
SMER28 Attenuates PI3K/mTOR Signaling by Direct Inhibition of PI3K p110 Delta

 ,
,  , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibodies and Chemical Compounds
2.2. Bacterial Strain and Cell Line Culture
2.3. Cell Lysate, Protein Measurements and Western Blotting
2.4. RT-qPCR
2.5. Cell Proliferation Assay
2.6. Annexin V Apoptosis and Necrosis Assay
2.7. Cell Viability Assay
2.8. Cell Cycle Analysis
2.9. Cell Scattering Assay
2.10. Growth Factor Response Assay
2.11. PI3 Kinase Inhibitor Assay
2.12. Gentamycin Protection Assay
2.13. Planktonic Bacterial Growth Assay
2.14. Immunofluorescence
2.15. Image Acquisition
2.16. Data Processing and Statistical Analyses
3. Results
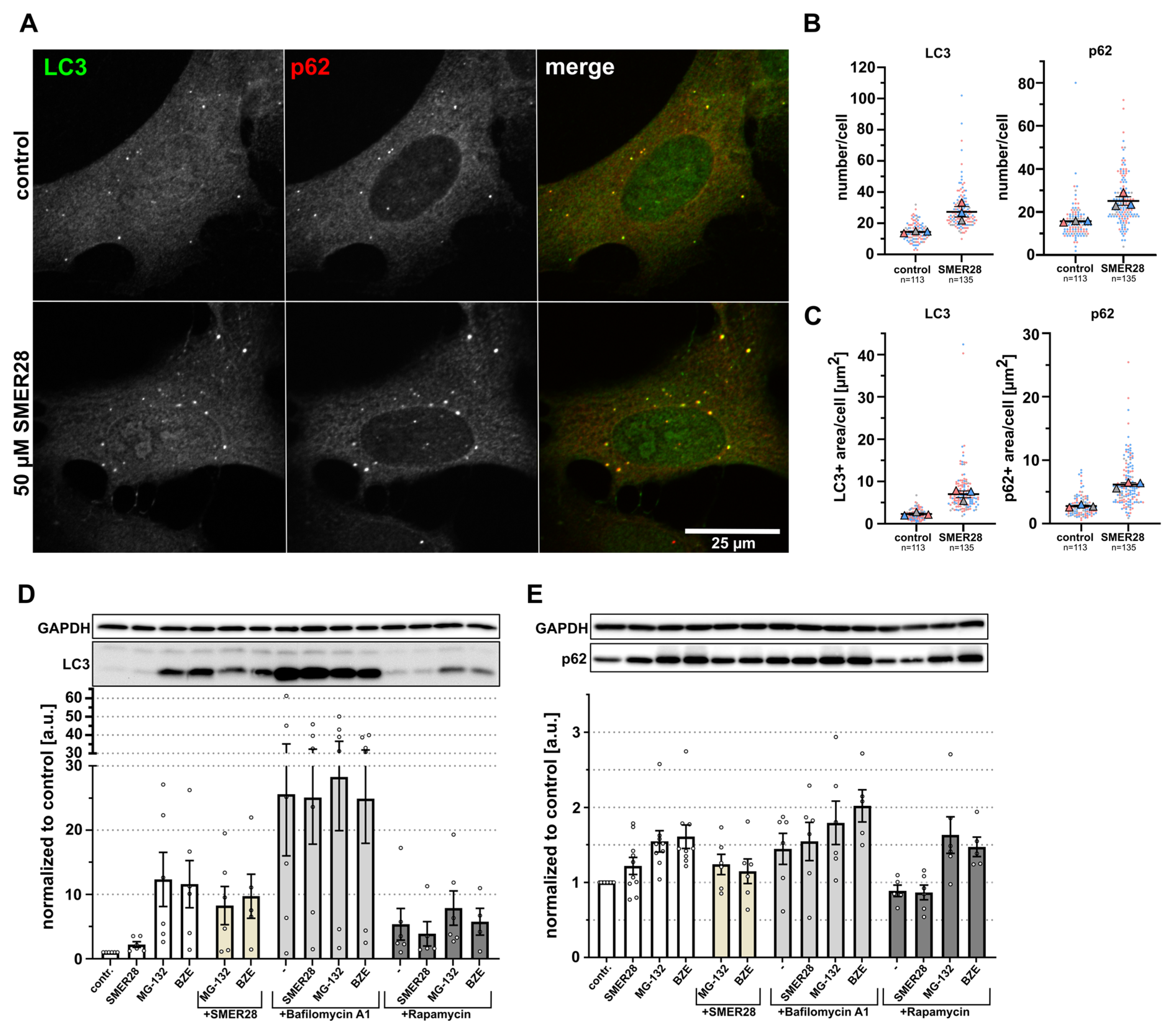
3.1. SMER28 Modestly Increases Autophagy
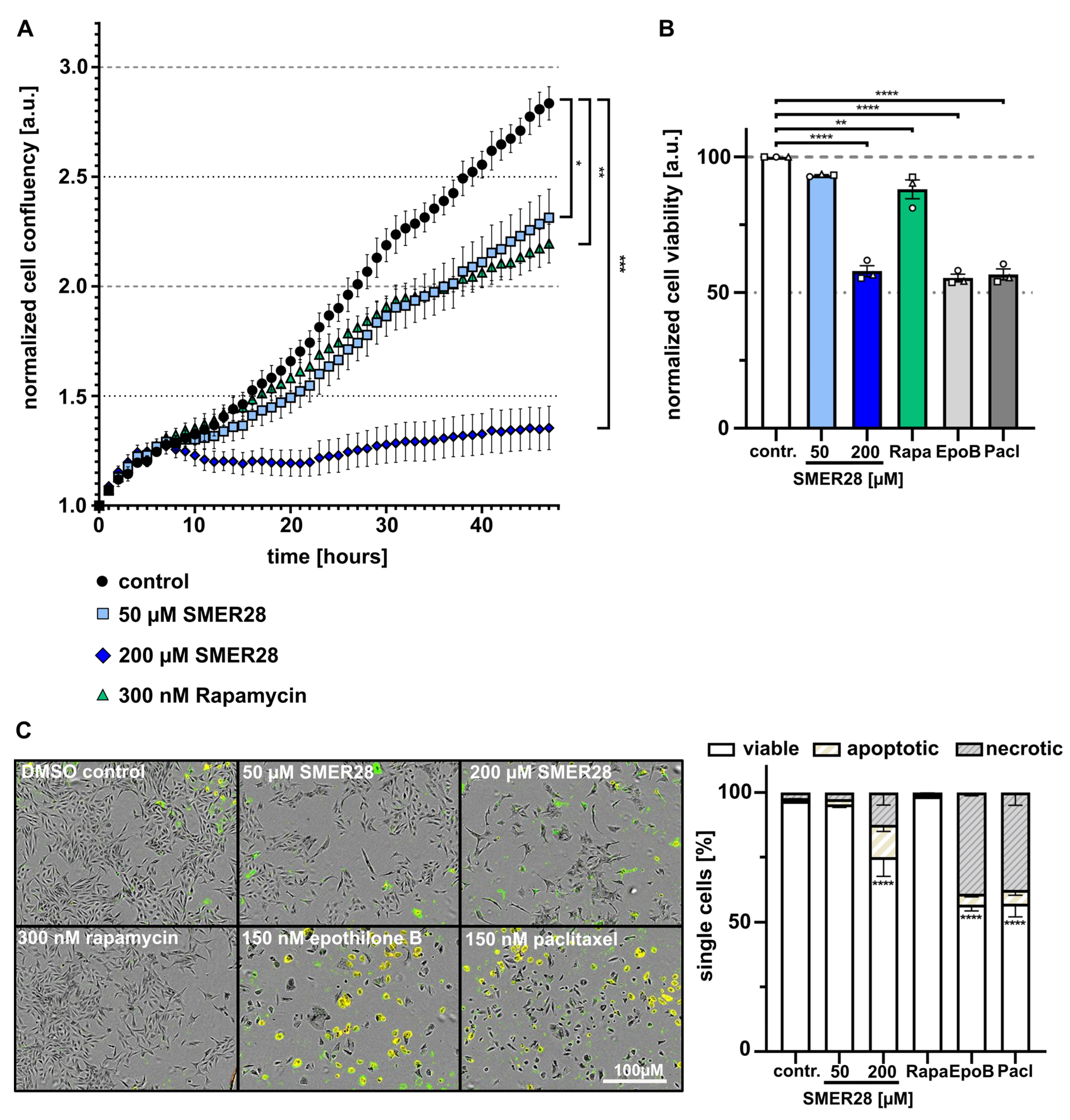
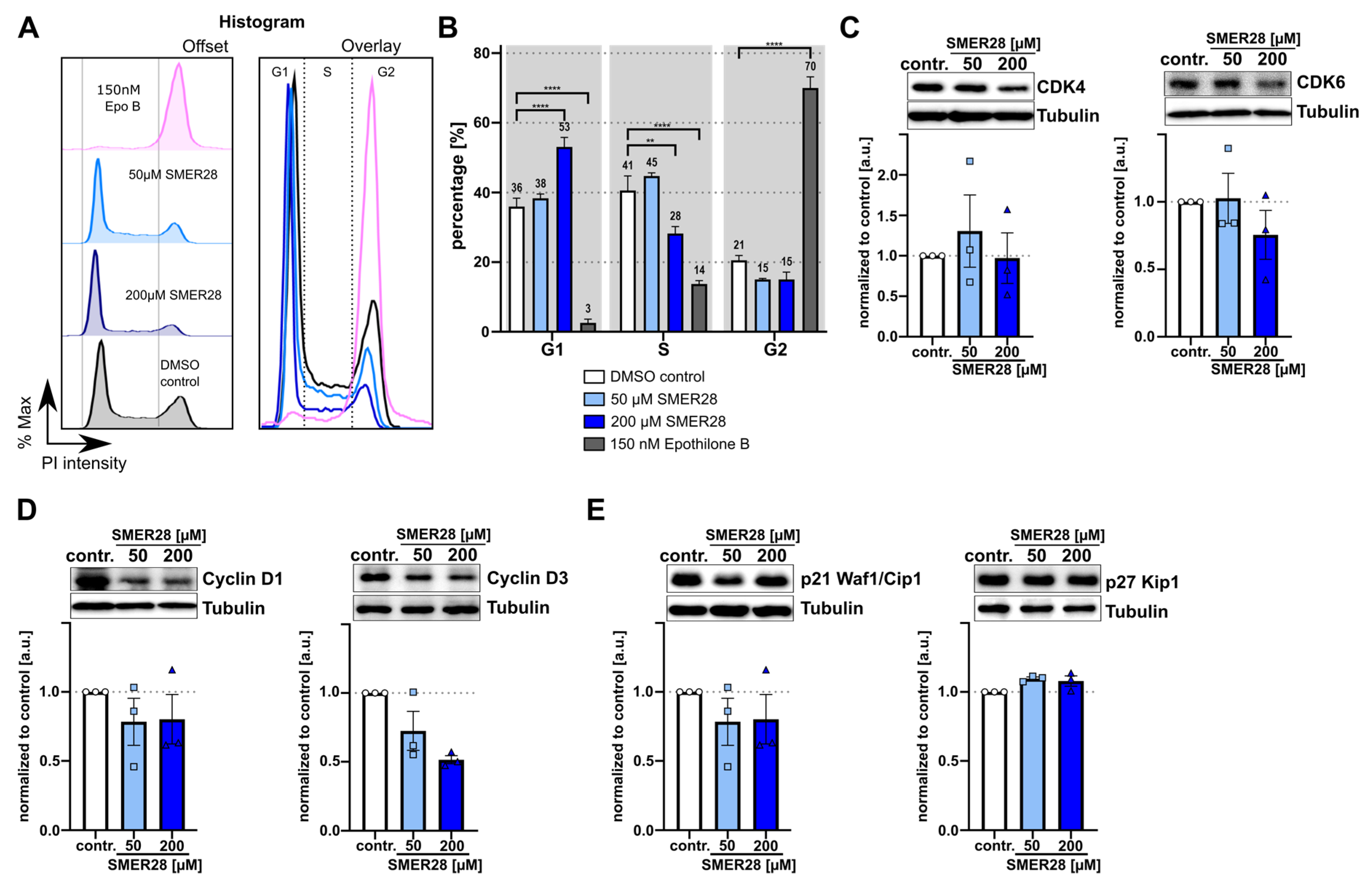
3.2. SMER28 Treatment Leads to Growth Arrest
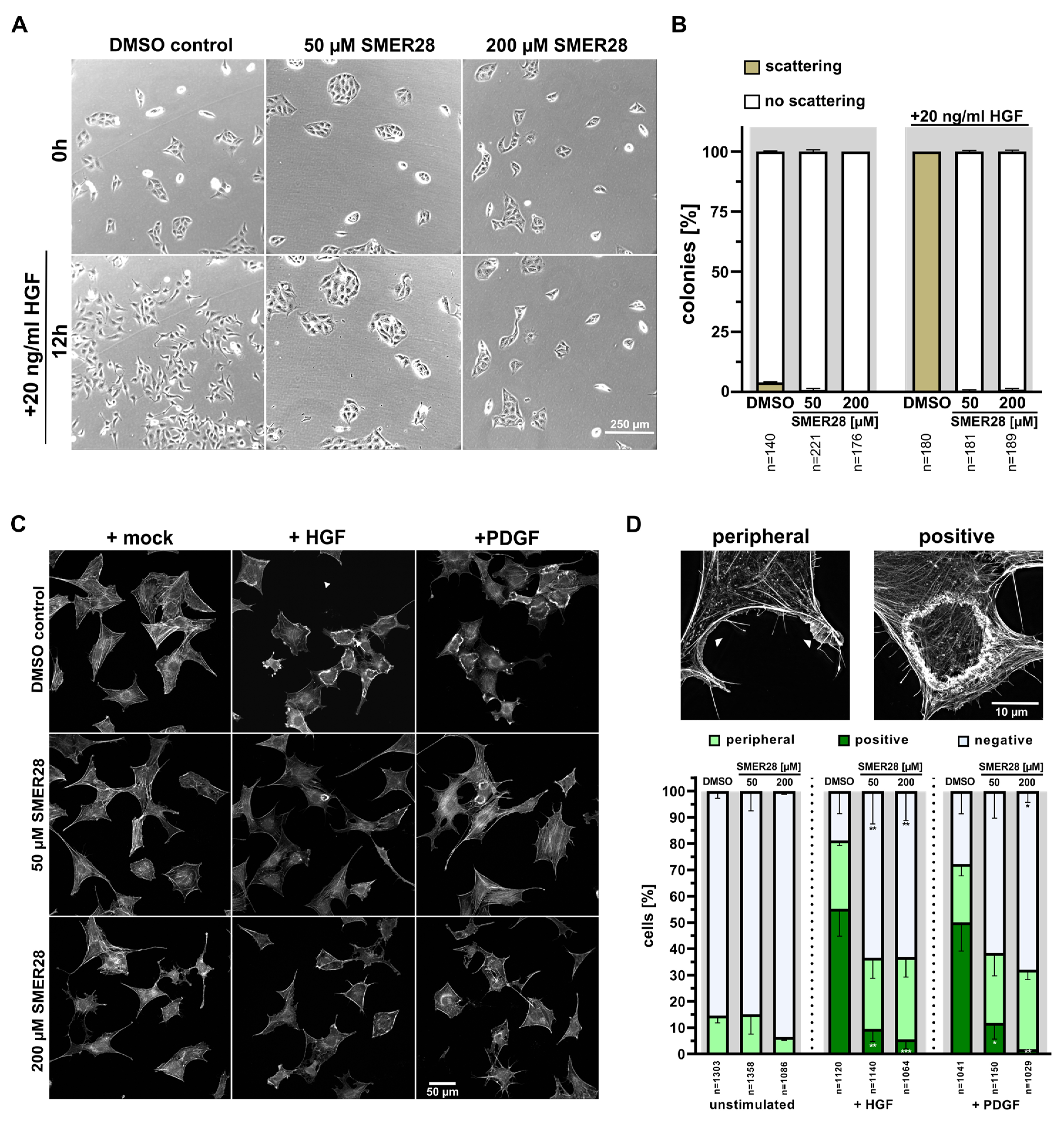
3.3. SMER28 Interferes with Receptor Tyrosine Kinase Signaling
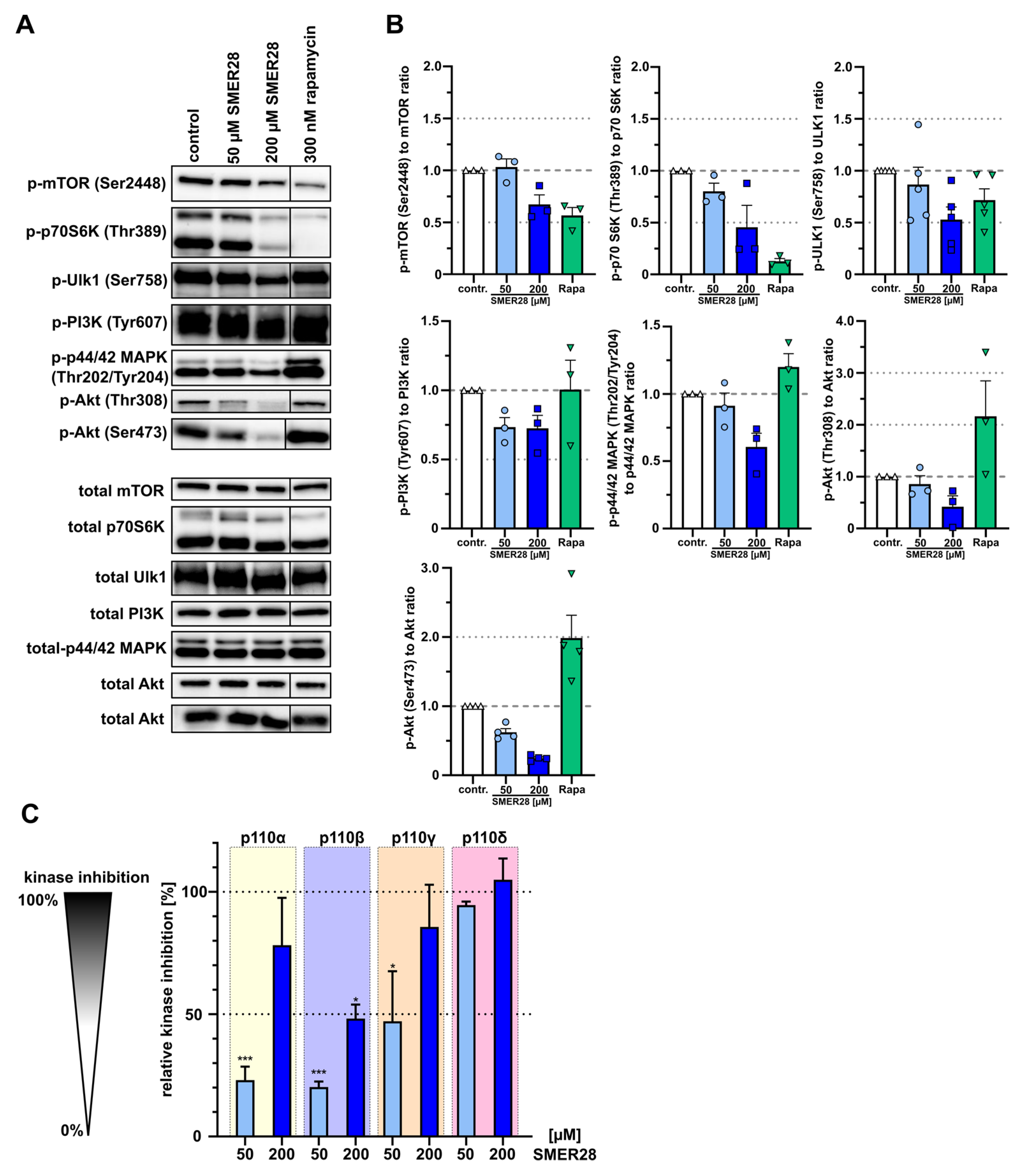
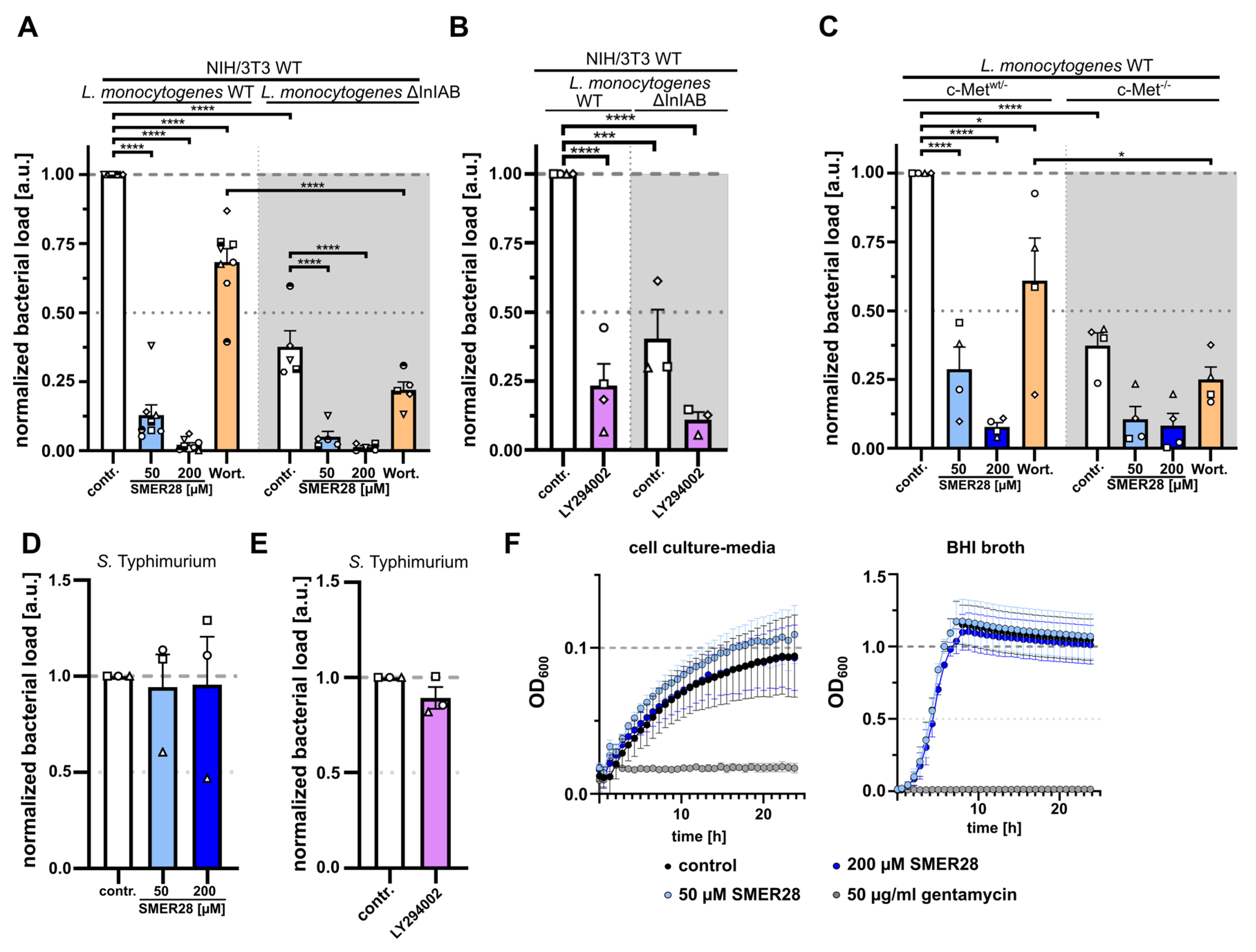
3.4. SMER28 Directly Targets PI3K Subunits Gamma and Delta
3.5. SMER28 Significantly Affects Viability of B Cell Lymphoma Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dibble, C.C.; Cantley, L.C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015, 25, 545–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Codogno, P.; Mehrpour, M.; Proikas-Cezanne, T. Canonical and non-canonical autophagy: Variations on a common theme of self-eating? Nat. Rev. Mol. Cell Biol. 2011, 13, 7–12. [Google Scholar] [CrossRef] [PubMed]
- McEwan, D.G. Host-pathogen interactions and subversion of autophagy. Essays Biochem. 2017, 61, 687–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorpe, L.M.; Yuzugullu, H.; Zhao, J.J. PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 2015, 15, 7–24. [Google Scholar] [CrossRef]
- Poillet-Perez, L.; Sarry, J.E.; Joffre, C. Autophagy is a major metabolic regulator involved in cancer therapy resistance. Cell Rep. 2021, 36, 109528. [Google Scholar] [CrossRef]
- Cianfanelli, V.; Fuoco, C.; Lorente, M.; Salazar, M.; Quondamatteo, F.; Gherardini, P.F.; De Zio, D.; Nazio, F.; Antonioli, M.; D’Orazio, M.; et al. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat. Cell Biol. 2015, 17, 20–30. [Google Scholar] [CrossRef] [Green Version]
- Wijshake, T.; Zou, Z.; Chen, B.; Zhong, L.; Xiao, G.; Xie, Y.; Doench, J.G.; Bennett, L.; Levine, B. Tumor-suppressor function of Beclin 1 in breast cancer cells requires E-cadherin. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.; Chen, H.Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gelinas, C.; et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009, 137, 1062–1075. [Google Scholar] [CrossRef] [Green Version]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strohecker, A.M.; Guo, J.Y.; Karsli-Uzunbas, G.; Price, S.M.; Chen, G.J.; Mathew, R.; McMahon, M.; White, E. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov. 2013, 3, 1272–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumont, R.A.; Tamma, M.; Braun, F.; Borkowski, S.; Reubi, J.C.; Maecke, H.; Weber, W.A.; Mansi, R. Targeted radiotherapy of prostate cancer with a gastrin-releasing peptide receptor antagonist is effective as monotherapy and in combination with rapamycin. J. Nucl. Med. 2013, 54, 762–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eshleman, J.S.; Carlson, B.L.; Mladek, A.C.; Kastner, B.D.; Shide, K.L.; Sarkaria, J.N. Inhibition of the mammalian target of rapamycin sensitizes U87 xenografts to fractionated radiation therapy. Cancer Res. 2002, 62, 7291–7297. [Google Scholar]
- Kim, K.W.; Hwang, M.; Moretti, L.; Jaboin, J.J.; Cha, Y.I.; Lu, B. Autophagy upregulation by inhibitors of caspase-3 and mTOR enhances radiotherapy in a mouse model of lung cancer. Autophagy 2008, 4, 659–668. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [Green Version]
- Tewari, D.; Patni, P.; Bishayee, A.; Sah, A.N.; Bishayee, A. Natural products targeting the PI3K-Akt-mTOR signaling pathway in cancer: A novel therapeutic strategy. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef]
- Popova, N.V.; Jucker, M. The Role of mTOR Signaling as a Therapeutic Target in Cancer. Int. J. Mol. Sci. 2021, 22, 1743. [Google Scholar] [CrossRef]
- Sarkar, S.; Perlstein, E.O.; Imarisio, S.; Pineau, S.; Cordenier, A.; Maglathlin, R.L.; Webster, J.A.; Lewis, T.A.; O’Kane, C.J.; Schreiber, S.L.; et al. Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nat. Chem. Biol. 2007, 3, 331–338. [Google Scholar] [CrossRef] [Green Version]
- Bladt, F.; Riethmacher, D.; Isenmann, S.; Aguzzi, A.; Birchmeier, C. Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nature 1995, 376, 768–771. [Google Scholar] [CrossRef]
- Jat, P.S.; Sharp, P.A. Large T antigens of simian virus 40 and polyomavirus efficiently establish primary fibroblasts. J. Virol. 1986, 59, 746–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steffen, A.; Kage, F.; Rottner, K. Imaging the Molecular Machines That Power Cell Migration. Methods Mol. Biol. 2018, 1749, 257–277. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Lord, S.J.; Velle, K.B.; Mullins, R.D.; Fritz-Laylin, L.K. SuperPlots: Communicating reproducibility and variability in cell biology. J Cell Biol 2020, 219, 6. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]
- Kisselev, A.F.; van der Linden, W.A.; Overkleeft, H.S. Proteasome inhibitors: An expanding army attacking a unique target. Chem. Biol. 2012, 19, 99–115. [Google Scholar] [CrossRef] [Green Version]
- Bao, W.; Gu, Y.; Ta, L.; Wang, K.; Xu, Z. Induction of autophagy by the MG132 proteasome inhibitor is associated with endoplasmic reticulum stress in MCF7 cells. Mol. Med. Rep. 2016, 13, 796–804. [Google Scholar] [CrossRef] [Green Version]
- Ji, M.M.; Lee, J.M.; Mon, H.; Xu, J.; Tatsuke, T.; Kusakabe, T. Proteasome inhibitor MG132 impairs autophagic flux through compromising formation of autophagosomes in Bombyx cells. Biochem. Biophys. Res. Commun. 2016, 479, 690–696. [Google Scholar] [CrossRef]
- Benjamin, D.; Colombi, M.; Moroni, C.; Hall, M.N. Rapamycin passes the torch: A new generation of mTOR inhibitors. Nat. Rev. Drug Discov. 2011, 10, 868–880. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Elazar, Z.; Seglen, P.O.; Rubinsztein, D.C. Does bafilomycin A1 block the fusion of autophagosomes with lysosomes? Autophagy 2008, 4, 849–850. [Google Scholar] [CrossRef] [Green Version]
- Gerth, K.; Bedorf, N.; Hofle, G.; Irschik, H.; Reichenbach, H. Epothilons A and B: Antifungal and cytotoxic compounds from Sorangium cellulosum (Myxobacteria). Production, physico-chemical and biological properties. J. Antibiot. 1996, 49, 560–563. [Google Scholar] [CrossRef] [PubMed]
- Wani, M.C.; Taylor, H.L.; Wall, M.E.; Coggon, P.; McPhail, A.T. Plant antitumor agents. VI. The isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J. Am. Chem. Soc. 1971, 93, 2325–2327. [Google Scholar] [CrossRef] [PubMed]
- Reichenbach, H.; Hofle, G. Discovery and development of the epothilones: A novel class of antineoplastic drugs. Drugs R D 2008, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Musgrove, E.A.; Caldon, C.E.; Barraclough, J.; Stone, A.; Sutherland, R.L. Cyclin D as a therapeutic target in cancer. Nat. Rev. Cancer 2011, 11, 558–572. [Google Scholar] [CrossRef]
- Yu, J.S.; Cui, W. Proliferation, survival and metabolism: The role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development 2016, 143, 3050–3060. [Google Scholar] [CrossRef] [Green Version]
- Hoon, J.L.; Wong, W.K.; Koh, C.G. Functions and regulation of circular dorsal ruffles. Mol. Cell Biol. 2012, 32, 4246–4257. [Google Scholar] [CrossRef] [Green Version]
- Steffen, A.; Ladwein, M.; Dimchev, G.A.; Hein, A.; Schwenkmezger, L.; Arens, S.; Ladwein, K.I.; Margit Holleboom, J.; Schur, F.; Victor Small, J.; et al. Rac function is crucial for cell migration but is not required for spreading and focal adhesion formation. J. Cell Sci. 2013, 126, 4572–4588. [Google Scholar] [CrossRef] [Green Version]
- Chai, M.; Jiang, M.; Vergnes, L.; Fu, X.; de Barros, S.C.; Doan, N.B.; Huang, W.; Chu, J.; Jiao, J.; Herschman, H.; et al. Stimulation of Hair Growth by Small Molecules that Activate Autophagy. Cell Rep. 2019, 27, 3413–3421.e3. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.A.; Pacold, M.E.; Cervantes, C.L.; Lim, D.; Lou, H.J.; Ottina, K.; Gray, N.S.; Turk, B.E.; Yaffe, M.B.; Sabatini, D.M. mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science 2013, 341, 1236566. [Google Scholar] [CrossRef] [Green Version]
- Castellano, E.; Downward, J. RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes Cancer 2011, 2, 261–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacinto, E.; Facchinetti, V.; Liu, D.; Soto, N.; Wei, S.; Jung, S.Y.; Huang, Q.; Qin, J.; Su, B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 2006, 127, 125–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alessi, D.R.; James, S.R.; Downes, C.P.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; Cohen, P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr. Biol. 1997, 7, 261–269. [Google Scholar] [CrossRef] [Green Version]
- Han, E.K.; Leverson, J.D.; McGonigal, T.; Shah, O.J.; Woods, K.W.; Hunter, T.; Giranda, V.L.; Luo, Y. Akt inhibitor A-443654 induces rapid Akt Ser-473 phosphorylation independent of mTORC1 inhibition. Oncogene 2007, 26, 5655–5661. [Google Scholar] [CrossRef] [Green Version]
- Breuleux, M.; Klopfenstein, M.; Stephan, C.; Doughty, C.A.; Barys, L.; Maira, S.M.; Kwiatkowski, D.; Lane, H.A. Increased AKT S473 phosphorylation after mTORC1 inhibition is rictor dependent and does not predict tumor cell response to PI3K/mTOR inhibition. Mol. Cancer Ther. 2009, 8, 742–753. [Google Scholar] [CrossRef] [Green Version]
- Lecuit, M.; Dramsi, S.; Gottardi, C.; Fedor-Chaiken, M.; Gumbiner, B.; Cossart, P. A single amino acid in E-cadherin responsible for host specificity towards the human pathogen Listeria monocytogenes. EMBO J. 1999, 18, 3956–3963. [Google Scholar] [CrossRef] [Green Version]
- Rottner, K.; Stradal, T.E.; Wehland, J. Bacteria-host-cell interactions at the plasma membrane: Stories on actin cytoskeleton subversion. Dev. Cell 2005, 9, 3–17. [Google Scholar] [CrossRef] [Green Version]
- Jiwani, S.; Wang, Y.; Dowd, G.C.; Gianfelice, A.; Pichestapong, P.; Gavicherla, B.; Vanbennekom, N.; Ireton, K. Identification of components of the host type IA phosphoinositide 3-kinase pathway that promote internalization of Listeria monocytogenes. Infect. Immun. 2012, 80, 1252–1266. [Google Scholar] [CrossRef] [Green Version]
- Tang, P.; Sutherland, C.L.; Gold, M.R.; Finlay, B.B. Listeria monocytogenes invasion of epithelial cells requires the MEK-1/ERK-2 mitogen-activated protein kinase pathway. Infect. Immun. 1998, 66, 1106–1112. [Google Scholar] [CrossRef] [Green Version]
- Bosse, T.; Ehinger, J.; Czuchra, A.; Benesch, S.; Steffen, A.; Wu, X.; Schloen, K.; Niemann, H.H.; Scita, G.; Stradal, T.E.; et al. Cdc42 and phosphoinositide 3-kinase drive Rac-mediated actin polymerization downstream of c-Met in distinct and common pathways. Mol. Cell Biol. 2007, 27, 6615–6628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, E.H.; Pacold, M.E.; Perisic, O.; Stephens, L.; Hawkins, P.T.; Wymann, M.P.; Williams, R.L. Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol. Cell 2000, 6, 909–919. [Google Scholar] [CrossRef]
- Cooper, K.G.; Winfree, S.; Malik-Kale, P.; Jolly, C.; Ireland, R.; Knodler, L.A.; Steele-Mortimer, O. Activation of Akt by the bacterial inositol phosphatase, SopB, is wortmannin insensitive. PLoS ONE 2011, 6, e22260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roppenser, B.; Kwon, H.; Canadien, V.; Xu, R.; Devreotes, P.N.; Grinstein, S.; Brumell, J.H. Multiple host kinases contribute to Akt activation during Salmonella infection. PLoS ONE 2013, 8, e71015. [Google Scholar] [CrossRef] [Green Version]
- Madsen, R.R.; Vanhaesebroeck, B. Cracking the context-specific PI3K signaling code. Sci. Signal. 2020, 13, 2940. [Google Scholar] [CrossRef] [Green Version]
- Okkenhaug, K.; Graupera, M.; Vanhaesebroeck, B. Targeting PI3K in Cancer: Impact on Tumor Cells, Their Protective Stroma, Angiogenesis, and Immunotherapy. Cancer Discov. 2016, 6, 1090–1105. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, H.; Hideshima, T.; Fulciniti, M.; Perrone, G.; Miura, N.; Yasui, H.; Okawa, Y.; Kiziltepe, T.; Santo, L.; Vallet, S.; et al. PI3K/p110{delta} is a novel therapeutic target in multiple myeloma. Blood 2010, 116, 1460–1468. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Modi, P.; Newcomb, T.; Queva, C.; Gandhi, V. Idelalisib: First-in-Class PI3K Delta Inhibitor for the Treatment of Chronic Lymphocytic Leukemia, Small Lymphocytic Leukemia, and Follicular Lymphoma. Clin. Cancer Res. 2015, 21, 1537–1542. [Google Scholar] [CrossRef] [Green Version]
- von Keudell, G.; Moskowitz, A.J. The Role of PI3K Inhibition in Lymphoid Malignancies. Curr. Hematol. Malig. Rep. 2019, 14, 405–413. [Google Scholar] [CrossRef]
- Lannutti, B.J.; Meadows, S.A.; Herman, S.E.; Kashishian, A.; Steiner, B.; Johnson, A.J.; Byrd, J.C.; Tyner, J.W.; Loriaux, M.M.; Deininger, M.; et al. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood 2011, 117, 591–594. [Google Scholar] [CrossRef] [Green Version]
- Lampson, B.L.; Kasar, S.N.; Matos, T.R.; Morgan, E.A.; Rassenti, L.; Davids, M.S.; Fisher, D.C.; Freedman, A.S.; Jacobson, C.A.; Armand, P.; et al. Idelalisib given front-line for treatment of chronic lymphocytic leukemia causes frequent immune-mediated hepatotoxicity. Blood 2016, 128, 195–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, T.J.; Michot, J.M.; Ribrag, V. Can Next-Generation PI3K Inhibitors Unlock the Full Potential of the Class in Patients With B-Cell Lymphoma? Clin. Lymphoma Myeloma Leuk 2021, 21, 8–20.e3. [Google Scholar] [CrossRef] [PubMed]
- Koukourakis, M.I.; Giatromanolaki, A.; Fylaktakidou, K.; Sivridis, E.; Zois, C.E.; Kalamida, D.; Mitrakas, A.; Pouliliou, S.; Karagounis, I.V.; Simopoulos, K.; et al. SMER28 is a mTOR-independent small molecule enhancer of autophagy that protects mouse bone marrow and liver against radiotherapy. Investig. New Drugs 2018, 36, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, A.K.; Gohr, C.M.; Mitton-Fitzgerald, E.; Grewal, R.; Ninomiya, J.; Coyne, C.B.; Jackson, W.T. Autophagy modulates articular cartilage vesicle formation in primary articular chondrocytes. J. Biol. Chem. 2015, 290, 13028–13038. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Bustos, V.; Flajolet, M.; Greengard, P. A small-molecule enhancer of autophagy decreases levels of Abeta and APP-CTF via Atg5-dependent autophagy pathway. FASEB J. 2011, 25, 1934–1942. [Google Scholar] [CrossRef] [Green Version]
- Onishi, K.; Higuchi, M.; Asakura, T.; Masuyama, N.; Gotoh, Y. The PI3K-Akt pathway promotes microtubule stabilization in migrating fibroblasts. Genes Cells 2007, 12, 535–546. [Google Scholar] [CrossRef]
- Kirchenwitz, M.; Stahnke, S.; Melcher, L.; Ham, M.; Rottner, K.; Steffen, A.; Stradal, T.E. The autophagy inducer SMER28 attenuates microtubule dynamics mediating neuroprotection. Sci. Rep. under revision. 2022. [Google Scholar]
- Saeed, M.F.; Kolokoltsov, A.A.; Freiberg, A.N.; Holbrook, M.R.; Davey, R.A. Phosphoinositide-3 kinase-Akt pathway controls cellular entry of Ebola virus. PLoS Pathog. 2008, 4, e1000141. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Yang, L.; Fikrig, E.; Wang, P. An essential role of PI3K in the control of West Nile virus infection. Sci. Rep. 2017, 7, 3724. [Google Scholar] [CrossRef] [Green Version]
- Dubich, T.; Dittrich, A.; Bousset, K.; Geffers, R.; Busche, G.; Koster, M.; Hauser, H.; Schulz, T.F.; Wirth, D. 3D culture conditions support Kaposi’s sarcoma herpesvirus (KSHV) maintenance and viral spread in endothelial cells. J. Mol. Med. 2021, 99, 425–438. [Google Scholar] [CrossRef]
- Carpier, J.M.; Lucas, C.L. Epstein-Barr Virus Susceptibility in Activated PI3Kdelta Syndrome (APDS) Immunodeficiency. Front. Immunol. 2017, 8, 2005. [Google Scholar] [CrossRef] [Green Version]
- Palma, G.; Pasqua, T.; Silvestri, G.; Rocca, C.; Gualtieri, P.; Barbieri, A.; De Bartolo, A.; De Lorenzo, A.; Angelone, T.; Avolio, E.; et al. PI3Kdelta Inhibition as a Potential Therapeutic Target in COVID-19. Front. Immunol. 2020, 11, 2094. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kirchenwitz, M.; Stahnke, S.; Prettin, S.; Borowiak, M.; Menke, L.; Sieben, C.; Birchmeier, C.; Rottner, K.; Stradal, T.E.B.; Steffen, A. SMER28 Attenuates PI3K/mTOR Signaling by Direct Inhibition of PI3K p110 Delta. Cells 2022, 11, 1648. https://doi.org/10.3390/cells11101648
Kirchenwitz M, Stahnke S, Prettin S, Borowiak M, Menke L, Sieben C, Birchmeier C, Rottner K, Stradal TEB, Steffen A. SMER28 Attenuates PI3K/mTOR Signaling by Direct Inhibition of PI3K p110 Delta. Cells. 2022; 11(10):1648. https://doi.org/10.3390/cells11101648
Chicago/Turabian StyleKirchenwitz, Marco, Stephanie Stahnke, Silvia Prettin, Malgorzata Borowiak, Laura Menke, Christian Sieben, Carmen Birchmeier, Klemens Rottner, Theresia E. B. Stradal, and Anika Steffen. 2022. "SMER28 Attenuates PI3K/mTOR Signaling by Direct Inhibition of PI3K p110 Delta" Cells 11, no. 10: 1648. https://doi.org/10.3390/cells11101648
APA StyleKirchenwitz, M., Stahnke, S., Prettin, S., Borowiak, M., Menke, L., Sieben, C., Birchmeier, C., Rottner, K., Stradal, T. E. B., & Steffen, A. (2022). SMER28 Attenuates PI3K/mTOR Signaling by Direct Inhibition of PI3K p110 Delta. Cells, 11(10), 1648. https://doi.org/10.3390/cells11101648