
Parkinson’s Disease-Specific Autoantibodies against the Neuroprotective Co-Chaperone STIP1

 ,
,  , ,
, , 
 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Recruitment of Parkinson’s Disease Patients and Healthy Controls
2.3. Blood Processing and Generation of Dopaminergic Neurons Derived from Induced Pluripotent Stem Cells
2.4. Cell Culture and Neuronal Differentiation
2.5. Immunofluorescence
2.6. STIP1-Derived Peptide Library
2.7. Production of Full-Length STIP1 Protein
2.8. Primary Hippocampal Neuronal Cell Culture
2.9. Peptide/Protein-Based ELISA
2.10. Cell Death Assay
2.11. T Cell ELISpot
2.12. Cancer Outlier Profile Analysis (COPA)
2.13. Statistical Analysis
3. Results
3.1. STIP1 Ameliorates Staurosporine-Induced Neurotoxicity on Dopaminergic Neurons Derived from hiPSC
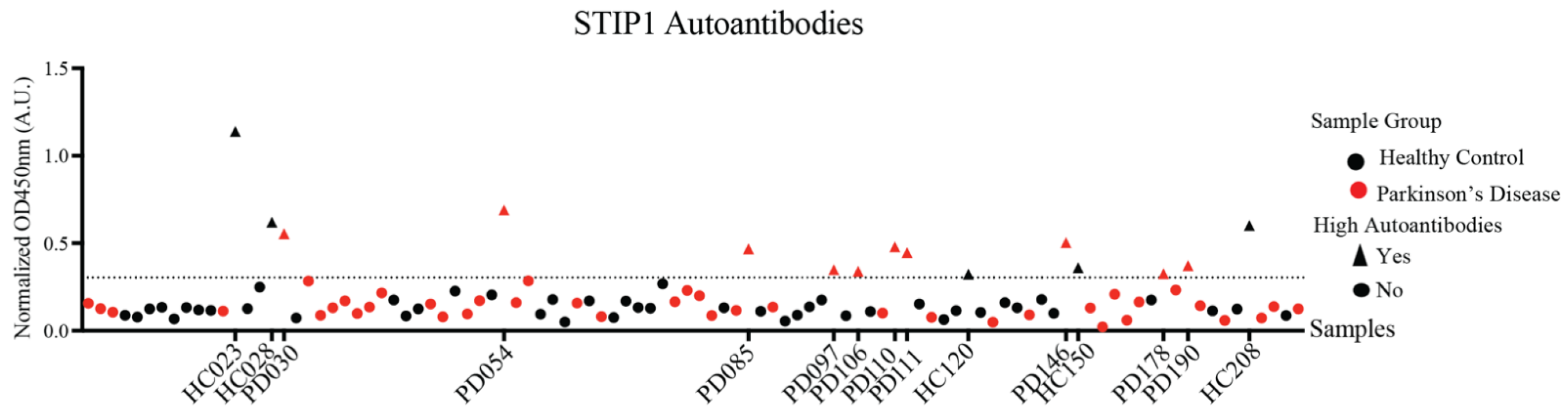
3.2. STIP1-Specific Autoantibodies in Parkinson’s Disease Patients and Healthy Controls
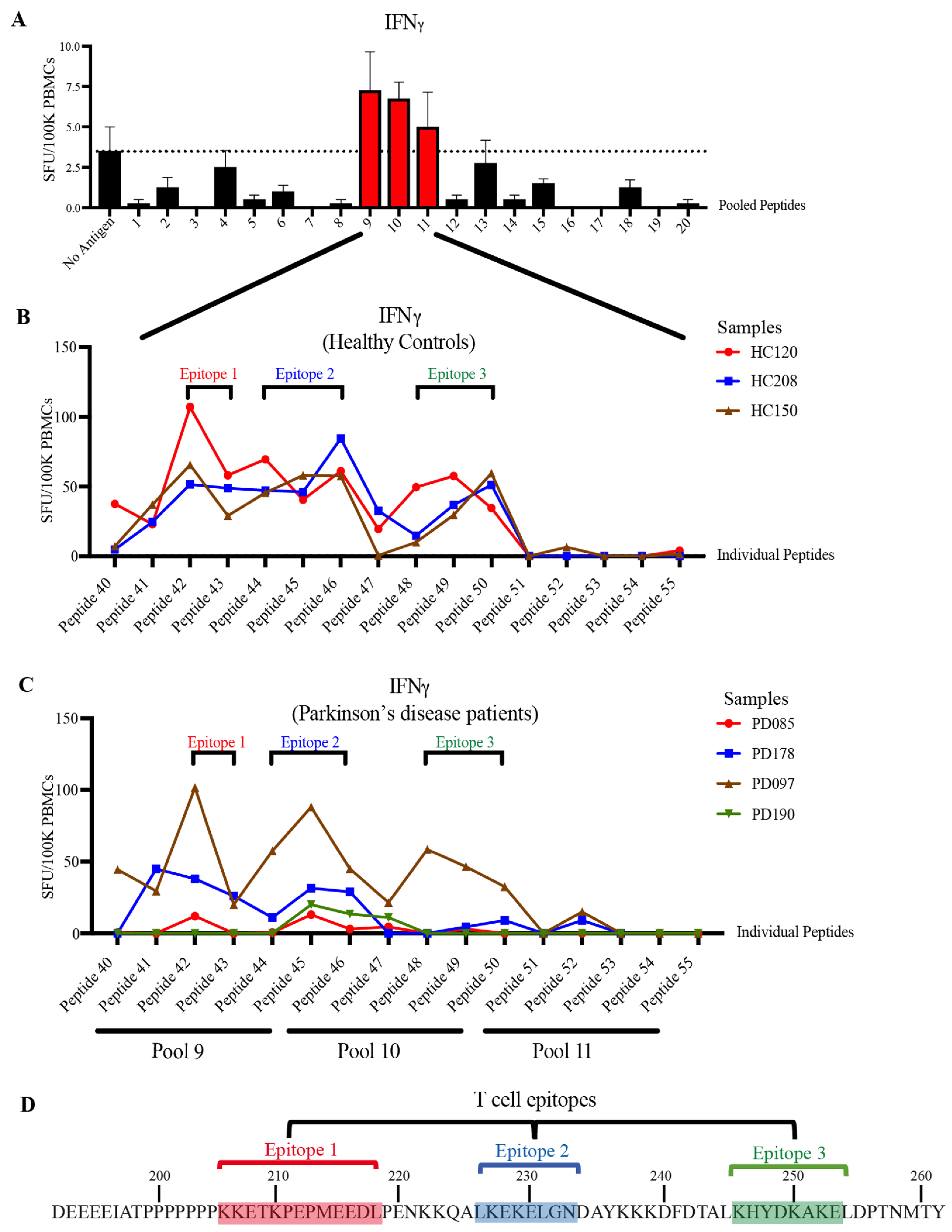
3.3. STIP1-Specific T Cell Epitopes Cluster around the PrPC Binding Site
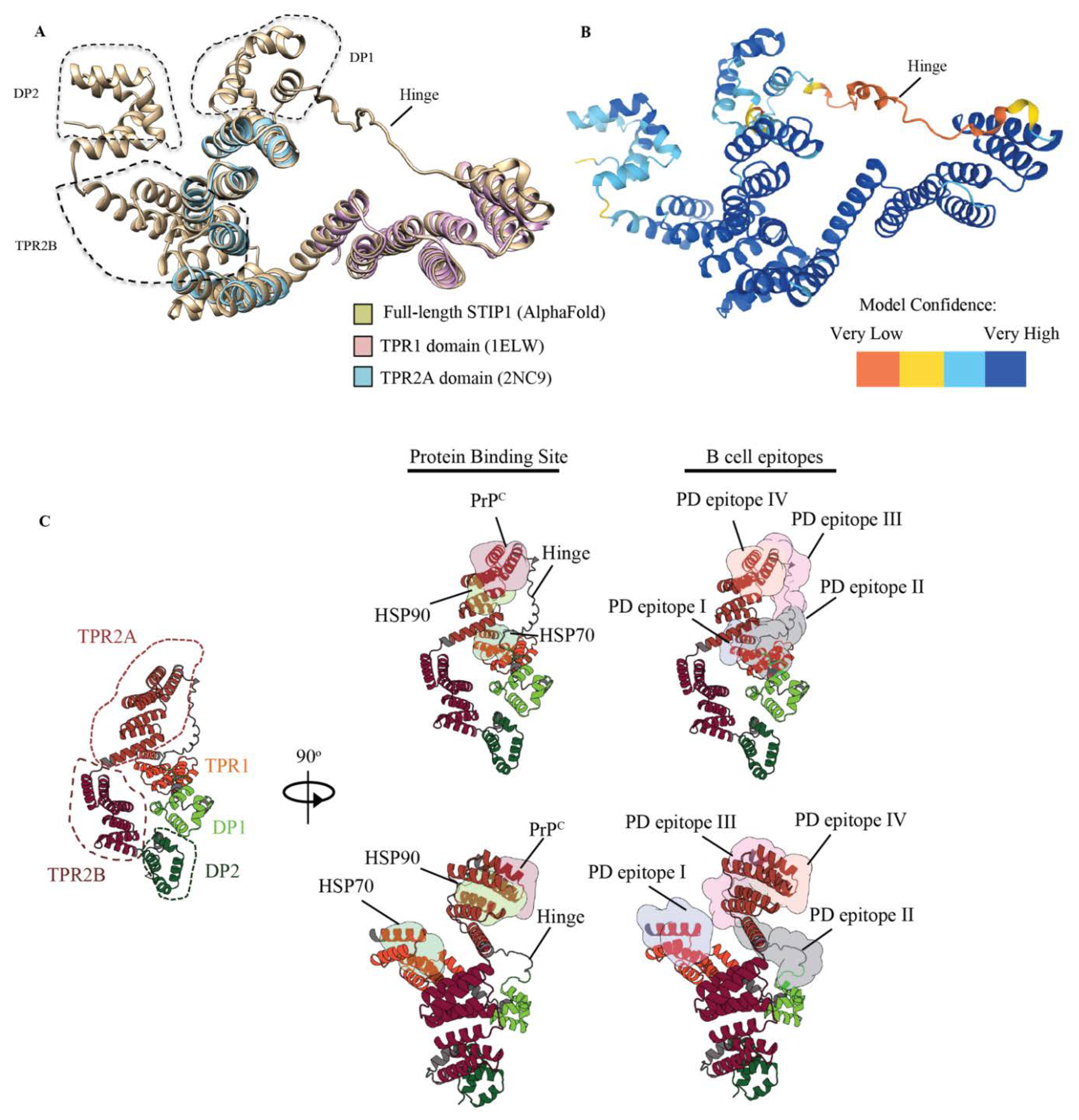
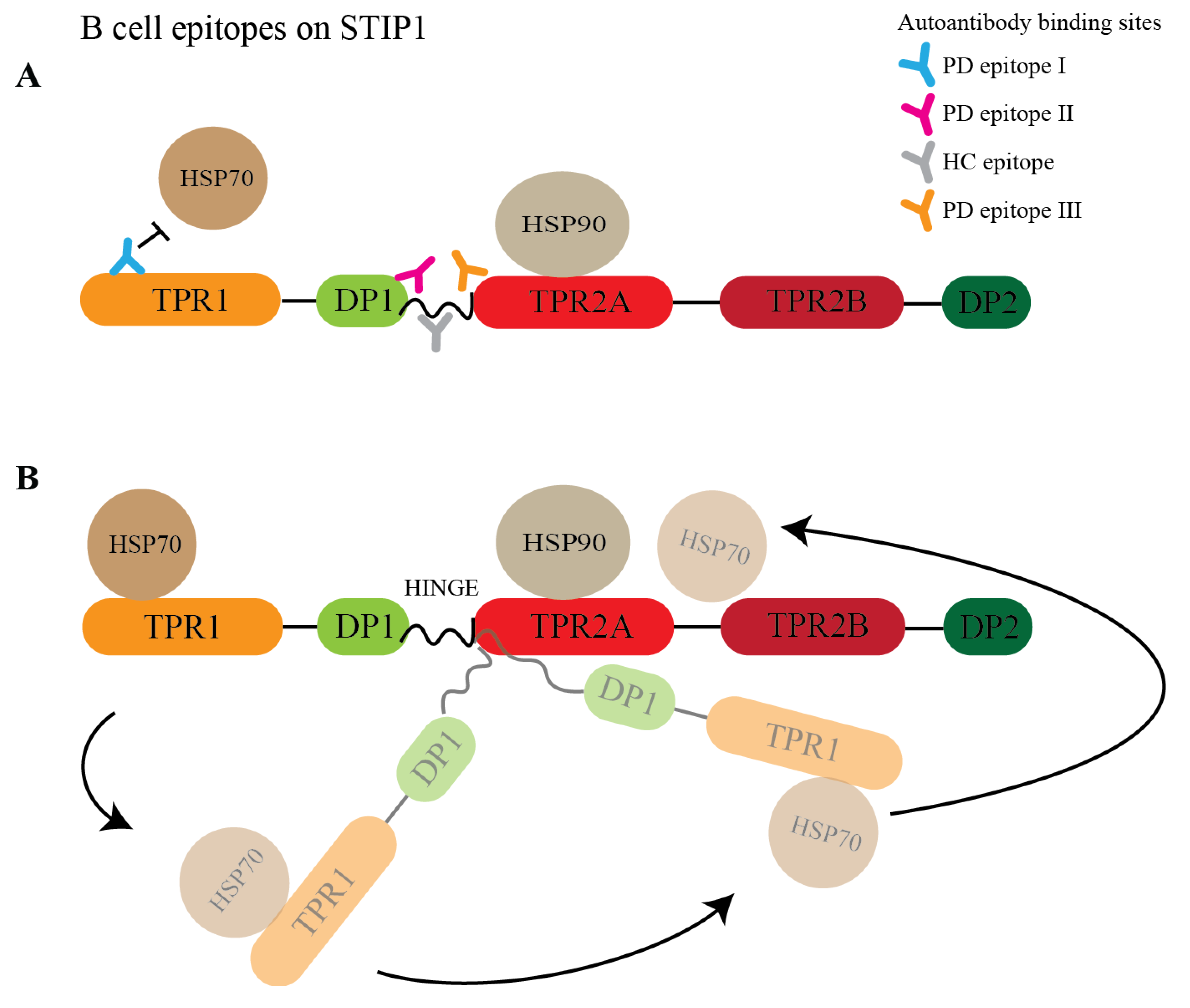
3.4. STIP1 Autoantibody Binding Sites Overlap with the HSP70 and PrPC Association Regions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rocca, W.A. The burden of Parkinson’s disease: A worldwide perspective. Lancet Neurol. 2018, 17, 928–929. [Google Scholar] [CrossRef]
- Giguère, N.; Burke Nanni, S.; Trudeau, L.-E. On Cell Loss and Selective Vulnerability of Neuronal Populations in Parkinson’s Disease. Front. Neurol. 2018, 9, 455. [Google Scholar] [CrossRef]
- Pellicano, C.; Benincasa, D.; Pisani, V.; Buttarelli, F.R.; Giovannelli, M.; Pontieri, F.E. Prodromal non-motor symptoms of Parkinson’s disease. Neuropsychiatr. Dis. Treat. 2007, 3, 145–152. [Google Scholar] [CrossRef]
- Grosch, J.; Winkler, J.; Kohl, Z. Early Degeneration of Both Dopaminergic and Serotonergic Axons—A Common Mechanism in Parkinson’s Disease. Front. Cell. Neurosci. 2016, 10, 293. [Google Scholar] [CrossRef]
- Honoré, B.; Leffers, H.; Madsen, P.; Rasmussen, H.H.; Vandekerckhove, J.; Celis, J.E. Molecular cloning and expression of a transformation-sensitive human protein containing the TPR motif and sharing identity to the stress-inducible yeast protein STI1. J. Biol. Chem. 1992, 267, 8485–8491. [Google Scholar] [CrossRef]
- Carvalho da Fonseca, A.C.; Wang, H.; Fan, H.; Chen, X.; Zhang, I.; Zhang, L.; Lima, F.R.; Badie, B. Increased expression of stress inducible protein 1 in glioma-associated microglia/macrophages. J. Neuroimmunol. 2014, 274, 71–77. [Google Scholar] [CrossRef]
- Hajj, G.N.; Arantes, C.P.; Dias, M.V.; Roffé, M.; Costa-Silva, B.; Lopes, M.H.; Porto-Carreiro, I.; Rabachini, T.; Lima, F.R.; Beraldo, F.H.; et al. The unconventional secretion of stress-inducible protein 1 by a heterogeneous population of extracellular vesicles. Cell. Mol. Life Sci. CMLS 2013, 70, 3211–3227. [Google Scholar] [CrossRef]
- Chao, A.; Lai, C.H.; Tsai, C.L.; Hsueh, S.; Hsueh, C.; Lin, C.Y.; Chou, H.H.; Lin, Y.J.; Chen, H.W.; Chang, T.C.; et al. Tumor stress-induced phosphoprotein1 (STIP1) as a prognostic biomarker in ovarian cancer. PLoS ONE 2013, 8, e57084. [Google Scholar] [CrossRef]
- Beraldo, F.H.; Soares, I.N.; Goncalves, D.F.; Fan, J.; Thomas, A.A.; Santos, T.G.; Mohammad, A.H.; Roffé, M.; Calder, M.D.; Nikolova, S.; et al. Stress-inducible phosphoprotein 1 has unique cochaperone activity during development and regulates cellular response to ischemia via the prion protein. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 3594–3607. [Google Scholar] [CrossRef]
- Lopes, M.H.; Hajj, G.N.M.; Muras, A.G.; Mancini, G.L.; Castro, R.M.P.S.; Ribeiro, K.C.B.; Brentani, R.R.; Linden, R.; Martins, V.R. Interaction of Cellular Prion and Stress-Inducible Protein 1 Promotes Neuritogenesis and Neuroprotection by Distinct Signaling Pathways. J. Neurosci. 2005, 25, 11330–11339. [Google Scholar] [CrossRef]
- Beraldo, F.H.; Arantes, C.P.; Santos, T.G.; Queiroz, N.G.T.; Young, K.; Rylett, R.J.; Markus, R.P.; Prado, M.A.M.; Martins, V.R. Role of alpha7 nicotinic acetylcholine receptor in calcium signaling induced by prion protein interaction with stress-inducible protein 1. J. Biol. Chem. 2010, 285, 36542–36550. [Google Scholar] [CrossRef]
- Zanata, S.M.; Lopes, M.H.; Mercadante, A.F.; Hajj, G.N.M.; Chiarini, L.B.; Nomizo, R.; Freitas, A.R.O.; Cabral, A.L.B.; Lee, K.S.; Juliano, M.A.; et al. Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO J. 2002, 21, 3307–3316. [Google Scholar] [CrossRef]
- Maciejewski, A.; Ostapchenko, V.G.; Beraldo, F.H.; Prado, V.F.; Prado, M.A.; Choy, W.Y. Domains of STIP1 responsible for regulating PrPC-dependent amyloid-β oligomer toxicity. Biochem. J. 2016, 473, 2119–2130. [Google Scholar] [CrossRef]
- Ostapchenko, V.G.; Beraldo, F.H.; Mohammad, A.H.; Xie, Y.F.; Hirata, P.H.; Magalhaes, A.C.; Lamour, G.; Li, H.; Maciejewski, A.; Belrose, J.C.; et al. The prion protein ligand, stress-inducible phosphoprotein 1, regulates amyloid-β oligomer toxicity. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 16552–16564. [Google Scholar] [CrossRef]
- Beraldo, F.H.; Thomas, A.; Kolisnyk, B.; Hirata, P.H.; De Jaeger, X.; Martyn, A.C.; Fan, J.; Goncalves, D.F.; Cowan, M.F.; Masood, T.; et al. Hyperactivity and attention deficits in mice with decreased levels of stress-inducible phosphoprotein 1 (STIP1). Dis. Models Mech. 2015, 8, 1457–1466. [Google Scholar] [CrossRef]
- Lackie, R.E.; Razzaq, A.R.; Farhan, S.M.K.; Qiu, L.R.; Moshitzky, G.; Beraldo, F.H.; Lopes, M.H.; Maciejewski, A.; Gros, R.; Fan, J.; et al. Modulation of hippocampal neuronal resilience during aging by the Hsp70/Hsp90 co-chaperone STI1. J. Neurochem. 2020, 153, 727–758. [Google Scholar] [CrossRef]
- Braunschweig, D.; Krakowiak, P.; Duncanson, P.; Boyce, R.; Hansen, R.L.; Ashwood, P.; Hertz-Picciotto, I.; Pessah, I.N.; Van de Water, J. Autism-specific maternal autoantibodies recognize critical proteins in developing brain. Transl. Psychiatry 2013, 3, e277. [Google Scholar] [CrossRef]
- Vural, B.; Uğurel, E.; Tüzün, E.; Kürtüncü, M.; Zuliani, L.; Cavuş, F.; Içöz, S.; Erdağ, E.; Gül, A.; Güre, A.O.; et al. Anti-neuronal and stress-induced-phosphoprotein 1 antibodies in neuro-Behçet’s disease. J. Neuroimmunol. 2011, 239, 91–97. [Google Scholar] [CrossRef]
- Hughes, A.J.; Daniel, S.E.; Kilford, L.; Lees, A.J. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: A clinico-pathological study of 100 cases. J. Neurol. Neurosurg. Psychiatry 1992, 55, 181–184. [Google Scholar] [CrossRef]
- Ma, D.; Ng, S.H.; Zeng, L.; Zhao, Y.; Tan, E.K. Generation of a human induced pluripotent stem cell (iPSC) line carrying the Parkinson’s disease linked LRRK2 variant S1647T. Stem Cell Res. 2017, 18, 54–56. [Google Scholar] [CrossRef]
- Zhang, X.-M.; Yin, M.; Zhang, M.-H. Cell-based assays for Parkinson’s disease using differentiated human LUHMES cells. Acta Pharmacol. Sin. 2014, 35, 945–956. [Google Scholar] [CrossRef]
- Amrun, S.N.; Yee, W.-X.; Abu Bakar, F.; Lee, B.; Kam, Y.-W.; Lum, F.-M.; Tan, J.J.; Lim, V.W.; Watthanaworawit, W.; Ling, C.; et al. Novel differential linear B-cell epitopes to identify Zika and dengue virus infections in patients. Clin. Transl. Immunol. 2019, 8, e1066. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Belmokhtar, C.A.; Hillion, J.; Ségal-Bendirdjian, E. Staurosporine induces apoptosis through both caspase-dependent and caspase-independent mechanisms. Oncogene 2001, 20, 3354–3362. [Google Scholar] [CrossRef]
- Edmiston, E.; Jones, K.L.; Vu, T.; Ashwood, P.; Van de Water, J. Identification of the antigenic epitopes of maternal autoantibodies in autism spectrum disorders. Brain Behav. Immun. 2018, 69, 399–407. [Google Scholar] [CrossRef]
- Röhl, A.; Wengler, D.; Madl, T.; Lagleder, S.; Tippel, F.; Herrmann, M.; Hendrix, J.; Richter, K.; Hack, G.; Schmid, A.B.; et al. Hsp90 regulates the dynamics of its cochaperone Sti1 and the transfer of Hsp70 between modules. Nat. Commun. 2015, 6, 6655. [Google Scholar] [CrossRef]
- Scheufler, C.; Brinker, A.; Bourenkov, G.; Pegoraro, S.; Moroder, L.; Bartunik, H.; Hartl, F.U.; Moarefi, I. Structure of TPR domain-peptide complexes: Critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell 2000, 101, 199–210. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr.; Travers, P.; Walport, M.; Shlomchik, M.J. Immunobiology: The Immune System in Health and Disease; Garland Science: New York, NY, USA, 2001. [Google Scholar]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Tomasello, G.; Armenia, I.; Molla, G. The Protein Imager: A full-featured online molecular viewer interface with server-side HQ-rendering capabilities. Bioinformatics 2020, 36, 2909–2911. [Google Scholar] [CrossRef]
- Corbett, G.T.; Wang, Z.; Hong, W.; Colom-Cadena, M.; Rose, J.; Liao, M.; Asfaw, A.; Hall, T.C.; Ding, L.; DeSousa, A.; et al. PrP is a central player in toxicity mediated by soluble aggregates of neurodegeneration-causing proteins. Acta Neuropathol. 2020, 139, 503–526. [Google Scholar] [CrossRef] [PubMed]
- Urrea, L.; Ferrer, I.; Gavín, R.; Del Río, J.A. The cellular prion protein (PrP(C)) as neuronal receptor for α-synuclein. Prion 2017, 11, 226–233. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ferreira, D.G.; Temido-Ferreira, M.; Vicente Miranda, H.; Batalha, V.L.; Coelho, J.E.; Szegö, É.M.; Marques-Morgado, I.; Vaz, S.H.; Rhee, J.S.; Schmitz, M.; et al. α-synuclein interacts with PrP(C) to induce cognitive impairment through mGluR5 and NMDAR2B. Nat. Neurosci. 2017, 20, 1569–1579. [Google Scholar] [CrossRef] [PubMed]
- Aulić, S.; Masperone, L.; Narkiewicz, J.; Isopi, E.; Bistaffa, E.; Ambrosetti, E.; Pastore, B.; De Cecco, E.; Scaini, D.; Zago, P.; et al. α-Synuclein Amyloids Hijack Prion Protein to Gain Cell Entry, Facilitate Cell-to-Cell Spreading and Block Prion Replication. Sci. Rep. 2017, 7, 10050. [Google Scholar] [CrossRef]
- Wolfe, K.J.; Ren, H.Y.; Trepte, P.; Cyr, D.M. The Hsp70/90 cochaperone, Sti1, suppresses proteotoxicity by regulating spatial quality control of amyloid-like proteins. Mol. Biol. Cell 2013, 24, 3588–3602. [Google Scholar] [CrossRef]
- Xu, F.; Kula-Eversole, E.; Iwanaszko, M.; Hutchison, A.L.; Dinner, A.; Allada, R. Circadian Clocks Function in Concert with Heat Shock Organizing Protein to Modulate Mutant Huntingtin Aggregation and Toxicity. Cell Rep. 2019, 27, 59–70.e54. [Google Scholar] [CrossRef]
- Lackie, R.E.; Marques-Lopes, J.; Ostapchenko, V.G.; Good, S.; Choy, W.-Y.; van Oosten-Hawle, P.; Pasternak, S.H.; Prado, V.F.; Prado, M.A.M. Increased levels of Stress-inducible phosphoprotein-1 accelerates amyloid-β deposition in a mouse model of Alzheimer’s disease. Acta Neuropathol. Commun. 2020, 8, 143. [Google Scholar] [CrossRef]
- Daturpalli, S.; Waudby, C.A.; Meehan, S.; Jackson, S.E. Hsp90 inhibits α-synuclein aggregation by interacting with soluble oligomers. J. Mol. Biol. 2013, 425, 4614–4628. [Google Scholar] [CrossRef]
- Bhattacharya, K.; Weidenauer, L.; Luengo, T.M.; Pieters, E.C.; Echeverría, P.C.; Bernasconi, L.; Wider, D.; Sadian, Y.; Koopman, M.B.; Villemin, M.; et al. The Hsp70-Hsp90 co-chaperone Hop/Stip1 shifts the proteostatic balance from folding towards degradation. Nat. Commun. 2020, 11, 5975. [Google Scholar] [CrossRef]
- Martín-Peña, A.; Rincón-Limas, D.E.; Fernandez-Fúnez, P. Engineered Hsp70 chaperones prevent Aβ42-induced memory impairments in a Drosophila model of Alzheimer’s disease. Sci. Rep. 2018, 8, 9915. [Google Scholar] [CrossRef]
- Fernandez-Funez, P.; Sanchez-Garcia, J.; de Mena, L.; Zhang, Y.; Levites, Y.; Khare, S.; Golde, T.E.; Rincon-Limas, D.E. Holdase activity of secreted Hsp70 masks amyloid-β42 neurotoxicity in Drosophila. Proc. Natl. Acad. Sci. USA 2016, 113, E5212–E5221. [Google Scholar] [CrossRef] [PubMed]
- Foster, E.M.; Dangla-Valls, A.; Lovestone, S.; Ribe, E.M.; Buckley, N.J. Clusterin in Alzheimer’s Disease: Mechanisms, Genetics, and Lessons From Other Pathologies. Front. Neurosci. 2019, 13, 164. [Google Scholar] [CrossRef] [PubMed]
- Yuste-Checa, P.; Trinkaus, V.A.; Riera-Tur, I.; Imamoglu, R.; Schaller, T.F.; Wang, H.; Dudanova, I.; Hipp, M.S.; Bracher, A.; Hartl, F.U. The extracellular chaperone Clusterin enhances Tau aggregate seeding in a cellular model. Nat. Commun. 2021, 12, 4863. [Google Scholar] [CrossRef]
- Miyakoshi, L.M.; Marques-Coelho, D.; De Souza, L.E.R.; Lima, F.R.S.; Martins, V.R.; Zanata, S.M.; Hedin-Pereira, C. Evidence of a Cell Surface Role for Hsp90 Complex Proteins Mediating Neuroblast Migration in the Subventricular Zone. Front. Cell. Neurosci. 2017, 11, 138. [Google Scholar] [CrossRef]
- Sims, J.D.; McCready, J.; Jay, D.G. Extracellular heat shock protein (Hsp)70 and Hsp90α assist in matrix metalloproteinase-2 activation and breast cancer cell migration and invasion. PLoS ONE 2011, 6, e18848. [Google Scholar] [CrossRef]
- Galiano-Landeira, J.; Torra, A.; Vila, M.; Bové, J. CD8 T cell nigral infiltration precedes synucleinopathy in early stages of Parkinson’s disease. Brain J. Neurol. 2020, 143, 3717–3733. [Google Scholar] [CrossRef]
- Lindestam Arlehamn, C.S.; Dhanwani, R.; Pham, J.; Kuan, R.; Frazier, A.; Rezende Dutra, J.; Phillips, E.; Mallal, S.; Roederer, M.; Marder, K.S.; et al. α-Synuclein-specific T cell reactivity is associated with preclinical and early Parkinson’s disease. Nat. Commun. 2020, 11, 1875. [Google Scholar] [CrossRef]
- Matheoud, D.; Cannon, T.; Voisin, A.; Penttinen, A.M.; Ramet, L.; Fahmy, A.M.; Ducrot, C.; Laplante, A.; Bourque, M.J.; Zhu, L.; et al. Intestinal infection triggers Parkinson’s disease-like symptoms in Pink1(-/-) mice. Nature 2019, 571, 565–569. [Google Scholar] [CrossRef]
- Tansey, M.G.; Wallings, R.L.; Houser, M.C.; Herrick, M.K.; Keating, C.E.; Joers, V. Inflammation and immune dysfunction in Parkinson disease. Nat. Rev. Immunol. 2022. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Parameters | Healthy Controls | Parkinson’s Disease Patients |
|---|---|---|
| Age (mean ± standard deviation) | 63.1 (±8) | 62.94 (±8.48) |
| Gender (Sample size) | Males, n = 27 Females, n = 23 | Males, n = 27 Females, n = 23 |
| Hoehn and Yahr Staging (%) | Not applicable | ≤2 (45%) >2 (55%) |
| Ethnicity (Sample size) | Chinese, n = 50 | Chinese, n = 48 Eurasian, n = 1 Indian, n = 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, J.S.Y.; Lee, B.; Lim, J.; Ma, D.R.; Goh, J.X.; Goh, S.Y.; Gulam, M.Y.; Koh, S.M.; Lee, W.W.; Feng, L.; et al. Parkinson’s Disease-Specific Autoantibodies against the Neuroprotective Co-Chaperone STIP1. Cells 2022, 11, 1649. https://doi.org/10.3390/cells11101649
Tan JSY, Lee B, Lim J, Ma DR, Goh JX, Goh SY, Gulam MY, Koh SM, Lee WW, Feng L, et al. Parkinson’s Disease-Specific Autoantibodies against the Neuroprotective Co-Chaperone STIP1. Cells. 2022; 11(10):1649. https://doi.org/10.3390/cells11101649
Chicago/Turabian StyleTan, Jolene Su Yi, Bernett Lee, Jackwee Lim, Dong Rui Ma, Jia Xin Goh, Suh Yee Goh, Muhammad Yaaseen Gulam, Ser Mei Koh, Weiling Wendy Lee, Lei Feng, and et al. 2022. "Parkinson’s Disease-Specific Autoantibodies against the Neuroprotective Co-Chaperone STIP1" Cells 11, no. 10: 1649. https://doi.org/10.3390/cells11101649
APA StyleTan, J. S. Y., Lee, B., Lim, J., Ma, D. R., Goh, J. X., Goh, S. Y., Gulam, M. Y., Koh, S. M., Lee, W. W., Feng, L., Wang, Q., Chao, Y., Rötzschke, O., & Tan, E. K. (2022). Parkinson’s Disease-Specific Autoantibodies against the Neuroprotective Co-Chaperone STIP1. Cells, 11(10), 1649. https://doi.org/10.3390/cells11101649