F.; Diamond, C.; Zeitler, M.; Gomez, A.I.; Baroja-Mazo, A.; Bagnall, J.; Spiller, D.; White, M.; Daniels, M.J.; Mortellaro, A.; et al. Inflammasome-dependent IL-1beta release depends upon membrane permeabilisation. Cell Death Differ. 2016, 23, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, M.D.; Laird, R.E.; Brown, R.D.; Long, C.S. IL-1beta stimulates rat cardiac fibroblast migration via MAP kinase pathways. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1139–H1147. [Google Scholar] [CrossRef] [PubMed]
- Bronnum, H.; Eskildsen, T.; Andersen, D.C.; Schneider, M.; Sheikh, S.P. IL-1beta suppresses TGF-beta-mediated myofibroblast differentiation in cardiac fibroblasts. Growth Factors 2013, 31, 81–89. [Google Scholar] [CrossRef]
- Bageghni, S.A.; Hemmings, K.E.; Yuldasheva, N.Y.; Maqbool, A.; Gamboa-Esteves, F.O.; Humphreys, N.E.; Jackson, M.S.; Denton, C.P.; Francis, S.; Porter, K.E.; et al. Fibroblast-specific deletion of interleukin-1 receptor-1 reduces adverse cardiac remodeling following myocardial infarction. JCI Insight 2019, 5. [Google Scholar] [CrossRef]
- Eun, S.Y.; Ko, Y.S.; Park, S.W.; Chang, K.C.; Kim, H.J. IL-1beta enhances vascular smooth muscle cell proliferation and migration via P2Y2 receptor-mediated RAGE expression and HMGB1 release. Vascul. Pharmacol. 2015, 72, 108–117. [Google Scholar] [CrossRef]
- Schultz, K.; Murthy, V.; Tatro, J.B.; Beasley, D. Endogenous interleukin-1 alpha promotes a proliferative and proinflammatory phenotype in human vascular smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2927–H2934. [Google Scholar] [CrossRef]
- Clarke, M.C.; Talib, S.; Figg, N.L.; Bennett, M.R. Vascular smooth muscle cell apoptosis induces interleukin-1-directed inflammation: Effects of hyperlipidemia-mediated inhibition of phagocytosis. Circ. Res. 2010, 106, 363–372. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Everett, B.M.; Cornel, J.H.; Lainscak, M.; Anker, S.D.; Abbate, A.; Thuren, T.; Libby, P.; Glynn, R.J.; Ridker, P.M. Anti-Inflammatory Therapy With Canakinumab for the Prevention of Hospitalization for Heart Failure. Circulation 2019, 139, 1289–1299. [Google Scholar] [CrossRef]
- Chia, Y.C.; Kieneker, L.M.; van Hassel, G.; Binnenmars, S.H.; Nolte, I.M.; van Zanden, J.J.; van der Meer, P.; Navis, G.; Voors, A.A.; Bakker, S.J.L.; et al. Interleukin 6 and Development of Heart Failure With Preserved Ejection Fraction in the General Population. J. Am. Heart Assoc. 2021, 10, e018549. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Devalaraja, M.; Baeres, F.M.M.; Engelmann, M.D.M.; Hovingh, G.K.; Ivkovic, M.; Lo, L.; Kling, D.; Pergola, P.; Raj, D.; et al. IL-6 inhibition with ziltivekimab in patients at high atherosclerotic risk (RESCUE): A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2021, 397, 2060–2069. [Google Scholar] [CrossRef]
- Villar-Fincheira, P.; Sanhueza-Olivares, F.; Norambuena-Soto, I.; Cancino-Arenas, N.; Hernandez-Vargas, F.; Troncoso, R.; Gabrielli, L.; Chiong, M. Role of Interleukin-6 in Vascular Health and Disease. Front. Mol. Biosci. 2021, 8, 79. [Google Scholar] [CrossRef]
- Roth, M.; Nauck, M.; Tamm, M.; Perruchoud, A.P.; Ziesche, R.; Block, L.H. Intracellular interleukin 6 mediates platelet-derived growth factor-induced proliferation of nontransformed cells. Proc. Natl. Acad. Sci. USA 1995, 92, 1312–1316. [Google Scholar] [CrossRef] [PubMed]
- Loppnow, H.; Libby, P. Proliferating or interleukin 1-activated human vascular smooth muscle cells secrete copious interleukin 6. J. Clin. Investig. 1990, 85, 731–738. [Google Scholar] [CrossRef]
- Zampetaki, A.; Zhang, Z.; Hu, Y.; Xu, Q. Biomechanical stress induces IL-6 expression in smooth muscle cells via Ras/Rac1-p38 MAPK-NF-kappaB signaling pathways. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2946–H2954. [Google Scholar] [CrossRef]
- Liu, Z.; Dronadula, N.; Rao, G.N. A novel role for nuclear factor of activated T cells in receptor tyrosine kinase and G protein-coupled receptor agonist-induced vascular smooth muscle cell motility. J. Biol. Chem. 2004, 279, 41218–41226. [Google Scholar] [CrossRef]
- Elias, J.A.; Lentz, V.; Cummings, P.J. Transforming growth factor-beta regulation of IL-6 production by unstimulated and IL-1-stimulated human fibroblasts. J. Immunol. 1991, 146, 3437–3443. [Google Scholar]
- Muller, J.; Gorressen, S.; Grandoch, M.; Feldmann, K.; Kretschmer, I.; Lehr, S.; Ding, Z.; Schmitt, J.P.; Schrader, J.; Garbers, C.; et al. Interleukin-6-dependent phenotypic modulation of cardiac fibroblasts after acute myocardial infarction. Basic Res. Cardiol. 2014, 109, 1–13. [Google Scholar] [CrossRef]
- Bageghni, S.A.; Hemmings, K.E.; Zava, N.; Denton, C.P.; Porter, K.E.; Ainscough, J.F.X.; Drinkhill, M.J.; Turner, N.A. Cardiac fibroblast-specific p38alpha MAP kinase promotes cardiac hypertrophy via a putative paracrine interleukin-6 signaling mechanism. FASEB J. 2018, 32, 4941–4954. [Google Scholar] [CrossRef]
- Wang, J.H.; Zhao, L.; Pan, X.; Chen, N.N.; Chen, J.; Gong, Q.L.; Su, F.; Yan, J.; Zhang, Y.; Zhang, S.H. Hypoxia-stimulated cardiac fibroblast production of IL-6 promotes myocardial fibrosis via the TGF-beta1 signaling pathway. Lab. Investig. 2016, 96, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Li, Y.; Jia, L.; Han, Y.; Cheng, J.; Li, H.; Qi, Y.; Du, J. Macrophage-stimulated cardiac fibroblast production of IL-6 is essential for TGF beta/Smad activation and cardiac fibrosis induced by angiotensin II. PLoS ONE 2012, 7, e35144. [Google Scholar] [CrossRef]
- Fischer, P.; Hilfiker-Kleiner, D. Survival pathways in hypertrophy and heart failure: The gp130-STAT axis. Basic Res. Cardiol. 2007, 102, 393–411. [Google Scholar] [CrossRef] [PubMed]
- Diao, H.; Kohanawa, M. Endogenous interleukin-6 plays a crucial protective role in streptococcal toxic shock syndrome via suppression of tumor necrosis factor alpha production. Infect. Immun. 2005, 73, 3745–3748. [Google Scholar] [CrossRef]
- Hilfiker-Kleiner, D.; Shukla, P.; Klein, G.; Schaefer, A.; Stapel, B.; Hoch, M.; Muller, W.; Scherr, M.; Theilmeier, G.; Ernst, M.; et al. Continuous glycoprotein-130-mediated signal transducer and activator of transcription-3 activation promotes inflammation, left ventricular rupture, and adverse outcome in subacute myocardial infarction. Circulation 2010, 122, 145–155. [Google Scholar] [CrossRef]
- Fuchs, M.; Hilfiker, A.; Kaminski, K.; Hilfiker-Kleiner, D.; Guener, Z.; Klein, G.; Podewski, E.; Schieffer, B.; Rose-John, S.; Drexler, H. Role of interleukin-6 for LV remodeling and survival after experimental myocardial infarction. FASEB J. 2003, 17, 2118–2120. [Google Scholar] [CrossRef]
- Morimoto, S.; Nabata, T.; Koh, E.; Shiraishi, T.; Fukuo, K.; Imanaka, S.; Kitano, S.; Miyashita, Y.; Ogihara, T. Interleukin-6 stimulates proliferation of cultured vascular smooth muscle cells independently of interleukin-1 beta. J. Cardiovasc. Pharmacol. 1991, 17 (Suppl. S2), S117–S118. [Google Scholar] [CrossRef]
- Wang, Z.; Newman, W.H. Smooth muscle cell migration stimulated by interleukin 6 is associated with cytoskeletal reorganization. J. Surg. Res. 2003, 111, 261–266. [Google Scholar] [CrossRef]
- Wassmann, S.; Stumpf, M.; Strehlow, K.; Schmid, A.; Schieffer, B.; Bohm, M.; Nickenig, G. Interleukin-6 induces oxidative stress and endothelial dysfunction by overexpression of the angiotensin II type 1 receptor. Circ. Res. 2004, 94, 534–541. [Google Scholar] [CrossRef]
- Dong, X.; Wu, D.; Zhang, Y.; Jia, L.; Pan, X.; Sun, J.; Pan, L.L. Cathelicidin Modulates Vascular Smooth Muscle Cell Phenotypic Switching through ROS/IL-6 Pathway. Antioxidants 2020, 9, 491. [Google Scholar] [CrossRef]
- Kokje, V.B.C.; Gabel, G.; Koole, D.; Northoff, B.H.; Holdt, L.M.; Hamming, J.F.; Lindeman, J.H.N. IL-6: A Janus-like factor in abdominal aortic aneurysm disease. Atherosclerosis 2016, 251, 139–146. [Google Scholar] [CrossRef]
- Takeda, N.; Manabe, I.; Shindo, T.; Iwata, H.; Iimuro, S.; Kagechika, H.; Shudo, K.; Nagai, R. Synthetic retinoid Am80 reduces scavenger receptor expression and atherosclerosis in mice by inhibiting IL-6. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.A.; Sakkinen, P.; Conze, D.; Hardin, N.; Tracy, R. Interleukin-6 exacerbates early atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2364–2367. [Google Scholar] [CrossRef] [PubMed]
- Loppnow, H.; Buerke, M.; Werdan, K.; Rose-John, S. Contribution of vascular cell-derived cytokines to innate and inflammatory pathways in atherogenesis. J. Cell. Mol. Med. 2011, 15, 484–500. [Google Scholar] [CrossRef] [PubMed]
- Schieffer, B.; Selle, T.; Hilfiker, A.; Hilfiker-Kleiner, D.; Grote, K.; Tietge, U.J.; Trautwein, C.; Luchtefeld, M.; Schmittkamp, C.; Heeneman, S.; et al. Impact of interleukin-6 on plaque development and morphology in experimental atherosclerosis. Circulation 2004, 110, 3493–3500. [Google Scholar] [CrossRef] [PubMed]
- Elhage, R.; Clamens, S.; Besnard, S.; Mallat, Z.; Tedgui, A.; Arnal, J.; Maret, A.; Bayard, F. Involvement of interleukin-6 in atherosclerosis but not in the prevention of fatty streak formation by 17beta-estradiol in apolipoprotein E-deficient mice. Atherosclerosis 2001, 156, 315–320. [Google Scholar] [CrossRef]
- Ridker, P.M. From RESCUE to ZEUS: Will interleukin-6 inhibition with ziltivekimab prove effective for cardiovascular event reduction? Cardiovasc. Res. 2021, 117, e138–e140. [Google Scholar] [CrossRef]
- Klouche, M.; Bhakdi, S.; Hemmes, M.; Rose-John, S. Novel path to activation of vascular smooth muscle cells: Up-regulation of gp130 creates an autocrine activation loop by IL-6 and its soluble receptor. J. Immunol. 1999, 163, 4583–4589. [Google Scholar]
- Dzialo, E.; Czepiel, M.; Tkacz, K.; Siedlar, M.; Kania, G.; Blyszczuk, P. WNT/beta-Catenin Signaling Promotes TGF-beta-Mediated Activation of Human Cardiac Fibroblasts by Enhancing IL-11 Production. Int. J. Mol. Sci. 2021, 22, 10072. [Google Scholar] [CrossRef]
- Widjaja, A.A.; Singh, B.K.; Adami, E.; Viswanathan, S.; Dong, J.; D’Agostino, G.A.; Ng, B.; Lim, W.W.; Tan, J.; Paleja, B.S.; et al. Inhibiting Interleukin 11 Signaling Reduces Hepatocyte Death and Liver Fibrosis, Inflammation, and Steatosis in Mouse Models of Nonalcoholic Steatohepatitis. Gastroenterology 2019, 157, 777–792.e714. [Google Scholar] [CrossRef]
- Ng, B.; Dong, J.; D’Agostino, G.; Viswanathan, S.; Widjaja, A.A.; Lim, W.W.; Ko, N.S.J.; Tan, J.; Chothani, S.P.; Huang, B.; et al. Interleukin-11 is a therapeutic target in idiopathic pulmonary fibrosis. Sci. Transl. Med. 2019, 11, eaaw1237. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.W.; Corden, B.; Ng, B.; Vanezis, K.; D’Agostino, G.; Widjaja, A.A.; Song, W.H.; Xie, C.; Su, L.; Kwek, X.Y.; et al. Interleukin-11 is important for vascular smooth muscle phenotypic switching and aortic inflammation, fibrosis and remodeling in mouse models. Sci. Rep. 2020, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Taki, H.; Sakai, T.; Sugiyama, E.; Mino, T.; Kuroda, A.; Taki, K.; Hamazaki, T.; Koizumi, H.; Kobayashi, M. Monokine stimulation of interleukin-11 production by human vascular smooth muscle cells in vitro. Atherosclerosis 1999, 144, 375–380. [Google Scholar] [CrossRef]
- Zimmerman, M.A.; Selzman, C.H.; Reznikov, L.L.; Raeburn, C.D.; Barsness, K.; McIntyre, R.C., Jr.; Hamiel, C.R.; Harken, A.H. Interleukin-11 attenuates human vascular smooth muscle cell proliferation. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H175–H180. [Google Scholar] [CrossRef] [PubMed]
- Gong, F.; Liu, Z.; Liu, J.; Zhou, P.; Liu, Y.; Lu, X. The paradoxical role of IL-17 in atherosclerosis. Cell. Immunol. 2015, 297, 33–39. [Google Scholar] [CrossRef]
- Zhou, S.F.; Yuan, J.; Liao, M.Y.; Xia, N.; Tang, T.T.; Li, J.J.; Jiao, J.; Dong, W.Y.; Nie, S.F.; Zhu, Z.F.; et al. IL-17A promotes ventricular remodeling after myocardial infarction. J. Mol. Med. (Berl.) 2014, 92, 1105–1116. [Google Scholar] [CrossRef]
- Liu, Y.; Zhu, H.; Su, Z.; Sun, C.; Yin, J.; Yuan, H.; Sandoghchian, S.; Jiao, Z.; Wang, S.; Xu, H. IL-17 contributes to cardiac fibrosis following experimental autoimmune myocarditis by a PKCbeta/Erk1/2/NF-kappaB-dependent signaling pathway. Int. Immunol. 2012, 24, 605–612. [Google Scholar] [CrossRef]
- Gaffen, S.L. An overview of IL-17 function and signaling. Cytokine 2008, 43, 402–407. [Google Scholar] [CrossRef]
- de Boer, O.J.; van der Meer, J.J.; Teeling, P.; van der Loos, C.M.; Idu, M.M.; van Maldegem, F.; Aten, J.; van der Wal, A.C. Differential expression of interleukin-17 family cytokines in intact and complicated human atherosclerotic plaques. J. Pathol. 2010, 220, 499–508. [Google Scholar] [CrossRef]
- Yue, E.; Yu, Y.; Wang, X.; Liu, B.; Bai, Y.; Yang, B. Anthocyanin Protects Cardiac Function and Cardiac Fibroblasts From High-Glucose Induced Inflammation and Myocardial Fibrosis by Inhibiting IL-17. Front. Pharmacol. 2020, 11, 2289. [Google Scholar] [CrossRef]
- Li, Y.; Wu, Y.; Zhang, C.; Li, P.; Cui, W.; Hao, J.; Ma, X.; Yin, Z.; Du, J. gammadeltaT Cell-derived interleukin-17A via an interleukin-1beta-dependent mechanism mediates cardiac injury and fibrosis in hypertension. Hypertension 2014, 64, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Valente, A.J.; Yoshida, T.; Gardner, J.D.; Somanna, N.; Delafontaine, P.; Chandrasekar, B. Interleukin-17A stimulates cardiac fibroblast proliferation and migration via negative regulation of the dual-specificity phosphatase MKP-1/DUSP-1. Cell. Signal. 2012, 24, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Tang, X. Analysis of interleukin-17 and interleukin-18 levels in animal models of atherosclerosis. Exp. Ther. Med. 2019, 18, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Gao, Q.; Zhu, F.; Guo, C.; Wang, Q.; Gao, F.; Zhang, L. Th17 cells and IL-17 are involved in the disruption of vulnerable plaques triggered by short-term combination stimulation in apolipoprotein E-knockout mice. Cell. Mol. Immunol. 2013, 10, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Erbel, C.; Dengler, T.J.; Wangler, S.; Lasitschka, F.; Bea, F.; Wambsganss, N.; Hakimi, M.; Bockler, D.; Katus, H.A.; Gleissner, C.A. Expression of IL-17A in human atherosclerotic lesions is associated with increased inflammation and plaque vulnerability. Basic Res. Cardiol. 2011, 106, 125–134. [Google Scholar] [CrossRef]
- Gistera, A.; Robertson, A.K.; Andersson, J.; Ketelhuth, D.F.; Ovchinnikova, O.; Nilsson, S.K.; Lundberg, A.M.; Li, M.O.; Flavell, R.A.; Hansson, G.K. Transforming growth factor-beta signaling in T cells promotes stabilization of atherosclerotic plaques through an interleukin-17-dependent pathway. Sci. Transl. Med. 2013, 5, 196ra100. [Google Scholar] [CrossRef]
- Orejudo, M.; Garcia-Redondo, A.B.; Rodrigues-Diez, R.R.; Rodrigues-Diez, R.; Santos-Sanchez, L.; Tejera-Munoz, A.; Egido, J.; Selgas, R.; Salaices, M.; Briones, A.M.; et al. Interleukin-17A induces vascular remodeling of small arteries and blood pressure elevation. Clin. Sci. (Lond.) 2020, 134, 513–527. [Google Scholar] [CrossRef]
- Patel, D.N.; King, C.A.; Bailey, S.R.; Holt, J.W.; Venkatachalam, K.; Agrawal, A.; Valente, A.J.; Chandrasekar, B. Interleukin-17 stimulates C-reactive protein expression in hepatocytes and smooth muscle cells via p38 MAPK and ERK1/2-dependent NF-kappaB and C/EBPbeta activation. J. Biol. Chem. 2007, 282, 27229–27238. [Google Scholar] [CrossRef]
- Cheng, G.; Wei, L.; Xiurong, W.; Xiangzhen, L.; Shiguang, Z.; Songbin, F. IL-17 stimulates migration of carotid artery vascular smooth muscle cells in an MMP-9 dependent manner via p38 MAPK and ERK1/2-dependent NF-kappaB and AP-1 activation. Cell. Mol. Neurobiol. 2009, 29, 1161–1168. [Google Scholar] [CrossRef]
- O’Brien, L.C.; Mezzaroma, E.; Van Tassell, B.W.; Marchetti, C.; Carbone, S.; Abbate, A.; Toldo, S. Interleukin-18 as a therapeutic target in acute myocardial infarction and heart failure. Mol. Med. 2014, 20, 221–229. [Google Scholar] [CrossRef]
- Fix, C.; Bingham, K.; Carver, W. Effects of interleukin-18 on cardiac fibroblast function and gene expression. Cytokine 2011, 53, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Chen, Y.; Chen, Q.; Hong, T.; Zhong, Z.; He, J.; Ni, C. Curcumin Ameliorates Cardiac Fibrosis by Regulating Macrophage-Fibroblast Crosstalk via IL18-P-SMAD2/3 Signaling Pathway Inhibition. Front. Pharmacol. 2021, 12, 784041. [Google Scholar] [CrossRef]
- Gerdes, N.; Sukhova, G.K.; Libby, P.; Reynolds, R.S.; Young, J.L.; Schonbeck, U. Expression of interleukin (IL)-18 and functional IL-18 receptor on human vascular endothelial cells, smooth muscle cells, and macrophages: Implications for atherogenesis. J. Exp. Med. 2002, 195, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Mallat, Z.; Corbaz, A.; Scoazec, A.; Besnard, S.; Leseche, G.; Chvatchko, Y.; Tedgui, A. Expression of interleukin-18 in human atherosclerotic plaques and relation to plaque instability. Circulation 2001, 104, 1598–1603. [Google Scholar] [CrossRef] [PubMed]
- de Nooijer, R.; von der Thusen, J.H.; Verkleij, C.J.; Kuiper, J.; Jukema, J.W.; van der Wall, E.E.; van Berkel, J.C.; Biessen, E.A. Overexpression of IL-18 decreases intimal collagen content and promotes a vulnerable plaque phenotype in apolipoprotein-E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2313–2319. [Google Scholar] [CrossRef]
- Badimon, L. Interleukin-18: A potent pro-inflammatory cytokine in atherosclerosis. Cardiovasc. Res. 2012, 96, 172–175; discussion 176–180. [Google Scholar] [CrossRef]
- Pigarevskii, P.V.; Maltseva, S.V.; Snegova, V.A.; Davydova, N.G. Role of interleukin-18 in destabilization of the atherosclerotic plaque in humans. Bull. Exp. Biol. Med. 2014, 157, 821–824. [Google Scholar] [CrossRef]
- Sahar, S.; Dwarakanath, R.S.; Reddy, M.A.; Lanting, L.; Todorov, I.; Natarajan, R. Angiotensin II enhances interleukin-18 mediated inflammatory gene expression in vascular smooth muscle cells: A novel cross-talk in the pathogenesis of atherosclerosis. Circ. Res. 2005, 96, 1064–1071. [Google Scholar] [CrossRef]
- Chandrasekar, B.; Mummidi, S.; Mahimainathan, L.; Patel, D.N.; Bailey, S.R.; Imam, S.Z.; Greene, W.C.; Valente, A.J. Interleukin-18-induced human coronary artery smooth muscle cell migration is dependent on NF-kappaB- and AP-1-mediated matrix metalloproteinase-9 expression and is inhibited by atorvastatin. J. Biol. Chem. 2006, 281, 15099–15109. [Google Scholar] [CrossRef]
- Chandrasekar, B.; Mummidi, S.; Valente, A.J.; Patel, D.N.; Bailey, S.R.; Freeman, G.L.; Hatano, M.; Tokuhisa, T.; Jensen, L.E. The pro-atherogenic cytokine interleukin-18 induces CXCL16 expression in rat aortic smooth muscle cells via MyD88, interleukin-1 receptor-associated kinase, tumor necrosis factor receptor-associated factor 6, c-Src, phosphatidylinositol 3-kinase, Akt, c-Jun N-terminal kinase, and activator protein-1 signaling. J. Biol. Chem. 2005, 280, 26263–26277. [Google Scholar] [CrossRef]
- Valente, A.J.; Yoshida, T.; Murthy, S.N.; Sakamuri, S.S.; Katsuyama, M.; Clark, R.A.; Delafontaine, P.; Chandrasekar, B. Angiotensin II enhances AT1-Nox1 binding and stimulates arterial smooth muscle cell migration and proliferation through AT1, Nox1, and interleukin-18. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H282–H296. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Zhang, Y.; Feng, W.; Chen, R.; Chen, J.; Touyz, R.M.; Wang, J.; Huang, H. Interleukin-18 Enhances Vascular Calcification and Osteogenic Differentiation of Vascular Smooth Muscle Cells Through TRPM7 Activation. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1933–1943. [Google Scholar] [CrossRef]
- Schelski, N.; Luong, T.T.D.; Lang, F.; Pieske, B.; Voelkl, J.; Alesutan, I. SGK1-dependent stimulation of vascular smooth muscle cell osteo-/chondrogenic transdifferentiation by interleukin-18. Pflugers Arch. 2019, 471, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Stewart, L.; Turner, N.A. Channelling the Force to Reprogram the Matrix: Mechanosensitive Ion Channels in Cardiac Fibroblasts. Cells 2021, 10, 990. [Google Scholar] [CrossRef] [PubMed]
- Hahn, C.; Schwartz, M.A. Mechanotransduction in vascular physiology and atherogenesis. Nat. Rev. Mol. Cell Biol. 2009, 10, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Mantella, L.E.; Quan, A.; Verma, S. Variability in vascular smooth muscle cell stretch-induced responses in 2D culture. Vasc. Cell 2015, 7, 1–9. [Google Scholar] [CrossRef]
- Herum, K.M.; Lunde, I.G.; Skrbic, B.; Florholmen, G.; Behmen, D.; Sjaastad, I.; Carlson, C.R.; Gomez, M.F.; Christensen, G. Syndecan-4 signaling via NFAT regulates extracellular matrix production and cardiac myofibroblast differentiation in response to mechanical stress. J. Mol. Cell. Cardiol. 2013, 54, 73–81. [Google Scholar] [CrossRef]
- Ploeg, M.C.; Munts, C.; Prinzen, F.W.; Turner, N.A.; van Bilsen, M.; van Nieuwenhoven, F.A. Piezo1 Mechanosensitive Ion Channel Mediates Stretch-Induced Nppb Expression in Adult Rat Cardiac Fibroblasts. Cells 2021, 10, 1745. [Google Scholar] [CrossRef]
- Miralles, F.; Posern, G.; Zaromytidou, A.I.; Treisman, R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 2003, 113, 329–342. [Google Scholar] [CrossRef]
- Amano, M.; Nakayama, M.; Kaibuchi, K. Rho-kinase/ROCK: A key regulator of the cytoskeleton and cell polarity. Cytoskeleton (Hoboken) 2010, 67, 545–554. [Google Scholar] [CrossRef]
- Sarrazy, V.; Koehler, A.; Chow, M.L.; Zimina, E.; Li, C.X.; Kato, H.; Caldarone, C.A.; Hinz, B. Integrins alphavbeta5 and alphavbeta3 promote latent TGF-beta1 activation by human cardiac fibroblast contraction. Cardiovasc. Res. 2014, 102, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.X.; Han, Y.; Jiang, Z.L. Mechanobiology and Vascular Remodeling: From Membrane to Nucleus. Adv. Exp. Med. Biol. 2018, 1097, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.D.; Holmes, J.W.; Saucerman, J.J.; Richardson, W.J. Mechano-chemo signaling interactions modulate matrix production by cardiac fibroblasts. Matrix Biol. Plus 2021, 10, 100055. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.P.; Zhang, P.; Qi, Y.X.; Chen, S.G.; Shen, B.R.; Han, Y.; Yan, Z.Q.; Jiang, Z.L. The role of SIRT6 in the differentiation of vascular smooth muscle cells in response to cyclic strain. Int. J. Biochem. Cell Biol. 2014, 49, 98–104. [Google Scholar] [CrossRef]
- Henderson, N.C.; Rieder, F.; Wynn, T.A. Fibrosis: From mechanisms to medicines. Nature 2020, 587, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Nagaraju, C.K.; Robinson, E.L.; Abdesselem, M.; Trenson, S.; Dries, E.; Gilbert, G.; Janssens, S.; Van Cleemput, J.; Rega, F.; Meyns, B.; et al. Myofibroblast Phenotype and Reversibility of Fibrosis in Patients With End-Stage Heart Failure. J. Am. Coll. Cardiol. 2019, 73, 2267–2282. [Google Scholar] [CrossRef]
- Khan, S.; Joyce, J.; Margulies, K.B.; Tsuda, T. Enhanced bioactive myocardial transforming growth factor-beta in advanced human heart failure. Circ. J. 2014, 78, 2711–2718. [Google Scholar] [CrossRef]
- Almendral, J.L.; Shick, V.; Rosendorff, C.; Atlas, S.A. Association between transforming growth factor-beta(1) and left ventricular mass and diameter in hypertensive patients. J. Am. Soc. Hypertens. 2010, 4, 135–141. [Google Scholar] [CrossRef]
- Aziz, T.M.; Burgess, M.I.; Haselton, P.S.; Yonan, N.A.; Hutchinson, I.V. Transforming growth factor beta and diastolic left ventricular dysfunction after heart transplantation: Echocardiographic and histologic evidence. J. Heart Lung Transplant. 2003, 22, 663–673. [Google Scholar] [CrossRef]
- Kuwahara, F.; Kai, H.; Tokuda, K.; Kai, M.; Takeshita, A.; Egashira, K.; Imaizumi, T. Transforming growth factor-beta function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats. Circulation 2002, 106, 130–135. [Google Scholar] [CrossRef]
- Engebretsen, K.V.; Skardal, K.; Bjornstad, S.; Marstein, H.S.; Skrbic, B.; Sjaastad, I.; Christensen, G.; Bjornstad, J.L.; Tonnessen, T. Attenuated development of cardiac fibrosis in left ventricular pressure overload by SM16, an orally active inhibitor of ALK5. J. Mol. Cell. Cardiol. 2014, 76, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Lucas, J.A.; Zhang, Y.; Li, P.; Gong, K.; Miller, A.P.; Hassan, E.; Hage, F.; Xing, D.; Wells, B.; Oparil, S.; et al. Inhibition of transforming growth factor-beta signaling induces left ventricular dilation and dysfunction in the pressure-overloaded heart. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H424–H432. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, V.; di Marzo, L.; Sapienza, P.; Colasanti, M.; Moroni, E.; Cavallaro, A. Role of platelet-derived growth factor and transforming growth factor beta1 the in the regulation of metalloproteinase expressions. Surgery 2006, 140, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Lacouture, M.E.; Morris, J.C.; Lawrence, D.P.; Tan, A.R.; Olencki, T.E.; Shapiro, G.I.; Dezube, B.J.; Berzofsky, J.A.; Hsu, F.J.; Guitart, J. Cutaneous keratoacanthomas/squamous cell carcinomas associated with neutralization of transforming growth factor beta by the monoclonal antibody fresolimumab (GC1008). Cancer. Immunol. Immunother. 2015, 64, 437–446. [Google Scholar] [CrossRef]
- Rice, L.M.; Padilla, C.M.; McLaughlin, S.R.; Mathes, A.; Ziemek, J.; Goummih, S.; Nakerakanti, S.; York, M.; Farina, G.; Whitfield, M.L.; et al. Fresolimumab treatment decreases biomarkers and improves clinical symptoms in systemic sclerosis patients. J. Clin. Investig. 2015, 125, 2795–2807. [Google Scholar] [CrossRef]
- Vincenti, F.; Fervenza, F.C.; Campbell, K.N.; Diaz, M.; Gesualdo, L.; Nelson, P.; Praga, M.; Radhakrishnan, J.; Sellin, L.; Singh, A.; et al. A Phase 2, Double-Blind, Placebo-Controlled, Randomized Study of Fresolimumab in Patients With Steroid-Resistant Primary Focal Segmental Glomerulosclerosis. Kidney Int. Rep. 2017, 2, 800–810. [Google Scholar] [CrossRef] [PubMed]
- Mitra, M.S.; Lancaster, K.; Adedeji, A.O.; Palanisamy, G.S.; Dave, R.A.; Zhong, F.; Holdren, M.S.; Turley, S.J.; Liang, W.C.; Wu, Y.; et al. A Potent Pan-TGFbeta Neutralizing Monoclonal Antibody Elicits Cardiovascular Toxicity in Mice and Cynomolgus Monkeys. Toxicol. Sci. 2020, 175, 24–34. [Google Scholar] [CrossRef]
- Lopez-de la Mora, D.A.; Sanchez-Roque, C.; Montoya-Buelna, M.; Sanchez-Enriquez, S.; Lucano-Landeros, S.; Macias-Barragan, J.; Armendariz-Borunda, J. Role and New Insights of Pirfenidone in Fibrotic Diseases. Int. J. Med. Sci. 2015, 12, 840–847. [Google Scholar] [CrossRef]
- Aimo, A.; Cerbai, E.; Bartolucci, G.; Adamo, L.; Barison, A.; Lo Surdo, G.; Biagini, S.; Passino, C.; Emdin, M. Pirfenidone is a cardioprotective drug: Mechanisms of action and preclinical evidence. Pharmacol. Res. 2020, 155, 104694. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, Y.; Chen, J.; Zhao, S.; Li, H. Pirfenidone attenuates cardiac fibrosis in a mouse model of TAC-induced left ventricular remodeling by suppressing NLRP3 inflammasome formation. Cardiology 2013, 126, 1–11. [Google Scholar] [CrossRef]
- Nakazato, H.; Oku, H.; Yamane, S.; Tsuruta, Y.; Suzuki, R. A novel anti-fibrotic agent pirfenidone suppresses tumor necrosis factor-alpha at the translational level. Eur. J. Pharmacol. 2002, 446, 177–185. [Google Scholar] [CrossRef]
- Hale, M.L.; Margolin, S.B.; Krakauer, T.; Roy, C.J.; Stiles, B.G. Pirfenidone blocks the in vitro and in vivo effects of staphylococcal enterotoxin B. Infect. Immun. 2002, 70, 2989–2994. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Paz, K.; Flynn, R.; Vulic, A.; Robinson, T.M.; Lineburg, K.E.; Alexander, K.A.; Meng, J.; Roy, S.; Panoskaltsis-Mortari, A.; et al. Pirfenidone ameliorates murine chronic GVHD through inhibition of macrophage infiltration and TGF-beta production. Blood 2017, 129, 2570–2580. [Google Scholar] [CrossRef]
- Visner, G.A.; Liu, F.; Bizargity, P.; Liu, H.; Liu, K.; Yang, J.; Wang, L.; Hancock, W.W. Pirfenidone inhibits T-cell activation, proliferation, cytokine and chemokine production, and host alloresponses. Transplantation 2009, 88, 330–338. [Google Scholar] [CrossRef] [PubMed]
- King, T.E., Jr.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef] [PubMed]
- Nathan, S.D.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glaspole, I.; Glassberg, M.K.; Kardatzke, D.R.; Daigl, M.; Kirchgaessler, K.U.; Lancaster, L.H.; et al. Effect of pirfenidone on mortality: Pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir. Med. 2017, 5, 33–41. [Google Scholar] [CrossRef]
- Noble, P.W.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glassberg, M.K.; Kardatzke, D.; King, T.E., Jr.; Lancaster, L.; Sahn, S.A.; Szwarcberg, J.; et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): Two randomised trials. Lancet 2011, 377, 1760–1769. [Google Scholar] [CrossRef]
- Glassberg, M.K.; Nathan, S.D.; Lin, C.Y.; Morgenthien, E.A.; Stauffer, J.L.; Chou, W.; Noble, P.W. Cardiovascular Risks, Bleeding Risks, and Clinical Events from 3 Phase III Trials of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis. Adv. Ther. 2019, 36, 2910–2926. [Google Scholar] [CrossRef]
- Lewis, G.A.; Dodd, S.; Clayton, D.; Bedson, E.; Eccleson, H.; Schelbert, E.B.; Naish, J.H.; Jimenez, B.D.; Williams, S.G.; Cunnington, C.; et al. Pirfenidone in heart failure with preserved ejection fraction: A randomized phase 2 trial. Nat. Med. 2021, 27, 1477–1482. [Google Scholar] [CrossRef]
- Travers, J.G.; Tharp, C.A.; Rubino, M.; McKinsey, T.A. Therapeutic targets for cardiac fibrosis: From old school to next-gen. J. Clin. Investig. 2022, 132, 5. [Google Scholar] [CrossRef]
- Atlas, S.A. The renin-angiotensin aldosterone system: Pathophysiological role and pharmacologic inhibition. J. Manag. Care Pharm. 2007, 13, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.D.; Mehta, R.M.; Tripathi, R.; Reed, G.L.; Gladysheva, I.P. Renin Activity in Heart Failure with Reduced Systolic Function-New Insights. Int. J. Mol. Sci. 2019, 20, 3182. [Google Scholar] [CrossRef]
- Brilla, C.G.; Funck, R.C.; Rupp, H. Lisinopril-mediated regression of myocardial fibrosis in patients with hypertensive heart disease. Circulation 2000, 102, 1388–1393. [Google Scholar] [CrossRef] [PubMed]
- Lopez, B.; Querejeta, R.; Varo, N.; Gonzalez, A.; Larman, M.; Martinez Ubago, J.L.; Diez, J. Usefulness of serum carboxy-terminal propeptide of procollagen type I in assessment of the cardioreparative ability of antihypertensive treatment in hypertensive patients. Circulation 2001, 104, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Shimada, Y.J.; Passeri, J.J.; Baggish, A.L.; O’Callaghan, C.; Lowry, P.A.; Yannekis, G.; Abbara, S.; Ghoshhajra, B.B.; Rothman, R.D.; Ho, C.Y.; et al. Effects of losartan on left ventricular hypertrophy and fibrosis in patients with nonobstructive hypertrophic cardiomyopathy. JACC Heart Fail. 2013, 1, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, M.; Ito, H.; Onuki, T.; Miyoshi, F.; Watanabe, N.; Asano, T.; Tanno, K.; Kobayashi, Y. Candesartan decreases type III procollagen-N-peptide levels and inflammatory marker levels and maintains sinus rhythm in patients with atrial fibrillation. J. Cardiovasc. Pharmacol. 2010, 55, 511–517. [Google Scholar] [CrossRef]
- Kosmala, W.; Przewlocka-Kosmala, M.; Szczepanik-Osadnik, H.; Mysiak, A.; O’Moore-Sullivan, T.; Marwick, T.H. A randomized study of the beneficial effects of aldosterone antagonism on LV function, structure, and fibrosis markers in metabolic syndrome. JACC Cardiovasc. Imaging 2011, 4, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Mak, G.J.; Ledwidge, M.T.; Watson, C.J.; Phelan, D.M.; Dawkins, I.R.; Murphy, N.F.; Patle, A.K.; Baugh, J.A.; McDonald, K.M. Natural history of markers of collagen turnover in patients with early diastolic dysfunction and impact of eplerenone. J. Am. Coll. Cardiol. 2009, 54, 1674–1682. [Google Scholar] [CrossRef]
- Desai, A.S.; Liu, J.; Pfeffer, M.A.; Claggett, B.; Fleg, J.; Lewis, E.F.; McKinlay, S.; O’Meara, E.; Shah, S.J.; Sweitzer, N.K.; et al. Incident Hyperkalemia, Hypokalemia, and Clinical Outcomes During Spironolactone Treatment of Heart Failure With Preserved Ejection Fraction: Analysis of the TOPCAT Trial. J. Card. Fail. 2018, 24, 313–320. [Google Scholar] [CrossRef]
- Fang, L.; Murphy, A.J.; Dart, A.M. A Clinical Perspective of Anti-Fibrotic Therapies for Cardiovascular Disease. Front. Pharmacol. 2017, 8, 186. [Google Scholar] [CrossRef]
- McMurray, J.J.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 2014, 371, 993–1004. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.D.; Maass, F.U. The sociobiological ideas of Arthur Schopenhauer. Z. Psychol. Z. Angew. Psychol. 1989, 197, 341–350. [Google Scholar] [PubMed]
- Zile, M.R.; O’Meara, E.; Claggett, B.; Prescott, M.F.; Solomon, S.D.; Swedberg, K.; Packer, M.; McMurray, J.J.V.; Shi, V.; Lefkowitz, M.; et al. Effects of Sacubitril/Valsartan on Biomarkers of Extracellular Matrix Regulation in Patients With HFrEF. J. Am. Coll. Cardiol. 2019, 73, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, J.W.; Claggett, B.L.; O’Meara, E.; Prescott, M.F.; Pfeffer, M.A.; Shah, S.J.; Redfield, M.M.; Zannad, F.; Chiang, L.M.; Rizkala, A.R.; et al. Effect of Sacubitril/Valsartan on Biomarkers of Extracellular Matrix Regulation in Patients With HFpEF. J. Am. Coll. Cardiol. 2020, 76, 503–514. [Google Scholar] [CrossRef]
- Cleland, J.G.; Tendera, M.; Adamus, J.; Freemantle, N.; Polonski, L.; Taylor, J.; Investigators, P.-C. The perindopril in elderly people with chronic heart failure (PEP-CHF) study. Eur. Heart J. 2006, 27, 2338–2345. [Google Scholar] [CrossRef]
- Yusuf, S.; Pfeffer, M.A.; Swedberg, K.; Granger, C.B.; Held, P.; McMurray, J.J.; Michelson, E.L.; Olofsson, B.; Ostergren, J.; Investigators, C.; et al. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: The CHARM-Preserved Trial. Lancet 2003, 362, 777–781. [Google Scholar] [CrossRef]
- Massie, B.M.; Carson, P.E.; McMurray, J.J.; Komajda, M.; McKelvie, R.; Zile, M.R.; Anderson, S.; Donovan, M.; Iverson, E.; Staiger, C.; et al. Irbesartan in patients with heart failure and preserved ejection fraction. N. Engl. J. Med. 2008, 359, 2456–2467. [Google Scholar] [CrossRef]
- Solomon, S.D.; McMurray, J.J.V.; Anand, I.S.; Ge, J.; Lam, C.S.P.; Maggioni, A.P.; Martinez, F.; Packer, M.; Pfeffer, M.A.; Pieske, B.; et al. Angiotensin-Neprilysin Inhibition in Heart Failure with Preserved Ejection Fraction. N. Engl. J. Med. 2019, 381, 1609–1620. [Google Scholar] [CrossRef]
- Novartis. Novartis Entresto® Granted Expanded Indication in Chronic Heart Failure by FDA. Available online: https://www.novartis.com/news/media-releases/novartis-entresto-granted-expanded-indication-chronic-heart-failure-fda (accessed on 10 March 2022).
- Fox, K.M.; EURopean trial On reduction of cardiac events with Perindopril in stable coronary Artery disease Investigators. Efficacy of perindopril in reduction of cardiovascular events among patients with stable coronary artery disease: Randomised, double-blind, placebo-controlled, multicentre trial (the EUROPA study). Lancet 2003, 362, 782–788. [Google Scholar] [CrossRef]
- Heart Outcomes Prevention Evaluation Study, I.; Yusuf, S.; Sleight, P.; Pogue, J.; Bosch, J.; Davies, R.; Dagenais, G. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. N. Engl. J. Med. 2000, 342, 145–153. [Google Scholar] [CrossRef]
- Oyama, K.; El-Nachef, D.; MacLellan, W.R. Regeneration potential of adult cardiac myocytes. Cell Res. 2013, 23, 978–979. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Nguyen, N.B.; Pezhouman, A.; Ardehali, R. Cardiac fibrosis: Potential therapeutic targets. Transl. Res. 2019, 209, 121–137. [Google Scholar] [CrossRef] [PubMed]
- Bauersachs, J. Heart failure drug treatment: The fantastic four. Eur. Heart J. 2021, 42, 681–683. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.; Packer, M.; Filippatos, G.; Ferreira, J.P.; Zeller, C.; Schnee, J.; Brueckmann, M.; Pocock, S.J.; Zannad, F.; Anker, S.D. Effect of empagliflozin in patients with heart failure across the spectrum of left ventricular ejection fraction. Eur. Heart J. 2022, 43, 416–426. [Google Scholar] [CrossRef]
- Packer, M.; Zannad, F.; Anker, S.D. Heart Failure and a Preserved Ejection Fraction: A Side-by-Side Examination of the PARAGON-HF and EMPEROR-Preserved Trials. Circulation 2021, 144, 1193–1195. [Google Scholar] [CrossRef] [PubMed]
- Gevaert, A.B.; Kataria, R.; Zannad, F.; Sauer, A.J.; Damman, K.; Sharma, K.; Shah, S.J.; Van Spall, H.G.C. Heart failure with preserved ejection fraction: Recent concepts in diagnosis, mechanisms and management. Heart 2022. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bachmann, J.C.; Baumgart, S.J.; Uryga, A.K.; Bosteen, M.H.; Borghetti, G.; Nyberg, M.; Herum, K.M. Fibrotic Signaling in Cardiac Fibroblasts and Vascular Smooth Muscle Cells: The Dual Roles of Fibrosis in HFpEF and CAD. Cells 2022, 11, 1657. https://doi.org/10.3390/cells11101657
Bachmann JC, Baumgart SJ, Uryga AK, Bosteen MH, Borghetti G, Nyberg M, Herum KM. Fibrotic Signaling in Cardiac Fibroblasts and Vascular Smooth Muscle Cells: The Dual Roles of Fibrosis in HFpEF and CAD. Cells. 2022; 11(10):1657. https://doi.org/10.3390/cells11101657
Chicago/Turabian StyleBachmann, Julian C., Simon J. Baumgart, Anna K. Uryga, Markus H. Bosteen, Giulia Borghetti, Michael Nyberg, and Kate M. Herum. 2022. "Fibrotic Signaling in Cardiac Fibroblasts and Vascular Smooth Muscle Cells: The Dual Roles of Fibrosis in HFpEF and CAD" Cells 11, no. 10: 1657. https://doi.org/10.3390/cells11101657
APA StyleBachmann, J. C., Baumgart, S. J., Uryga, A. K., Bosteen, M. H., Borghetti, G., Nyberg, M., & Herum, K. M. (2022). Fibrotic Signaling in Cardiac Fibroblasts and Vascular Smooth Muscle Cells: The Dual Roles of Fibrosis in HFpEF and CAD. Cells, 11(10), 1657. https://doi.org/10.3390/cells11101657