
AUTS2 Gene: Keys to Understanding the Pathogenesis of Neurodevelopmental Disorders


{kind=link}
{kind=link}
Abstract
:1. Introduction
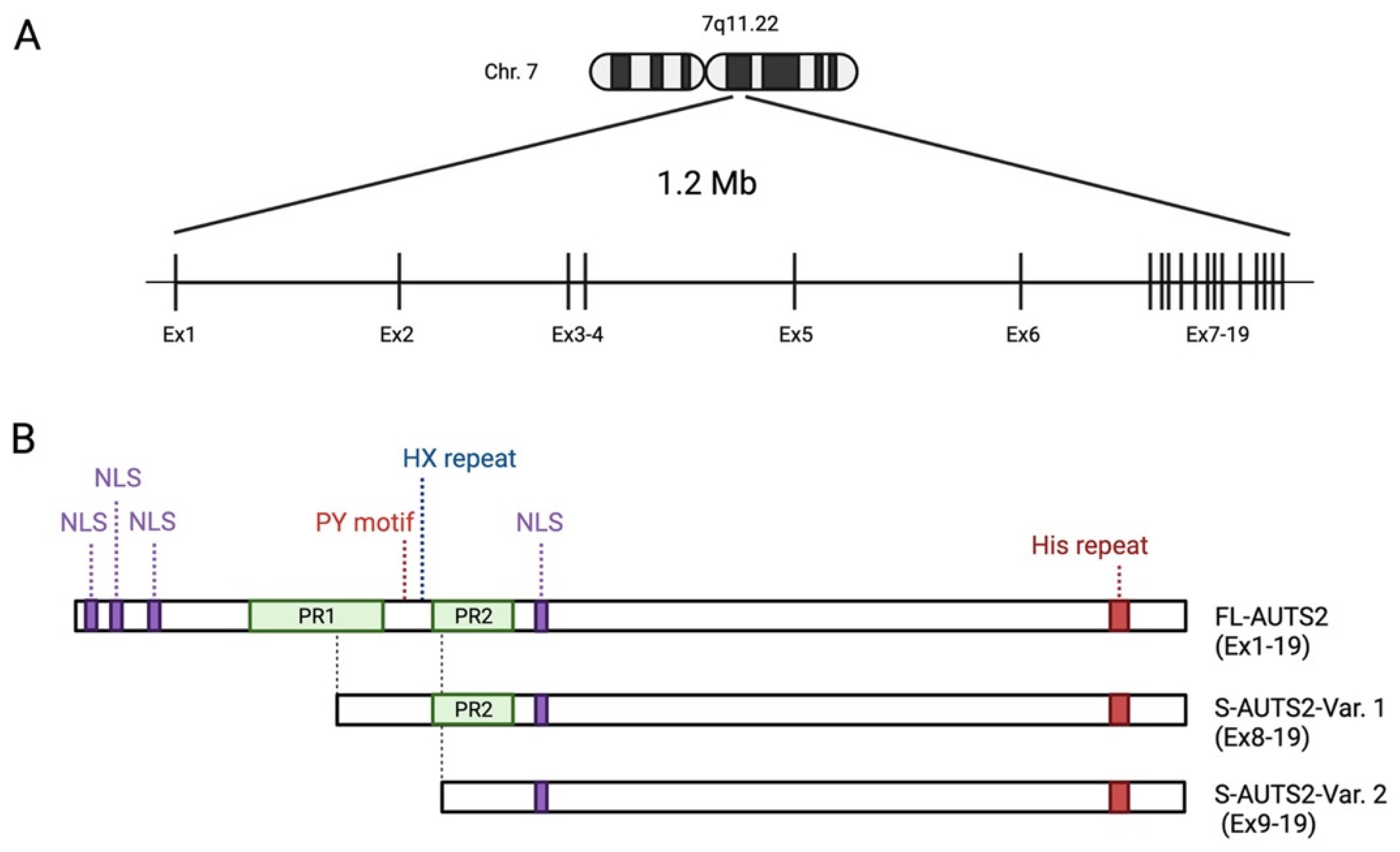
2. Structure and Expression of the AUTS2 Gene
3. Genotype–Phenotype Correlations of AUTS2 Syndrome
4. Protein Structure of AUTS2
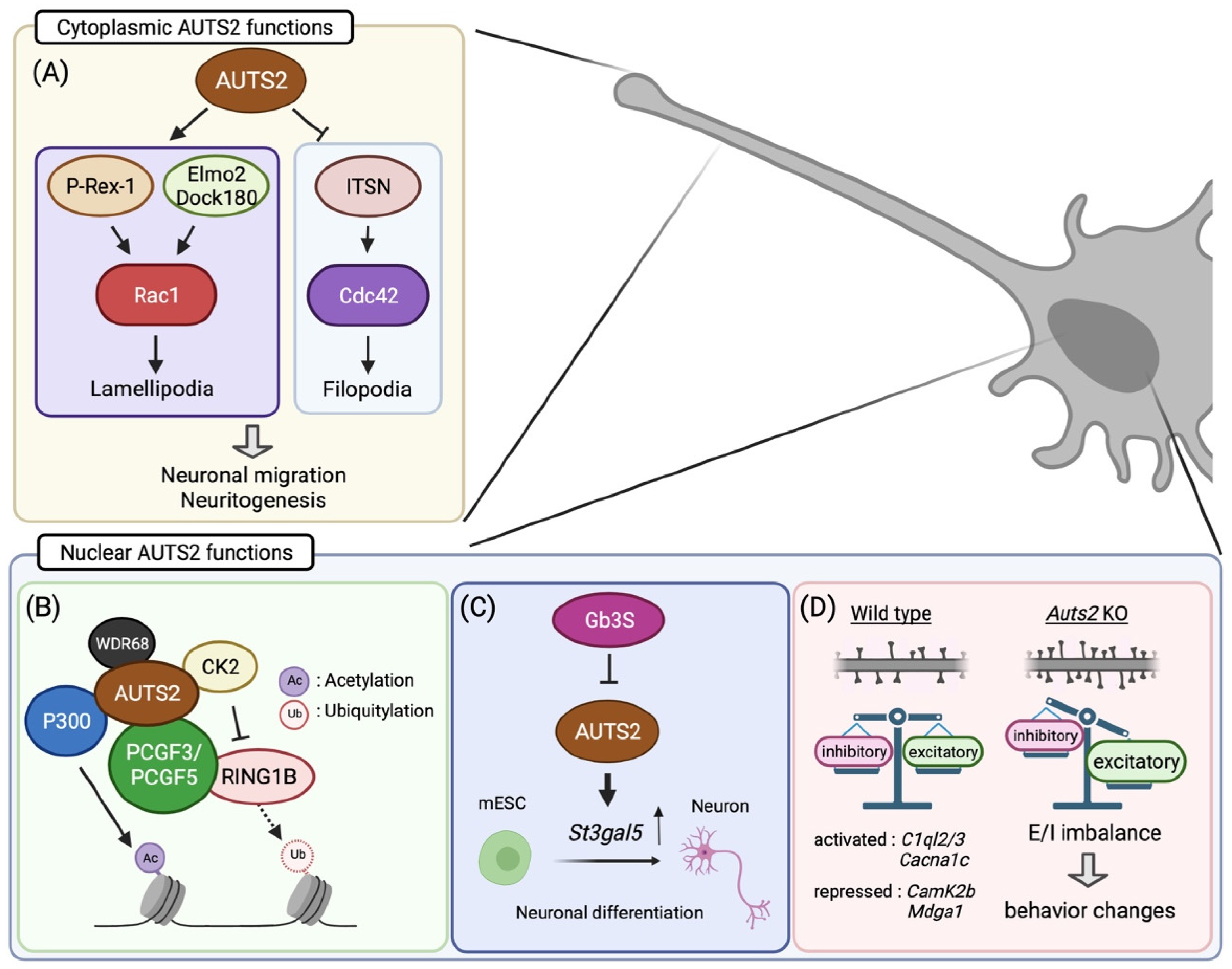
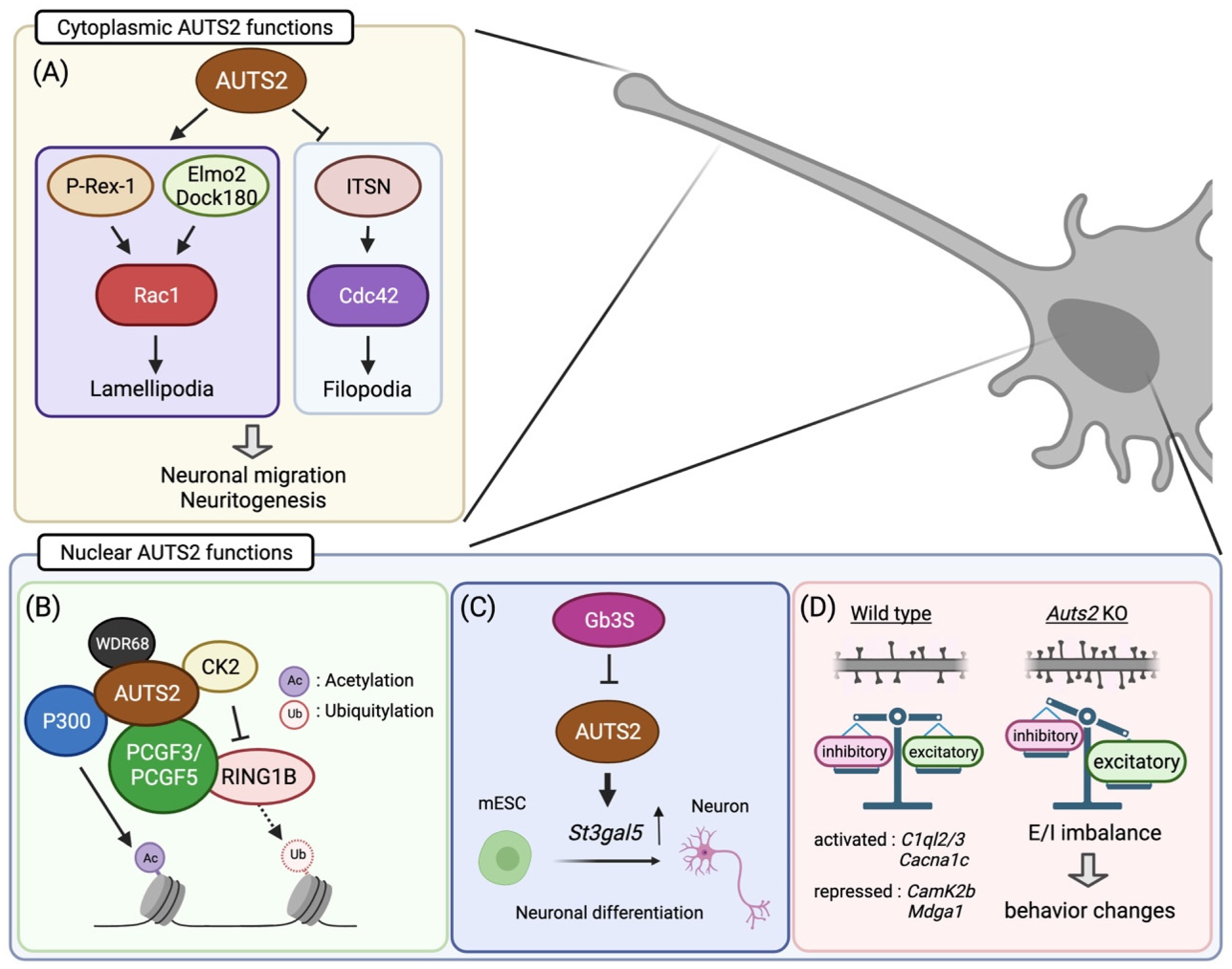
5. Cytoplasmic AUTS2 Functions in Cytoskeletal Organization
6. Transcriptional Regulation by Nuclear AUTS2
7. The Role of AUTS2 in Neurogenesis
8. AUTS2 Regulates Neuronal Migration and Neuritogenesis during Corticogenesis
9. AUTS2 Restricts the Number of Excitatory Synapses to Regulate the E/I Balance
10. AUTS2 Is Involved in Cerebellar Development
11. Behavioral Abnormalities in Auts2 Mutant Mice
12. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Parenti, I.; Rabaneda, L.G.; Schoen, H.; Novarino, G. Neurodevelopmental Disorders: From Genetics to Functional Pathways. Trends Neurosci. 2020, 43, 608–621. [Google Scholar] [CrossRef] [PubMed]
- Niemi, M.E.K.; Martin, H.C.; Rice, D.L.; Gallone, G.; Gordon, S.; Kelemen, M.; McAloney, K.; McRae, J.; Radford, E.J.; Yu, S.; et al. Common genetic variants contribute to risk of rare severe neurodevelopmental disorders. Nature 2018, 562, 268–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Washington, DC, USA, 2013. [Google Scholar]
- Tarlungeanu, D.C.; Novarino, G. Genomics in neurodevelopmental disorders: An avenue to personalized medicine. Exp. Mol. Med. 2018, 50, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mardis, E.R. A decade’s perspective on DNA sequencing technology. Nature 2011, 470, 198–203. [Google Scholar] [CrossRef]
- Ng, S.B.; Buckingham, K.J.; Lee, C.; Bigham, A.W.; Tabor, H.K.; Dent, K.M.; Huff, C.D.; Shannon, P.T.; Jabs, E.W.; Nickerson, D.A.; et al. Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 2010, 42, 30–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584. [Google Scholar] [CrossRef]
- Sanders, S.J.; He, X.; Willsey, A.J.; Ercan-Sencicek, A.G.; Samocha, K.E.; Cicek, A.E.; Murtha, M.T.; Bal, V.H.; Bishop, S.L.; Dong, S.; et al. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 2015, 87, 1215–1233. [Google Scholar] [CrossRef] [Green Version]
- Ben-David, E.; Shifman, S. Combined analysis of exome sequencing points toward a major role for transcription regulation during brain development in autism. Mol. Psychiatry 2013, 18, 1054–1056. [Google Scholar] [CrossRef] [Green Version]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef]
- Sultana, R.; Yu, C.E.; Yu, J.; Munson, J.; Chen, D.; Hua, W.; Estes, A.; Cortes, F.; de la Barra, F.; Yu, D.; et al. Identification of a novel gene on chromosome 7q11.2 interrupted by a translocation breakpoint in a pair of autistic twins. Genomics 2002, 80, 129–134. [Google Scholar] [CrossRef]
- Oksenberg, N.; Stevison, L.; Wall, J.D.; Ahituv, N. Function and regulation of AUTS2, a gene implicated in autism and human evolution. PLoS Genet. 2013, 9, e1003221. [Google Scholar] [CrossRef] [Green Version]
- Amarillo, I.E.; Li, W.L.; Li, X.; Vilain, E.; Kantarci, S. De novo single exon deletion of AUTS2 in a patient with speech and language disorder: A review of disrupted AUTS2 and further evidence for its role in neurodevelopmental disorders. Am. J. Med. Genet. A 2014, 164A, 958–965. [Google Scholar] [CrossRef]
- Beunders, G.; Voorhoeve, E.; Golzio, C.; Pardo, L.M.; Rosenfeld, J.A.; Talkowski, M.E.; Simonic, I.; Lionel, A.C.; Vergult, S.; Pyatt, R.E.; et al. Exonic deletions in AUTS2 cause a syndromic form of intellectual disability and suggest a critical role for the C terminus. Am. J. Hum. Genet. 2013, 92, 210–220. [Google Scholar] [CrossRef] [Green Version]
- Jolley, A.; Corbett, M.; McGregor, L.; Waters, W.; Brown, S.; Nicholl, J.; Yu, S. De novo intragenic deletion of the autism susceptibility candidate 2 (AUTS2) gene in a patient with developmental delay: A case report and literature review. Am. J. Med. Genet. A 2013, 161A, 1508–1512. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Xu, Y.H.; Wei, S.G.; Zhang, H.B.; Fu, D.K.; Feng, Z.F.; Guan, F.L.; Zhu, Y.S.; Li, S.B. Association Study Identifying a New Susceptibility Gene (AUTS2) for Schizophrenia. Int. J. Mol. Sci. 2014, 15, 19406–19416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elia, J.; Gai, X.; Xie, H.M.; Perin, J.C.; Geiger, E.; Glessner, J.T.; D'Arcy, M.; deBerardinis, R.; Frackelton, E.; Kim, C.; et al. Rare structural variants found in attention-deficit hyperactivity disorder are preferentially associated with neurodevelopmental genes. Mol. Psychiatry 2010, 15, 637–646. [Google Scholar] [CrossRef]
- Girirajan, S.; Brkanac, Z.; Coe, B.P.; Baker, C.; Vives, L.; Vu, T.H.; Shafer, N.; Bernier, R.; Ferrero, G.B.; Silengo, M.; et al. Relative burden of large CNVs on a range of neurodevelopmental phenotypes. PLoS Genet. 2011, 7, e1002334. [Google Scholar] [CrossRef] [PubMed]
- Mefford, H.C.; Muhle, H.; Ostertag, P.; von Spiczak, S.; Buysse, K.; Baker, C.; Franke, A.; Malafosse, A.; Genton, P.; Thomas, P.; et al. Genome-wide copy number variation in epilepsy: Novel susceptibility loci in idiopathic generalized and focal epilepsies. PLoS Genet. 2010, 6, e1000962. [Google Scholar] [CrossRef] [PubMed]
- Myung, W.; Kim, J.; Lim, S.W.; Shim, S.; Won, H.H.; Kim, S.; Kim, S.; Lee, M.S.; Chang, H.S.; Kim, J.W.; et al. A genome-wide association study of antidepressant response in Koreans. Transl. Psychiatry 2015, 5, e633. [Google Scholar] [CrossRef] [Green Version]
- Oksenberg, N.; Ahituv, N. The role of AUTS2 in neurodevelopment and human evolution. Trends Genet. 2013, 29, 600–608. [Google Scholar] [CrossRef] [Green Version]
- Beunders, G.; van de Kamp, J.; Vasudevan, P.; Morton, J.; Smets, K.; Kleefstra, T.; de Munnik, S.A.; Schuurs-Hoeijmakers, J.; Ceulemans, B.; Zollino, M.; et al. A detailed clinical analysis of 13 patients with AUTS2 syndrome further delineates the phenotypic spectrum and underscores the behavioural phenotype. J. Med. Genet. 2016, 53, 523–532. [Google Scholar] [CrossRef]
- Hori, K.; Hoshino, M. Neuronal Migration and AUTS2 Syndrome. Brain Sci. 2017, 7, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hori, K.; Nagai, T.; Shan, W.; Sakamoto, A.; Taya, S.; Hashimoto, R.; Hayashi, T.; Abe, M.; Yamazaki, M.; Nakao, K.; et al. Cytoskeletal regulation by AUTS2 in neuronal migration and neuritogenesis. Cell Rep. 2014, 9, 2166–2179. [Google Scholar] [CrossRef] [Green Version]
- Hori, K.; Yamashiro, K.; Nagai, T.; Shan, W.; Egusa, S.F.; Shimaoka, K.; Kuniishi, H.; Sekiguchi, M.; Go, Y.; Tatsumoto, S.; et al. AUTS2 Regulation of Synapses for Proper Synaptic Inputs and Social Communication. iScience 2020, 23, 101183. [Google Scholar] [CrossRef]
- Monderer-Rothkoff, G.; Tal, N.; Risman, M.; Shani, O.; Nissim-Rafinia, M.; Malki-Feldman, L.; Medvedeva, V.; Groszer, M.; Meshorer, E.; Shifman, S. AUTS2 isoforms control neuronal differentiation. Mol. Psychiatry 2019, 26, 666–681. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Lee, P.; Stafford, J.M.; von Schimmelmann, M.; Schaefer, A.; Reinberg, D. An AUTS2-Polycomb complex activates gene expression in the CNS. Nature 2014, 516, 349–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondrychyn, I.; Robra, L.; Thirumalai, V. Transcriptional Complexity and Distinct Expression Patterns of auts2 Paralogs in Danio rerio. G3 2017, 7, 2577–2593. [Google Scholar] [CrossRef] [Green Version]
- Bedogni, F.; Hodge, R.D.; Elsen, G.E.; Nelson, B.R.; Daza, R.A.; Beyer, R.P.; Bammler, T.K.; Rubenstein, J.L.; Hevner, R.F. Tbr1 regulates regional and laminar identity of postmitotic neurons in developing neocortex. Proc. Natl. Acad. Sci. USA 2010, 107, 13129–13134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Notwell, J.H.; Heavner, W.E.; Darbandi, S.F.; Katzman, S.; McKenna, W.L.; Ortiz-Londono, C.F.; Tastad, D.; Eckler, M.J.; Rubenstein, J.L.; McConnell, S.K.; et al. TBR1 regulates autism risk genes in the developing neocortex. Genome Res. 2016, 26, 1013–1022. [Google Scholar] [CrossRef] [Green Version]
- Renthal, W.; Boxer, L.D.; Hrvatin, S.; Li, E.; Silberfeld, A.; Nagy, M.A.; Griffith, E.C.; Vierbuchen, T.; Greenberg, M.E. Characterization of human mosaic Rett syndrome brain tissue by single-nucleus RNA sequencing. Nat. Neurosci. 2018, 21, 1670–1679. [Google Scholar] [CrossRef]
- Ben-Shachar, S.; Chahrour, M.; Thaller, C.; Shaw, C.A.; Zoghbi, H.Y. Mouse models of MeCP2 disorders share gene expression changes in the cerebellum and hypothalamus. Hum. Mol. Genet. 2009, 18, 2431–2442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedogni, F.; Hodge, R.D.; Nelson, B.R.; Frederick, E.A.; Shiba, N.; Daza, R.A.; Hevner, R.F. Autism susceptibility candidate 2 (Auts2) encodes a nuclear protein expressed in developing brain regions implicated in autism neuropathology. Gene Expr. Patterns 2010, 10, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashiro, K.; Hori, K.; Lai, E.S.K.; Aoki, R.; Shimaoka, K.; Arimura, N.; Egusa, S.F.; Sakamoto, A.; Abe, M.; Sakimura, K.; et al. AUTS2 Governs Cerebellar Development, Purkinje Cell Maturation, Motor Function and Social Communication. iScience 2020, 23, 101820. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Jimeno, C.; Blanco-Kelly, F.; Lopez-Grondona, F.; Losada-Del Pozo, R.; Moreno, B.; Rodrigo-Moreno, M.; Martinez-Cayuelas, E.; Riveiro-Alvarez, R.; Fenollar-Cortes, M.; Ayuso, C.; et al. Attention Deficit Hyperactivity and Autism Spectrum Disorders as the Core Symptoms of AUTS2 Syndrome: Description of Five New Patients and Update of the Frequency of Manifestations and Genotype-Phenotype Correlation. Genes 2021, 12, 1360. [Google Scholar] [CrossRef]
- Beunders, G.; de Munnik, S.A.; Van der Aa, N.; Ceulemans, B.; Voorhoeve, E.; Groffen, A.J.; Nillesen, W.M.; Meijers-Heijboer, E.J.; Frank Kooy, R.; Yntema, H.G.; et al. Two male adults with pathogenic AUTS2 variants, including a two-base pair deletion, further delineate the AUTS2 syndrome. Eur. J. Hum. Genet. 2015, 23, 803–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yagi, R.; Chen, L.F.; Shigesada, K.; Murakami, Y.; Ito, Y. A WW domain-containing yes-associated protein (YAP) is a novel transcriptional co-activator. EMBO J. 1999, 18, 2551–2562. [Google Scholar] [CrossRef] [Green Version]
- Salichs, E.; Ledda, A.; Mularoni, L.; Alba, M.M.; de la Luna, S. Genome-wide analysis of histidine repeats reveals their role in the localization of human proteins to the nuclear speckles compartment. PLoS Genet. 2009, 5, e1000397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castanza, A.S.; Ramirez, S.; Tripathi, P.P.; Daza, R.A.M.; Kalume, F.K.; Ramirez, J.M.; Hevner, R.F. AUTS2 Regulates RNA Metabolism and Dentate Gyrus Development in Mice. Cereb. Cortex 2021, 31, 4808–4824. [Google Scholar] [CrossRef]
- Palumbo, P.; Di Muro, E.; Accadia, M.; Benvenuto, M.; Di Giacomo, M.C.; Castellana, S.; Mazza, T.; Castori, M.; Palumbo, O.; Carella, M. Whole Exome Sequencing Reveals a Novel AUTS2 In-Frame Deletion in a Boy with Global Developmental Delay, Absent Speech, Dysmorphic Features, and Cerebral Anomalies. Genes 2021, 12, 229. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Aldinger, K.A.; Cheng, C.V.; Kiyama, T.; Dave, M.; McNamara, H.K.; Zhao, W.; Caraffi, S.G.; Ivanovski, I.; Errichiello, E.; et al. NRF1 association with AUTS2-Polycomb mediates specific gene activation in the brain. Mol. Cell 2021, 81, P4663–P4676. [Google Scholar] [CrossRef]
- Oksenberg, N.; Haliburton, G.D.; Eckalbar, W.L.; Oren, I.; Nishizaki, S.; Murphy, K.; Pollard, K.S.; Birnbaum, R.Y.; Ahituv, N. Genome-wide distribution of Auts2 binding localizes with active neurodevelopmental genes. Transl. Psychiatry 2014, 4, e431. [Google Scholar] [CrossRef] [Green Version]
- Geng, Z.; Gao, Z. Mammalian PRC1 Complexes: Compositional Complexity and Diverse Molecular Mechanisms. Int. J. Mol. Sci. 2020, 21, 8594. [Google Scholar] [CrossRef]
- Wang, Q.; Geng, Z.; Gong, Y.; Warren, K.; Zheng, H.; Imamura, Y.; Gao, Z. WDR68 is essential for the transcriptional activation of the PRC1-AUTS2 complex and neuronal differentiation of mouse embryonic stem cells. Stem Cell Res 2018, 33, 206–214. [Google Scholar] [CrossRef]
- Weisner, P.A.; Chen, C.Y.; Sun, Y.; Yoo, J.; Kao, W.C.; Zhang, H.; Baltz, E.T.; Troy, J.M.; Stubbs, L. A Mouse Mutation That Dysregulates Neighboring Galnt17 and Auts2 Genes Is Associated with Phenotypes Related to the Human AUTS2 Syndrome. G3 2019, 9, 3891–3906. [Google Scholar] [CrossRef] [Green Version]
- Russo, D.; Della Ragione, F.; Rizzo, R.; Sugiyama, E.; Scalabri, F.; Hori, K.; Capasso, S.; Sticco, L.; Fioriniello, S.; De Gregorio, R.; et al. Glycosphingolipid metabolic reprogramming drives neural differentiation. EMBO J. 2018, 37, e97674. [Google Scholar] [CrossRef]
- Reiner, O.; Karzbrun, E.; Kshirsagar, A.; Kaibuchi, K. Regulation of neuronal migration, an emerging topic in autism spectrum disorders. J. Neurochem. 2016, 136, 440–456. [Google Scholar] [CrossRef]
- Davis, G.W. Homeostatic signaling and the stabilization of neural function. Neuron 2013, 80, 718–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tien, N.W.; Kerschensteiner, D. Homeostatic plasticity in neural development. Neural Dev. 2018, 13, 9. [Google Scholar] [CrossRef] [PubMed]
- Wefelmeyer, W.; Puhl, C.J.; Burrone, J. Homeostatic Plasticity of Subcellular Neuronal Structures: From Inputs to Outputs. Trends Neurosci. 2016, 39, 656–667. [Google Scholar] [CrossRef] [Green Version]
- Gao, R.; Penzes, P. Common mechanisms of excitatory and inhibitory imbalance in schizophrenia and autism spectrum disorders. Curr. Mol. Med. 2015, 15, 146–167. [Google Scholar] [CrossRef] [PubMed]
- Penzes, P.; Cahill, M.E.; Jones, K.A.; VanLeeuwen, J.E.; Woolfrey, K.M. Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 2011, 14, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Carta, I.; Chen, C.H.; Schott, A.L.; Dorizan, S.; Khodakhah, K. Cerebellar modulation of the reward circuitry and social behavior. Science 2019, 363, eaav0581. [Google Scholar] [CrossRef] [PubMed]
- Tsai, P.T.; Hull, C.; Chu, Y.; Greene-Colozzi, E.; Sadowski, A.R.; Leech, J.M.; Steinberg, J.; Crawley, J.N.; Regehr, W.G.; Sahin, M. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 2012, 488, 647–651. [Google Scholar] [CrossRef] [Green Version]
- Schmahmann, J.D.; Caplan, D. Cognition, emotion and the cerebellum. Brain 2006, 129, 290–292. [Google Scholar] [CrossRef]
- Van Overwalle, F.; Baetens, K.; Marien, P.; Vandekerckhove, M. Social cognition and the cerebellum: A meta-analysis of over 350 fMRI studies. Neuroimage 2014, 86, 554–572. [Google Scholar] [CrossRef]
- White, J.J.; Sillitoe, R.V. Development of the cerebellum: From gene expression patterns to circuit maps. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 149–164. [Google Scholar] [CrossRef]
- Xiao, L.; Bornmann, C.; Hatstatt-Burkle, L.; Scheiffele, P. Regulation of striatal cells and goal-directed behavior by cerebellar outputs. Nat. Commun. 2018, 9, 3133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, E.; Meng, F.; Fujita, H.; Morgado, F.; Kazemi, Y.; Rice, L.C.; Ren, C.; Escamilla, C.O.; Gibson, J.M.; Sajadi, S.; et al. Regulation of autism-relevant behaviors by cerebellar-prefrontal cortical circuits. Nat. Neurosci. 2020, 23, 1102–1110. [Google Scholar] [CrossRef] [PubMed]
- Dahmane, N.; Ruiz i Altaba, A. Sonic hedgehog regulates the growth and patterning of the cerebellum. Development 1999, 126, 3089–3100. [Google Scholar] [CrossRef]
- Donald, S.; Humby, T.; Fyfe, I.; Segonds-Pichon, A.; Walker, S.A.; Andrews, S.R.; Coadwell, W.J.; Emson, P.; Wilkinson, L.S.; Welch, H.C. P-Rex2 regulates Purkinje cell dendrite morphology and motor coordination. Proc. Natl. Acad. Sci. USA 2008, 105, 4483–4488. [Google Scholar] [CrossRef] [Green Version]
- Kano, M.; Watanabe, T.; Uesaka, N.; Watanabe, M. Multiple Phases of Climbing Fiber Synapse Elimination in the Developing Cerebellum. Cerebellum 2018, 17, 722–734. [Google Scholar] [CrossRef]
- Hashimoto, K.; Tsujita, M.; Miyazaki, T.; Kitamura, K.; Yamazaki, M.; Shin, H.S.; Watanabe, M.; Sakimura, K.; Kano, M. Postsynaptic P/Q-type Ca2+ channel in Purkinje cell mediates synaptic competition and elimination in developing cerebellum. Proc. Natl. Acad. Sci. USA 2011, 108, 9987–9992. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, T.; Hashimoto, K.; Shin, H.S.; Kano, M.; Watanabe, M. P/Q-type Ca2+ channel alpha1A regulates synaptic competition on developing cerebellar Purkinje cells. J. Neurosci. 2004, 24, 1734–1743. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, T.; Yamasaki, M.; Hashimoto, K.; Yamazaki, M.; Abe, M.; Usui, H.; Kano, M.; Sakimura, K.; Watanabe, M. Cav2.1 in cerebellar Purkinje cells regulates competitive excitatory synaptic wiring, cell survival, and cerebellar biochemical compartmentalization. J. Neurosci. 2012, 32, 1311–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaral, D.G.; Schumann, C.M.; Nordahl, C.W. Neuroanatomy of autism. Trends Neurosci. 2008, 31, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Bauman, M.L.; Kemper, T.L. Neuroanatomic observations of the brain in autism: A review and future directions. Int. J. Dev. Neurosci. 2005, 23, 183–187. [Google Scholar] [CrossRef]
- Hori, K.; Nagai, T.; Shan, W.; Sakamoto, A.; Abe, M.; Yamazaki, M.; Sakimura, K.; Yamada, K.; Hoshino, M. Heterozygous Disruption of Autism susceptibility candidate 2 Causes Impaired Emotional Control and Cognitive Memory. PLoS ONE 2015, 10, e0145979. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hori, K.; Shimaoka, K.; Hoshino, M. AUTS2 Gene: Keys to Understanding the Pathogenesis of Neurodevelopmental Disorders. Cells 2022, 11, 11. https://doi.org/10.3390/cells11010011
Hori K, Shimaoka K, Hoshino M. AUTS2 Gene: Keys to Understanding the Pathogenesis of Neurodevelopmental Disorders. Cells. 2022; 11(1):11. https://doi.org/10.3390/cells11010011
Chicago/Turabian StyleHori, Kei, Kazumi Shimaoka, and Mikio Hoshino. 2022. "AUTS2 Gene: Keys to Understanding the Pathogenesis of Neurodevelopmental Disorders" Cells 11, no. 1: 11. https://doi.org/10.3390/cells11010011
APA StyleHori, K., Shimaoka, K., & Hoshino, M. (2022). AUTS2 Gene: Keys to Understanding the Pathogenesis of Neurodevelopmental Disorders. Cells, 11(1), 11. https://doi.org/10.3390/cells11010011