
In Vitro and In Vivo Models to Study Nephropathic Cystinosis


Abstract
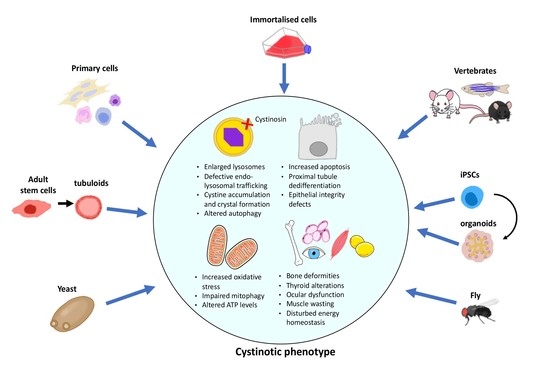
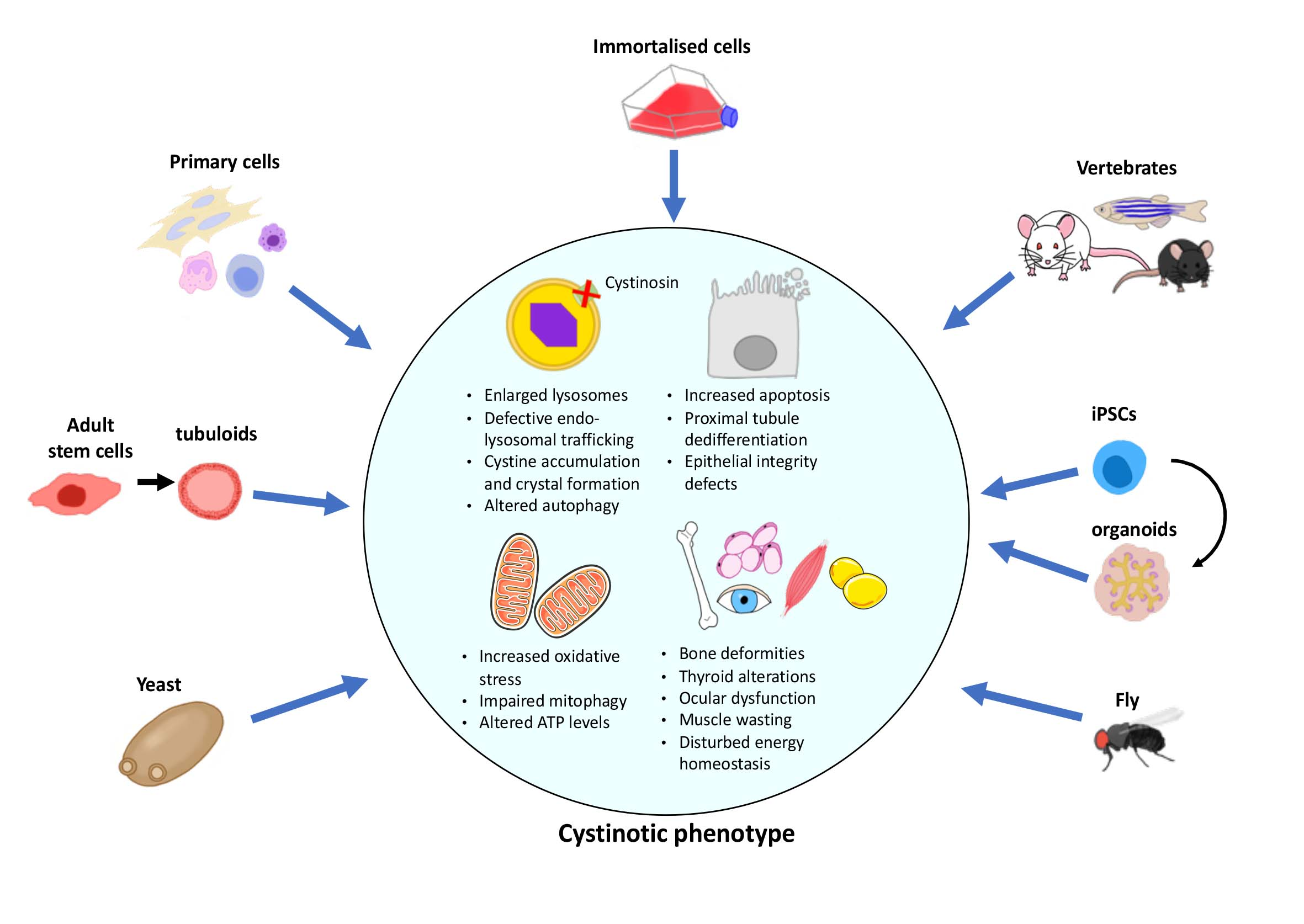
1. Introduction
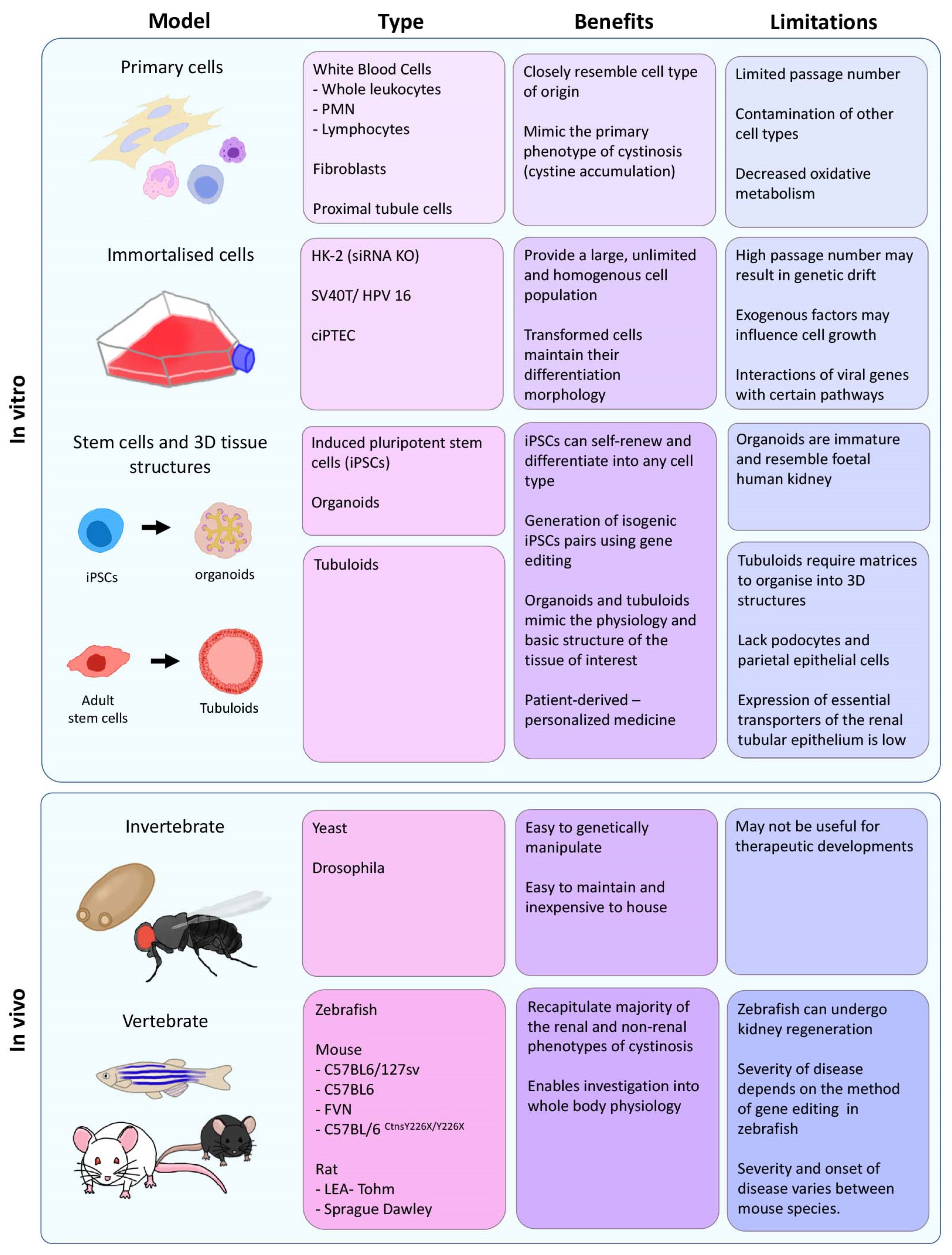
2. In Vitro Models
2.1. Primary Human Cells
2.2. Immortalised Cell Lines
2.3. Modelling Cystinosis by siRNA Knockdown
2.4. Induced Pluripotent Stem Cells and Kidney Organoids
2.5. Tubuloids
3. In Vivo Models
3.1. Yeast
3.2. Mouse
3.2.1. C57BL6/129sv-
3.2.2. C57BL/6 and FVB/N-
3.2.3. C57BL/6 CtnsY226X/Y226X Nonsense Mutant Mouse
3.3. Zebrafish
3.4. Rat
3.4.1. LEA/Tohm-
3.4.2. Sprague Dawley
3.5. Drosophila
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gahl, W.; Thoene, J.; Schneider, J. Cystinosis. N. Engl. J. Med. 2002, 347, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Town, M.; Jean, G.; Cherqui, S.; Attard, M.; Forestier, L.; Whitmore, S.A.; Callen, D.F.; Gribouval, O.; Broyer, M.; Bates, G.P.; et al. A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nat. Genet. 1998, 18, 319–324. [Google Scholar] [CrossRef]
- Kalatzis, V.; Cherqui, S.; Antignac, C.; Gasnier, B. Cystinosin, the protein defective in cystinosis, is a H+-driven lysosomal cystine transporter. EMBO J. 2001, 20, 5940–5949. [Google Scholar] [CrossRef]
- Forster, S.; Scarlett, L.; Lloyd, J.B. Mechanism of cystine reaccumulation by cystinotic fibroblasts in vitro. Biosci. Rep. 1990, 10, 225–229. [Google Scholar] [CrossRef]
- Cherqui, S.; Courtoy, P.J. The renal Fanconi syndrome in cystinosis: Pathogenic insights and therapeutic perspectives. Nat. Rev. Nephrol. 2017, 13, 115–131. [Google Scholar] [CrossRef]
- Bäumner, S.; Weber, L.T. Nephropathic cystinosis: Symptoms, treatment, and perspectives of a systemic disease. Front. Pediatrics 2018, 6, 1–8. [Google Scholar] [CrossRef]
- Schneider, J.A.; Bradley, K.; Seegmiller, J.E. Increased cystine in leukocytes from individuals homozygous and heterozygous for cystinosis. Science 1967, 157, 1321–1322. [Google Scholar] [CrossRef]
- Monnens, L.; Levtchenko, E. Evaluation of the proximal tubular function in hereditary renal Fanconi syndrome. Nephrol. Dial. Transpl. 2008, 23, 2719–2722. [Google Scholar] [CrossRef]
- Mahoney, C.P.; Striker, G.E. Early development of the renal lesions in infantile cystinosis. Pediatric Nephrol. 2000, 15, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Stokes, M.B.; Jernigan, S.; D’Agati, V.D. Infantile nephropathic cystinosis. Kidney Int. 2008, 73, 782–786. [Google Scholar] [CrossRef] [PubMed]
- Elmonem, M.A.; Veys, K.R.; Soliman, N.A.; Van Dyck, M.; Van Den Heuvel, L.P.; Levtchenko, E. Cystinosis: A review. Orphanet J. Rare Dis. 2016, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Trauner, D. Neurocognitive Complications of Cystinosis. J. Pediatrics 2017, 183, S15–S18. [Google Scholar] [CrossRef] [PubMed]
- Servais, A.; Morinière, V.; Grünfeld, J.P.; Noël, L.H.; Goujon, J.M.; Chadefaux-Vekemans, B.; Antignac, C. Late-onset nephropathic cystinosis: Clinical presentation, outcome, and genotyping. Clin. J. Am. Soc. Nephrol. 2008, 3, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Gahl, W.A.; Tietze, F.; De Butler, B.J.; Schulman, J.D. Cysteamine depletes cystinotic leucocyte granular fractions of cystine by the mechanism of disulphide interchange. Biochem. J. 1985, 228, 545–550. [Google Scholar] [CrossRef]
- Jézégou, A.; Llinares, E.; Anne, C.; Kieffer-jaquinod, S.; O’Regan, S.; Aupetit, J.; Chabli, A.; Sagńe, C.; Debacker, C.; Chadefaux-Vekemans, B.; et al. Heptahelical protein PQLC2 is a lysosomal cationic amino acid exporter underlying the action of cysteamine in cystinosis therapy. Proc. Natl. Acad. Sci. USA 2012, 109, E3434–E3443. [Google Scholar] [CrossRef]
- Pisoni, R.L.; Acker, T.L.; Lisowski, K.M.; Lemons, R.M.; Thoene, J.G. A cysteine-specific lysosomal transport system provides a major route for the delivery of thiol to human fibroblast lysosomes: Possible role in supporting lysosomal proteolysis. J. Cell Biol. 1990, 110, 327–335. [Google Scholar] [CrossRef]
- Van Stralen, K.J.; Emma, F.; Jager, K.J.; Verrina, E.; Schaefer, F.; Laube, G.F.; Lewis, M.A.; Levtchenko, E.N. Improvement in the renal prognosis in nephropathic cystinosis. Clin. J. Am. Soc. Nephrol. 2011, 6, 2485–2491. [Google Scholar] [CrossRef]
- Brodin-Sartorius, A.; Tête, M.J.; Niaudet, P.; Antignac, C.; Guest, G.; Ottolenghi, C.; Charbit, M.; Moyse, D.; Legendre, C.; Lesavre, P.; et al. Cysteamine therapy delays the progression of nephropathic cystinosis in late adolescents and adults. Kidney Int. 2012, 81, 179–189. [Google Scholar] [CrossRef]
- Gahl, W.A.; Balog, J.Z.; Kleta, R. Nephropathic cystinosis in adults: Natural history and effects of oral cysteamine therapy. Ann. Intern. Med. 2007, 147, 242–250. [Google Scholar] [CrossRef]
- Kleta, R.; Bernardini, I.; Ueda, M.; Varade, W.S.; Phornphutkul, C.; Krasnewich, D.; Gahl, W.A. Long-term follow-up of well-treated nephropathic cystinosis patients. J. Pediatr. 2004, 145, 555–560. [Google Scholar] [CrossRef]
- Markello, T.C.; Bernardini, I.M.; Gahl, W.A. Improved renal function in children with cystinosis treated with cysteamine. N. Engl. J. Med. 1993, 329, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Nesterova, G.; Williams, C.; Bernardini, I.; Gahl, W.A. Cystinosis: Renal glomerular and renal tubular function in relation to compliance with cystine-depleting therapy. Pediatric Nephrol. 2015, 30, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Gahl, W.A.; Reed, G.F.; Thoene, J.G.; Schulman, J.D.; Rizzo, W.B.; Jonas, A.J.; Denman, D.W.; Schlesselman, J.J.; Corden, B.J.; Schneider, J.A. Cysteamine therapy for children with nephropathic cystinosis. N. Engl. J. Med. 1987, 316, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Dohil, R.; Fidler, M.; Gangoiti, J.A.; Kaskel, F.; Schneider, J.A.; Barshop, B.A. Twice-daily cysteamine bitartrate therapy for children with cystinosis. J. Pediatr. 2010, 156, 71–75. [Google Scholar] [CrossRef]
- Dohil, R.; Cabrera, B.L. Treatment of cystinosis with delayed-release cysteamine: 6-year follow-up. Pediatr. Nephrol. 2013, 28, 507–510. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, G.; Johnson, J.L.; He, J.; Rocca, C.J.; Monfregola, J.; Pestonjamasp, K.; Cherqui, S.; Catz, S.D. Impairment of chaperone-mediated autophagy leads to selective lysosomal degradation defects in the lysosomal storage disease cystinosis. EMBO Mol. Med. 2015, 7, 158–174. [Google Scholar] [CrossRef]
- Rega, L.R.; Polishchuk, E.; Montefusco, S.; Napolitano, G.; Tozzi, G.; Zhang, J.; Bellomo, F.; Taranta, A.; Pastore, A.; Polishchuk, R.; et al. Activation of the transcription factor EB rescues lysosomal abnormalities in cystinotic kidney cells. Kidney Int. 2016, 89, 862–873. [Google Scholar] [CrossRef]
- Andrzejewska, Z.; Nevo, N.; Thomas, L.; Bailleux, A.; Chauvet, V.; Courtoy, P.J.; Chol, M.; Guerrera, I.C.; Antignac, C. Cystinosin is a Component of the Vacuolar H+-ATPase-Ragulator-Rag Complex Controlling Mammalian Target of Rapamycin Complex 1 Signaling. J. Am. Soc. Nephrol. 2016, 27, 1678–1688. [Google Scholar] [CrossRef]
- Hollywood, J.A.; Przepiorski, A.; D’Souza, R.F.; Sreebhavan, S.; Wolvetang, E.J.; Harrison, P.T.; Davidson, A.J.; Holm, T.M. Use of Human Induced Pluripotent Stem Cells and Kidney Organoids To Develop a Cysteamine/mTOR Inhibition Combination Therapy for Cystinosis. J. Am. Soc. Nephrol. 2020, 31, 962–982. [Google Scholar] [CrossRef]
- Schneider, J.A.; Rosenbloom, F.M.; Bradley, K.H.; Seegmiller, J.E. Increased free-cystine content of fibroblasts cultured from patients with cystinosis. Biochem. Biophys. Res. Commun. 1967, 29, 527–531. [Google Scholar] [CrossRef]
- Patrick, A.D.; Lake, B.D. Cystinosis: Electron microscopic evidence of lysosomal storage of cystine in lymph node. J. Clin. Pathol. 1968, 21, 571–575. [Google Scholar] [CrossRef]
- Reeves, J.P. Accumulation of amino acids by lysosomes incubated with amino acid methyl esters. J. Biol. Chem. 1979, 254, 8914–8921. [Google Scholar] [CrossRef]
- Gahl, W.A.; Bashan, N.; Tietze, F.; Bernardini, I.; Schulman, J.D. Cystine transport is defective in isolated leukocyte lysosomes from patients with cystinosis. Science 1982, 217, 1263–1265. [Google Scholar] [CrossRef] [PubMed]
- Gahl, W.A.; Tietze, F.; Bashan, N.; Steinherz, R.; Schulman, J.D. Defective cystine exodus from isolated lysosome-rich fractions of cystinotic leucocytes. J. Biol. Chem. 1982, 257, 9570–9575. [Google Scholar] [CrossRef]
- Jonas, A.J.; Greene, A.A.; Smith, M.L.; Schneider, J.A. Cystine accumulation and loss in normal, heterozygous, and cystinotic fibroblasts. Proc. Natl. Acad. Sci. USA. 1982, 79, 4442–4445. [Google Scholar] [CrossRef]
- Jonas, A.; Smith, M.; Allison, W.; Laikind, P.; Greene, A.; Schneider, J. Proton translocating ATPase and lysosomal cystine transport. J. Biol. Chem. 1983, 258, 11727–11730. [Google Scholar] [CrossRef]
- Wilmer, M.J.; Willems, P.H.; Verkaart, S.; Visch, H.J.; Graaf-Hess, D.E.; Blom, H.J.; Monnens, L.A.; van den Heuvel, L.P.; Levtchenko, E.N. Cystine dimethylester model of cystinosis: Still reliable? Pediatr. Res. 2007, 62, 151–155. [Google Scholar] [CrossRef]
- Sumayao, R.; Mcevoy, B.; Martin-Martin, N.; Mcmorrow, T.; Newsholme, P. Cystine dimethylester loading promotes oxidative stress and a reduction in ATP independent of lysosomal cystine accumulation in a human proximal tubular epithelial cell line. Exp. Physiol. 2013, 98, 1505–1517. [Google Scholar] [CrossRef]
- Thoene, J.G.; Lemons, R.M. Cystine accumulation in cystinotic fibroblasts from free and protein-linked cystine but not cysteine. Biochem. J. 1982, 208, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Adelmann, C.H.; Traunbauer, A.K.; Chen, B.; Condon, K.J.; Chan, S.H.; Kunchok, T.; Lewis, C.A.; Sabatini, D.M. MFSD12 mediates the import of cysteine into melanosomes and lysosomes. Nature 2020, 588, 699–704. [Google Scholar] [CrossRef]
- Kroll, W.A.; Schneider, J.A. Decrease in Free Cystine Content of Cultured Cystinotic Fibroblasts by Ascorbic Acid. Science 1974, 186, 1040–1042. [Google Scholar] [CrossRef] [PubMed]
- Thoene, J.G.; Oshima, R.G.; Crawhall, J.C. Intracellular cystine depletion by aminothiols in vitro and in vivo. J. Clin. Investig. 1976, 58, 180–189. [Google Scholar] [CrossRef]
- Levtchenko, E.; de Graaf-Hess, A.; Wilmer, M.; van den Heuvel, L.; Monnens, L.; Blom, H. Altered status of glutathione and its metabolites in cystinotic cells. Nephrol. Dial. Transplant. 2005, 20, 1828–1832. [Google Scholar] [CrossRef]
- Mannucci, L.; Pastore, A.; Rizzo, C.; Piemonte, F.; Rizzoni, G.; Emma, F. Impaired activity of the γ-glutamyl cycle in nephropathic cystinosis fibroblasts. Pediatric Res. 2006, 59, 332–335. [Google Scholar] [CrossRef] [PubMed]
- Vitvitsky, V.; Witcher, M.; Banerjee, R.; Thoene, J. The redox status of cystinotic fibroblasts. Mol. Genet. Metab. 2010, 99, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Trifillis, A.L.; Regec, A.L.; Trump, B.F. Isolation, culture and characterization of human renal tubular cells. J. Urol. 1985, 133, 324–329. [Google Scholar] [CrossRef]
- Pellett, O.L.; Smith, M.L.; Thoene, S.J.G.; Schneider, J.A.; Jonas, A.J. Renal cell culture using autopsy material from children with cystinosis. In Vitro 1984, 20, 53–58. [Google Scholar] [CrossRef]
- Racusen, L.C.; Fivush, B.A.; Andersson, H.; Gahl, W.A. Culture of Renal with Nephropathic from the Urine of Patients. J. Am. Soc. Nephrol. 1991, 1, 1028–1033. [Google Scholar] [CrossRef]
- Laube, G.F.; Haq, M.R.; van’t Hoff, W.G. Exfoliated human proximal tubular cells: A model of cystinosis and Fanconi syndrome. Pediatric Nephrol. 2005, 20, 136–140. [Google Scholar] [CrossRef]
- Laube, G.F.; Shah, V.; Stewart, V.C.; Hargreaves, I.P.; Haq, M.R.; Heales, S.J.R.; von’t Hoff, W.G. Glutathione depletion and increased apoptosis rate in human cystinotic proximal tubular cells. Pediatric Nephrol. 2006, 21, 503–509. [Google Scholar] [CrossRef]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef]
- Sansanwal, P.; Yen, B.; Gahl, W.A.; Ma, Y.; Ying, L.; Wong, L.C.; Sarwal, M.M. Mitochondrial autophagy promotes cellular injury in nephropathic cystinosis. J. Am. Soc. Nephrol. 2010, 21, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Sansanwal, P.; Sarwal, M.M. P62/SQSTM1 prominently accumulates in renal proximal tubules in nephropathic cystinosis. Pediatric Nephrol. 2012, 27, 2137–2144. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Johnson, D.W.; Vesey, D.A.; Pollock, C.A.; Chen, X. Isolation, propagation and characterization of primary tubule cell culture from human kidney. Nephrology 2007, 12, 155–159. [Google Scholar] [CrossRef]
- Dickman, K.G.; Mandel, L.J. Glycolytic and oxidative metabolism in primary renal proximal tubule cultures. Am. J. Physiol. 1989, 257, C333–C340. [Google Scholar] [CrossRef] [PubMed]
- Münger, K.; Howley, P.M. Human papillomavirus immortalization and transformation functions. Virus Res. 2002, 89, 213–228. [Google Scholar] [CrossRef]
- Ahuja, D.; Sáenz-Robles, M.T.; Pipas, J.M. SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene 2005, 24, 7729–7745. [Google Scholar] [CrossRef]
- Ryan, M.J.; Johnson, G.; Kirk, J.; Fuerstenberg, S.M.; Zager, R.A.; Torok-Storb, B. HK-2: An immortalized proximal tubule epithelial cell line from normal adult human kidney. Kidney Int. 1994, 45, 48–57. [Google Scholar] [CrossRef]
- Ivanova, E.A.; De Leo, M.G.; Van Den Heuvel, L.; Pastore, A.; Dijkman, H.; De Matteis, M.A.; Levtchenko, E.N. Endo-lysosomal dysfunction in human proximal tubular epithelial cells deficient for lysosomal cystine transporter cystinosin. PLoS ONE 2015, 10, 1–18. [Google Scholar] [CrossRef]
- Bellomo, F.; Signorile, A.; Tamma, G.; Ranieri, M.; Emma, F.; De Rasmo, D. Impact of atypical mitochondrial cyclic-AMP level in nephropathic cystinosis. Cell Mol. Life Sci. 2018, 75, 3411–3422. [Google Scholar] [CrossRef] [PubMed]
- Sumayao, R.; McEvoy, B.; Newsholme, P.; McMorrow, T. Lysosomal cystine accumulation promotes mitochondrial depolarization and induction of redox-sensitive genes in human kidney proximal tubular cells. J. Physiol. 2016, 594, 3353–3370. [Google Scholar] [CrossRef] [PubMed]
- Taranta, A.; Petrini, S.; Palma, A.; Mannucci, L.; Wilmer, M.J.; De Luca, V.; Diomedi-Camassei, F.; Corallini, S.; Bellomo, F.; van den Heuvel, L.P.; et al. Identification and subcellular localization of a new cystinosin isoform. Am. J. Physiol. Renal. Physiol. 2008, 294, F1101–F1108. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; He, J.; Johnson, J.L.; Rahman, F.; Gavathiotis, E.; Cuervo, A.M.; Catz, S.D. Chaperone-Mediated Autophagy Upregulation Rescues Megalin Expression and Localization in Cystinotic Proximal Tubule Cells. Front. Endocrinol. 2019, 10, 21. [Google Scholar] [CrossRef]
- Wilmer, M.J.G.; De Graaf-Hess, A.; Blom, H.J.; Dijkman, H.B.P.M.; Monnens, L.A.; Van Den Heuvel, L.P.; Levtchenko, E.N. Elevated oxidized glutathione in cystinotic proximal tubular epithelial cells. Biochem. Biophys. Res. Commun. 2005, 337, 610–614. [Google Scholar] [CrossRef]
- Chol, M.; Nevo, N.; Cherqui, S.; Antignac, C.; Rustin, P. Glutathione precursors replenish decreased glutathione pool in cystinotic cell lines. Biochem. Biophys. Res. Commun. 2004, 324, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Levtchenko, E.N.; Wilmer, M.J.G.; Janssen, A.J.M.; Koenderink, J.B.; Visch, H.J.; Willems, P.H.G.M.; de Graaf-Hess, A.; Blom, H.J.; van den Heuvel, L.P.; Monnens, L.A. Decreased intracellular ATP content and intact mitochondrial energy generating capacity in human cystinotic fibroblasts. Pediatric Res. 2006, 59, 287–292. [Google Scholar] [CrossRef]
- Racusen, L.C.; Wilson, P.D.; Hartz, P.A.; Fivush, B.A.; Burrow, C.R. Renal proximal tubular epithelium from patients with nephropathic cystinosis: Immortalized cell lines as in vitro model systems. Kidney Int. 1995, 48, 536–543. [Google Scholar] [CrossRef]
- Wilmer, M.J.; Saleem, M.A.; Masereeuw, R.; Ni, L.; van der Velden, T.J.; Russel, F.G.; Mathieson, P.W.; Monnens, L.A.; van den Heuvel, L.P.; Levtchenko, E.N. Novel conditionally immortalized human proximal tubule cell line expressing functional influx and efflux transporters. Cell Tissue Res. 2010, 339, 449–457. [Google Scholar] [CrossRef]
- Bens, M.; Vandewalle, A. Cell models for studying renal physiology. Pflug. Arch 2008, 457, 1–15. [Google Scholar] [CrossRef]
- Mattoscio, D.; Medda, A.; Chiocca, S. Human papilloma virus and autophagy. Int. J. Mol. Sci. 2018, 19, 1775. [Google Scholar] [CrossRef]
- Mattoscio, D.; Casadio, C.; Miccolo, C.; Maffini, F.; Raimondi, A.; Tacchetti, C.; Gheit, T.; Tagliabue, M.; Galimberti, V.E.; De Lorenzi, F.; et al. Autophagy regulates UBC9 levels during viral-mediated tumorigenesis. PLoS Pathog. 2017, 13, e1006262. [Google Scholar] [CrossRef]
- Bellomo, F.; Corallini, S.; Pastore, A.; Palma, A.; Laurenzi, C.; Emma, F.; Taranta, A. Modulation of CTNS gene expression by intracellular thiols. Free Radic. Biol. Med. 2010, 48, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Soto-Heredero, G.; Baixauli, F.; Mittelbrunn, M. Interorganelle Communication between Mitochondria and the Endolysosomal System. Front. Cell Dev. Biol. 2017, 7, 95. [Google Scholar] [CrossRef]
- Taub, M.L.; Springate, J.E.; Cutuli, F. Reduced phosphate transport in the renal proximal tubule cells in cystinosis is due to decreased expression of transporters rather than an energy defect. Biochem. Biophys. Res. Commun. 2011, 407, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Taub, M.; Cutuli, F. Activation of AMP kinase plays a role in the increased apoptosis in the renal proximal tubule in cystinosis. Biochem. Biophys. Res. Commun. 2012, 426, 516–521. [Google Scholar] [CrossRef]
- Okoshi, R.; Ozaki, T.; Yamamoto, H.; Ando, K.; Koida, N.; Ono, S.; Koda, T.; Kamijo, T.; Nakagawara, A.; Kizaki, H. Activation of AMP-activated protein kinase induces p53-dependent apoptotic cell death in response to energetic stress. J. Biol. Chem. 2008, 283, 3979–3987. [Google Scholar] [CrossRef]
- McEvoy, B.; Sumayao, R.; Slattery, C.; McMorrow, T.; Newsholme, P. Cystine accumulation attenuates insulin release from the pancreatic β-cell due to elevated oxidative stress and decreased ATP levels. J. Physiol. 2015, 593, 5167–5182. [Google Scholar] [CrossRef] [PubMed]
- Borger, D.K.; McMahon, B.; Lal, T.R.; Serra-Vinardell, J.; Aflaki, E.; Sidransky, E. Induced pluripotent stem cell models of lysosomal storage disorders. DMM Dis. Models Mech. 2017, 10, 691–704. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Song, B.; Smink, A.M.; Jones, C.V.; Callaghan, J.M.; Firth, S.D.; Bernard, C.A.; Laslett, A.L.; Kerr, P.G.; Ricardo, S.D. The Directed Differentiation of Human iPS Cells into Kidney Podocytes. PLoS ONE 2012, 7, 1–9. [Google Scholar] [CrossRef]
- Lam, A.Q.; Freedman, B.S.; Morizane, R.; Lerou, P.H.; Valerius, M.T.; Bonventre, J.V. Rapid and efficient differentiation of human pluripotent stem cells into intermediate mesoderm that forms tubules expressing kidney proximal tubular markers. J. Am. Soc. Nephrol. 2014, 25, 1211–1225. [Google Scholar] [CrossRef]
- Tian, P.; Lennon, R. The myriad possibility of kidney organoids. Curr. Opin. Nephrol. Hypertens. 2019, 28, 211–218. [Google Scholar] [CrossRef]
- Lau, Y.K.; Du, X.; Rayannavar, V.; Hopkins, B.; Shaw, J.; Bessler, E.; Thomas, T.; Pires, M.M.; Keniry, M.; Parsons, R.E.; et al. Metformin and erlotinib synergize to inhibit basal breast cancer. Oncotarget 2014, 5, 10503–10517. [Google Scholar] [CrossRef]
- Varum, S.; Rodrigues, A.S.; Moura, M.B.; Momcilovic, O.; Easley, C.A., 4th; Ramalho-Santos, J.; van Houten, B.; Schatten, G. Energy metabolism in human pluripotent stem cells and their differentiated counterparts. PLoS ONE 2011, 6, e20914. [Google Scholar] [CrossRef]
- Kimura, T.; Takabatake, Y.; Takahashi, A.; Kaimori, J.Y.; Matsui, I.; Namba, T.; Kitamura, H.; Niimura, F.; Matsusaka, T.; Soga, T.; et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J. Am. Soc. Nephrol. 2011, 22, 902–913. [Google Scholar] [CrossRef]
- Combes, A.N.; Zappia, L.; Er, P.X.; Oshlack, A.; Little, M.H. Single-cell analysis reveals congruence between kidney organoids and human fetal kidney. Genome Med. 2019, 11, 1–15. [Google Scholar] [CrossRef]
- Wu, H.; Uchimura, K.; Donnelly, E.L.; Kirita, Y.; Morris, S.A.; Humphreys, B.D. Comparative Analysis and Refinement of Human PSC-Derived Kidney Organoid Differentiation with Single-Cell Transcriptomics. Cell Stem. Cell 2018, 23, 869–881. [Google Scholar] [CrossRef]
- Przepiorski, A.; Sander, V.; Tran, T.; Hollywood, J.A.; Sorrenson, B.; Shih, J.H.; Wolvetang, E.J.; McMahon, A.P.; Holm, T.M.; Davidson, A.J. A Simple Bioreactor-Based Method to Generate Kidney Organoids from Pluripotent Stem Cells. Stem. Cell Rep. 2018, 11, 470–484. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, C.W.; Ritsma, L.; Avramut, M.C.; Wiersma, L.E.; van den Berg, B.M.; Leuning, D.G.; Lievers, E.; Koning, M.; Vanslambrouck, J.M.; Koster, A.J.; et al. Renal Subcapsular Transplantation of PSC-Derived Kidney Organoids Induces Neo-vasculogenesis and Significant Glomerular and Tubular Maturation In Vivo. Stem. Cell Rep. 2018, 10, 751–765. [Google Scholar] [CrossRef] [PubMed]
- Bantounas, I.; Ranjzad, P.; Tengku, F.; Silajdži’c, E.; Forster, D.; Asselin, M.C.; Lewis, P.; Lennon, R.; Plagge, A.; Wang, Q.; et al. Generation of Functioning Nephrons by Implanting Human Pluripotent Stem Cell-Derived Kidney Progenitors. Stem. Cell Rep. 2018, 10, 766–779. [Google Scholar] [CrossRef] [PubMed]
- Schutgens, F.; Rookmaaker, M.B.; Margaritis, T.; Rios, A.; Ammerlaan, C.; Jansen, J. Tubuloids derived from human adult kidney and urine for personalized disease modeling. Nat. Biotechnol. 2019, 37, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Yousef Yengej, F.A.; Jansen, J.; Rookmaaker, M.B.; Verhaar, M.C.; Clevers, H. Kidney Organoids and Tubuloids. Cells 2020, 9, 1326. [Google Scholar] [CrossRef] [PubMed]
- Jamalpoor, A.; van Gelder, C.A.; Yousef Yengej, F.A.; Zaal, E.A.; Berlingerio, S.P.; Veys, K.; Pou Casellas, C.; Voskuil, K.; Essa, K.; Ammerlaan, C.M.; et al. Cysteamine-bicalutamide combination therapy corrects proximal tubule phenotype in cystinosis. EMBO Mol. Med. 2021, 13, e13067. [Google Scholar] [CrossRef]
- Cherqui, S.; Sevin, C.; Hamard, G.; Kalatzis, V.; Sich, M.; Pequignot, M.; Gogat, K.; Abitbol, M.; Broyer, M.; Gubler, M.C.; et al. Intralysosomal Cystine Accumulation in Mice Lacking Cystinosin, the Protein Defective in Cystinosis. Mol. Cell. Biol. 2002, 22, 7622–7632. [Google Scholar] [CrossRef] [PubMed]
- Nevo, N.; Chol, M.; Bailleux, A.; Kalatzis, V.; Morisset, L.; Devuyst, O.; Gubler, M.C.; Antignac, C. Renal phenotype of the cystinosis mouse model is dependent upon genetic background. Nephrol. Dial. Transplant. 2010, 25, 1059–1066. [Google Scholar] [CrossRef]
- Gaide Chevronnay, H.P.; Janssens, V.; Van Der Smissen, P.; N’Kuli, F.; Nevo, N.; Guiot, Y.; Levtchenko, E.; Marbaix, E.; Pierreux, C.E. Time course of pathogenic and adaptation mechanisms in cystinotic mouse kidneys. J. Am. Soc. Nephrol. 2014, 25, 1256–1269. [Google Scholar] [CrossRef]
- Elmonem, M.A.; Khalil, R.; Khodaparast, L.; Khodaparast, L.; Arcolino, F.O.; Morgan, J.; Pastore, A.; Tylzanowski, P.; Ny, A.; Lowe, M.; et al. Cystinosis (ctns) zebrafish mutant shows pronephric glomerular and tubular dysfunction. Sci. Rep. 2017, 7, 1–17. [Google Scholar]
- Berlingerio, S.P.; He, J.; Groef, L.; De Taeter, H.; Norton, T.; Baatsen, P.; Cairoli, S.; Goffredo, B.; de Witte, P.; van den Heuvel, L.; et al. Renal and Extra Renal Manifestations in Adult Zebrafish Model of Cystinosis. Int. J. Mol. Sci. 2021, 22, 9398. [Google Scholar] [CrossRef]
- Festa, B.P.; Chen, Z.; Berquez, M.; Debaix, H.; Tokonami, N.; Prange, J.A.; van de Hoek, G.; Alessio, C.; Raimondi, A.; Nevo, N.; et al. Impaired autophagy bridges lysosomal storage disease and epithelial dysfunction in the kidney. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef]
- Shimizu, Y.; Yanobu-Takanashi, R.; Nakano, K.; Hamase, K.; Shimizu, T.; Okamura, T. A deletion in the Ctns gene causes renal tubular dysfunction and cystine accumulation in LEA/Tohm rats. Mamm. Genome 2019, 30, 23–33. [Google Scholar] [CrossRef]
- Hollywood, J.A.; Kallingappa, P.K.; Cheung, P.Y.; Martis, R.M.; Sreebhavan, S.; Chatterjee, A.; Buckels, E.J.; Mathews, B.G.; Lewis, P.M.; Davidson, A.J. Cystinosin deficient rats recaputulate the phenotype of nephropathic cytinosis. bioRxiv 2021. [Google Scholar] [CrossRef]
- Hardwick, K.G.; Pelham, H.R. ERS1 a seven transmembrane domain protein from Saccharomyces cerevisiae. Nucleic Acids Res. 1990, 25, 2177. [Google Scholar] [CrossRef][Green Version]
- Zhai, Y.; Heijne, W.H.; Smith, D.W.; Saier, M.H., Jr. Homologues of archaeal rhodopsins in plants, animals and fungi: Structural and functional predications for a putative fungal chaperone protein. Biochim. Biophys. Acta 2001, 1511, 206–223. [Google Scholar] [CrossRef]
- Gao, X.D.; Wang, J.; Keppler-Ross, S.; Dean, N. ERS1 encodes a functional homologue of the human lysosomal cystine transporter. FEBS J. 2005, 272, 2497–2511. [Google Scholar] [CrossRef] [PubMed]
- Simpkins, J.A.; Rickel, K.E.; Madeo, M.; Ahlers, B.A.; Carlisle, G.B.; Nelson, H.J.; Cardillo, A.L.; Weber, E.A.; Vitiello, P.F.; Pearce, D.A.; et al. Disruption of a cystine transporter downregulates expression of genes involved in sulfur regulation and cellular respiration. Biol. Open 2016, 5, 689–697. [Google Scholar] [CrossRef]
- Kalatzis, V.; Nevo, N.; Cherqui, S.; Gasnier, B.; Antignac, C. Molecular pathogenesis of cystinosis: Effect of CTNS mutations on the transport activity and subcellular localization of cystinosin. Hum. Mol. Genet. 2004, 13, 1361–1371. [Google Scholar] [CrossRef]
- Ratelade, J.; Lavin, T.A.; Muda, A.O.; Morisset, L.; Mollet, G.; Boyer, O.; Chen, D.S.; Henger, A.; Kretzler, M.; Hubner, N.; et al. Maternal environment interacts with modifier genes to influence progression of nephrotic syndrome. J. Am. Soc. Nephrol. 2008, 19, 1491–1499. [Google Scholar] [CrossRef]
- Raggi, C.; Luciani, A.; Nevo, N.; Antignac, C.; Terryn, S.; Devuyst, O. Dedifferentiation and aberrations of the endolysosomal compartment characterize the early stage of nephropathic cystinosis. Hum. Mol. Genet. 2014, 23, 2266–2278. [Google Scholar] [CrossRef]
- Sharma, A.; Gupta, R.; Sethi, S.K.; Bagga, A.; Dinda, A.K. Giant cell transformation of podocytes: A unique histological feature associated with cystinosis. Indian J. Nephrol. 2011, 21, 123–125. [Google Scholar]
- Janssens, V.; Gaide Chevronnay, H.P.; Marie, S.; Vincent, M.F.; Van Der Smissen, P.; Nevo, N.; Vainio, S.; Nielsen, R.; Christensen, E.I.; Jouret, F.; et al. Protection of Cystinotic Mice by Kidney-Specific Megalin Ablation Supports an Endocytosis-Based Mechanism for Nephropathic Cystinosis Progression. J. Am. Soc. Nephrol. 2019, 30, 2177–2190. [Google Scholar] [CrossRef]
- Battafarano, G.; Rossi, M.; Rega, L.R.; Di Giovamberardino, G.; Pastore, A.; D’Agostini, M.; Porzio, O.; Nebo, N.; Emma, F.; Taranta, A.; et al. Intrinsic Bone Defects in Cystinotic Mice. Am. J. Pathol. 2019, 189, 1053–1064. [Google Scholar] [CrossRef]
- Galarreta, C.I.; Forbes, M.S.; Thornhill, B.A.; Antignac, C.; Gubler, M.C.; Nevo, N.; Murphy, M.P.; Chevalier, R.L. The swan-neck lesion: Proximal tubular adaptation to oxidative stress in nephropathic cystinosis. Am. J. Physiol. Renal. Physiol. 2015, 308, F1155–F1166. [Google Scholar] [CrossRef]
- Yu, Y.; Kudchodkar, S.B.; Alwine, J.C. Effects of simian virus 40 large and small tumor antigens on mammalian target of rapamycin signaling: Small tumor antigen mediates hypophosphorylation of eIF4E-binding protein 1 late in infection. J. Virol. 2005, 79, 6882–6889. [Google Scholar] [CrossRef]
- Gaide Chevronnay, H.P.; Janssens, V.; Van Der Smissen, P.; Rocca, C.J.; Liao, X.H.; Refetoff, S.; Pierreux, C.E.; Cherqui, S.; Courtoy, P.J. Hematopoietic Stem Cells Transplantation Can Normalize Thyroid Function in a Cystinosis Mouse Model. Endocrinol. 2016, 157, 1363–1371. [Google Scholar] [CrossRef]
- Kalatzis, V.; Serratrice, N.; Hippert, C.; Payet, O.; Arndt, C.; Cazevieille, C.; Maurice, T.; Hamel, C.; Malecaze, F.; Antignac, C.; et al. The ocular anomalies in a cystinosis animal model mimic disease pathogenesis. Pediatr. Res. 2007, 62, 156–162. [Google Scholar] [CrossRef]
- Tsilou, E.T.; Rubin, B.I.; Reed, G.; Caruso, R.C.; Iwata, F.; Balog, J.; Gahl, W.A.; Kaiser-Kupfer, M.I. Nephropathic cystinosis: Posterior segment manifestations and effects of cysteamine therapy. Ophthalmology 2006, 113, 1002–1009. [Google Scholar] [CrossRef]
- Cheung, W.W.; Cherqui, S.; Ding, W.; Esparza, M.; Zhou, P.; Shao, J.; Lieber, R.L.; Mak, R.H. Muscle wasting and adipose tissue browning in infantile nephropathic cystinosis. Cachexia Sarcopenia Muscle 2016, 7, 152–164. [Google Scholar] [CrossRef]
- Cheung, W.W.; Hao, S.; Wang, Z.; Ding, W.; Zheng, R.; Gonzalez, A.; Zhan, J.Y.; Zhou, P.; Li, S.; Esparza, M.C.; et al. Vitamin D repletion ameliorates adipose tissue browning and muscle wasting in infantile nephropathic cystinosis-associated cachexia. J. Cachexia Sarcopenia Muscle. 2020, 11, 120–134. [Google Scholar] [CrossRef] [PubMed]
- Hippert, C.; Dubois, G.; Morin, C.; Disson, O.; Ibanes, S.; Jacquet, C.; Schwendener, R.; Antignac, C.; Kremer, E.J.; Kalatzis, V. Gene transfer may be preventive but not curative for a lysosomal transport disorder. Mol. Ther. 2008, 16, 1372–1381. [Google Scholar] [CrossRef] [PubMed]
- Syres, K.; Harrison, F.; Tadlock, M.; Jester, J.V.; Simpson, J.; Roy, S.; Salomon, D.R.; Cherqui, S. Successful treatment of the murine model of cystinosis using bone marrow cell transplantation. Blood 2009, 114, 2542–2552. [Google Scholar] [CrossRef] [PubMed]
- Yeagy, B.A.; Harrison, F.; Gubler, M.C.; Koziol, J.A.; Salomon, D.R.; Cherqui, S. Kidney preservation by bone marrow cell transplantation in hereditary nephropathy. Kidney Int. 2011, 79, 1198–1206. [Google Scholar] [CrossRef] [PubMed]
- Harrison, F.; Yeagy, B.A.; Rocca, C.J.; Kohn, D.B.; Salomon, D.R.; Cherqui, S. Hematopoietic stem cell gene therapy for the multisystemic lysosomal storage disorder cystinosis. Mol. Ther. 2013, 21, 433–444. [Google Scholar] [CrossRef]
- Naphade, S.; Sharma, J.; Gaide Chevronnay, H.P.; Shook, M.A.; Yeagy, B.A.; Rocca, C.J.; Ur, S.N.; Lau, A.J.; Courtoy, P.J.; Cherqui, S. Brief reports: Lysosomal cross-correction by hematopoietic stem cell-derived macrophages via tunneling nanotubes. Stem Cells 2015, 33, 301–309. [Google Scholar] [CrossRef]
- Brasell, E.J.; Chu, L.L.; Akpa, M.M.; Eshkar-Oren, I.; Alroy, I.; Corsini, R.; Gilfix, B.M.; Yamanaka, Y.; Huertas, P.; Goodyer, P. The novel aminoglycoside, ELX-02, permits CTNSW138X translational read-through and restores lysosomal cystine efflux in cystinosis. PLoS ONE 2019, 14, e0223954. [Google Scholar] [CrossRef]
- Schlegel, A.; Gut, P. Metabolic insights from zebrafish genetics, physiology, and chemical biology. Cell Mol. Life Sci. 2015, 72, 2249–2260. [Google Scholar] [CrossRef] [PubMed]
- Diep, C.Q.; Ma, D.; Deo, R.C.; Holm, T.M.; Naylor, R.W.; Arora, N.; Wingert, R.A.; Bollig, F.; Djordjevic, G.; Lichman, B.; et al. Identification of adult nephron progenitors capable of kidney regeneration in zebrafish. Nature 2011, 470, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Okamura, T.; Pei, X.Y.; Miyoshi, I.; Shimizu, Y.; Takanashi-Yanobu, R.; Mototani, Y.; Kanai, T.; Satoh, J.; Kimura, N.; Kasai, N. Phenotypic Characterization of LEA Rat: A New Rat Model of Nonobese Type 2 Diabetes. J. Diabetes Res. 2013, 2013, 986462. [Google Scholar] [CrossRef]
- Ponting, C.P.; Mott, R.; Bork, P.; Copley, R.R. Novel protein domains and repeats in Drosophila melanogaster: Insights into structure, function, and evolution. Genome Res. 2001, 11, 1996–2008. [Google Scholar] [CrossRef]
- Jouandin, P.; Marelja, Z.; Parkhitko, A.A.; Dambowsky, M.; Asara, J.M.; Nemazanyy, I.; Simons, M.; Perrimon, N. Lysosomal cystine efflux opposes mTORC1 reactivation through the TCA cycle. bioRxiv 2021. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Species | Strain | Mutation in Ctns | Phenotypes | Reference | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cystine Accumulation | Cystine Crystal | Renal Failure | Glomerulus Changes | PTC Lesions | PTC Dysfunction | Ocular Abnormalities | Bone Deformities | ||||
| Mouse | C57BL6 | IRES- βgal-neo cassette to remove the last 4 exon of Ctns | Yes | Yes | Yes (mild, onset at 10 months of age) | No | Yes (onset at 6 months of age) | Yes (partial, onset at 2 months of age) | Yes | Yes | [94,95,96] |
| FVN | IRES- βgal-neo cassette to remove the last 4 exon of Ctns | Yes | Yes (but mild) | No | No | No | No | N.I | N.I | [95] | |
| Zebrafish | larvae | homozygous nonsense mutation in exon 8 | Yes | No | Yes (decreased inulin clearance) | Yes (podocyte foot effacement) | No | Yes (loss of megalin expression in PTCs) | N.I | N.I | [97] |
| Adult | homozygous nonsense mutation in exon 8 | Yes | Yes | N.I | Partial | Partial | Suggested | Yes | N.I | [97,98] | |
| larvae | TALEN-drive 8 bp deletion in exon 3 | Yes | N.I | N.I | N.I | No | No | N.I | N.I | [99] | |
| Rat | F344 | 13-bp deletion in exon 7 | Yes | Yes (only in kidney cortex) | N.I | N.I | Yes | N.I (glucosuria was detected) | N.I | N.I | [100] |
| Sprague Dawley | Indel mutations exon 3 | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | [101] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheung, P.Y.; Harrison, P.T.; Davidson, A.J.; Hollywood, J.A. In Vitro and In Vivo Models to Study Nephropathic Cystinosis. Cells 2022, 11, 6. https://doi.org/10.3390/cells11010006
Cheung PY, Harrison PT, Davidson AJ, Hollywood JA. In Vitro and In Vivo Models to Study Nephropathic Cystinosis. Cells. 2022; 11(1):6. https://doi.org/10.3390/cells11010006
Chicago/Turabian StyleCheung, Pang Yuk, Patrick T. Harrison, Alan J. Davidson, and Jennifer A. Hollywood. 2022. "In Vitro and In Vivo Models to Study Nephropathic Cystinosis" Cells 11, no. 1: 6. https://doi.org/10.3390/cells11010006
APA StyleCheung, P. Y., Harrison, P. T., Davidson, A. J., & Hollywood, J. A. (2022). In Vitro and In Vivo Models to Study Nephropathic Cystinosis. Cells, 11(1), 6. https://doi.org/10.3390/cells11010006