
Regulation of TNF-Induced Osteoclast Differentiation


Abstract
:1. Introduction
2. Origin and Biological Characteristics of OC
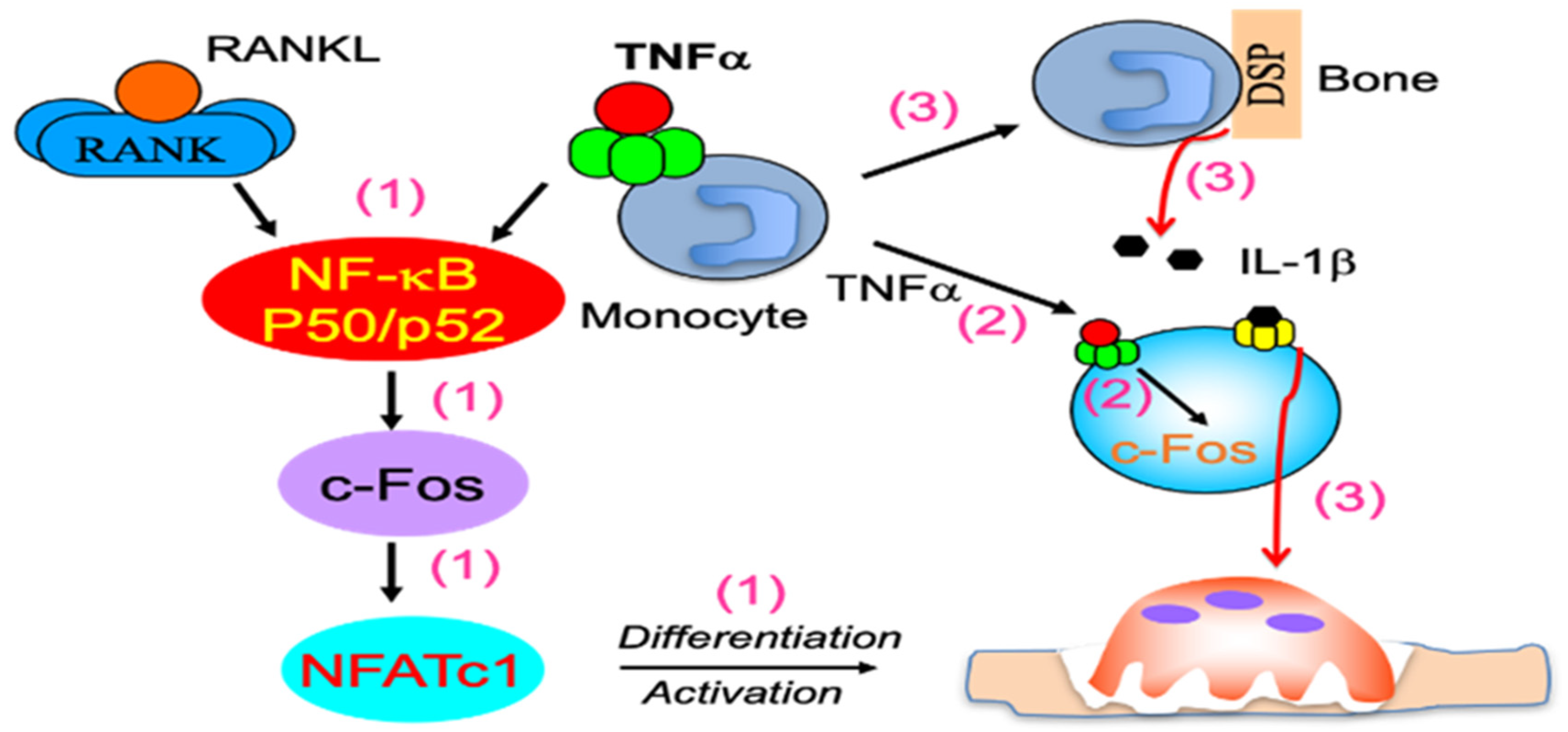
3. RANKL/OPG Signaling in Control of OC Differentiation
4. TNFα Induction of OC Formation Independent of RANKL Signaling
5. Intracellular Factors That Limit TNFα Induction of OC Differentiation
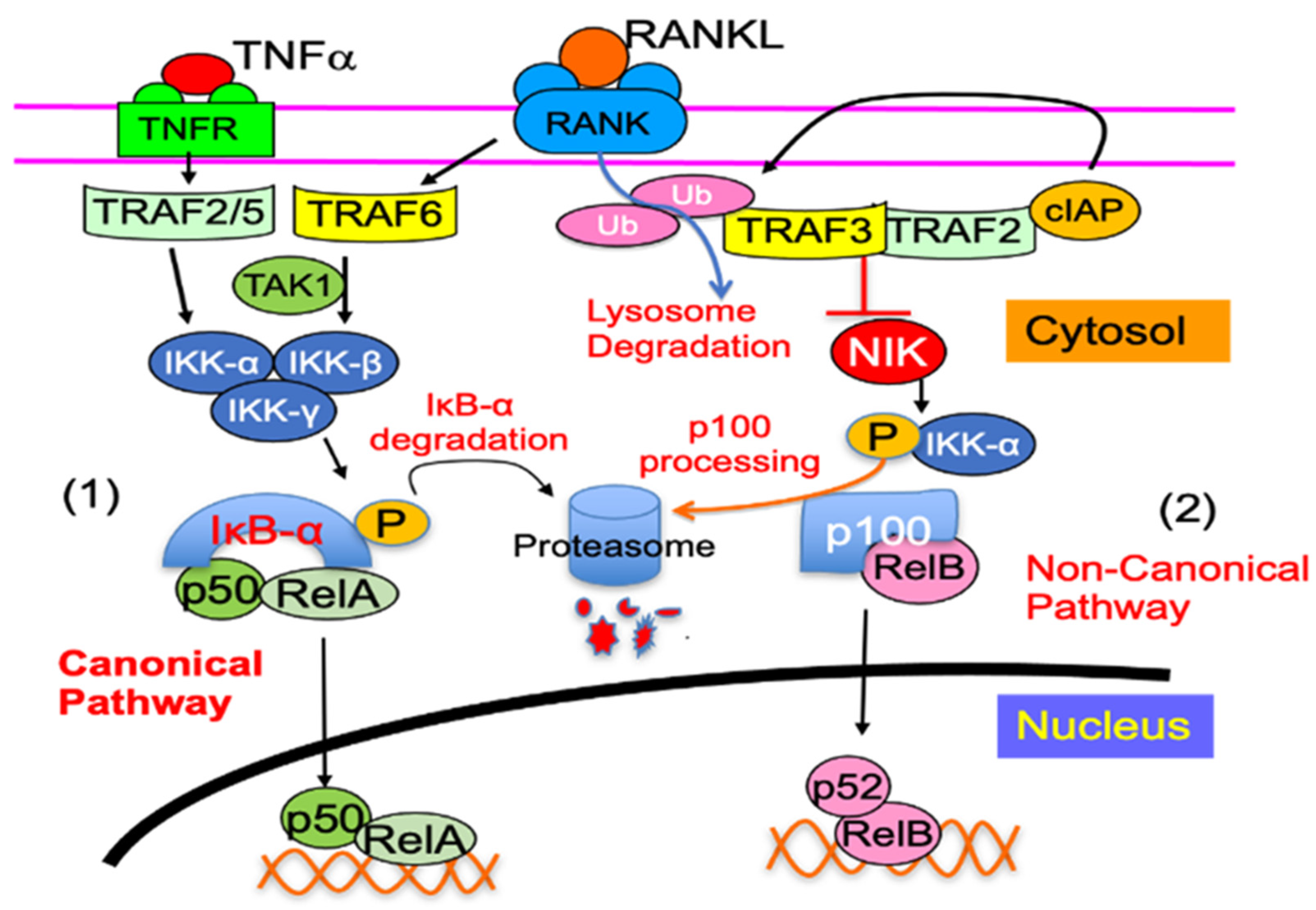
5.1. Non-Canonical NF-κB2 Signaling Proteins
5.1.1. NF-κB2 p100
5.1.2. TRAF3
5.1.3. RelB
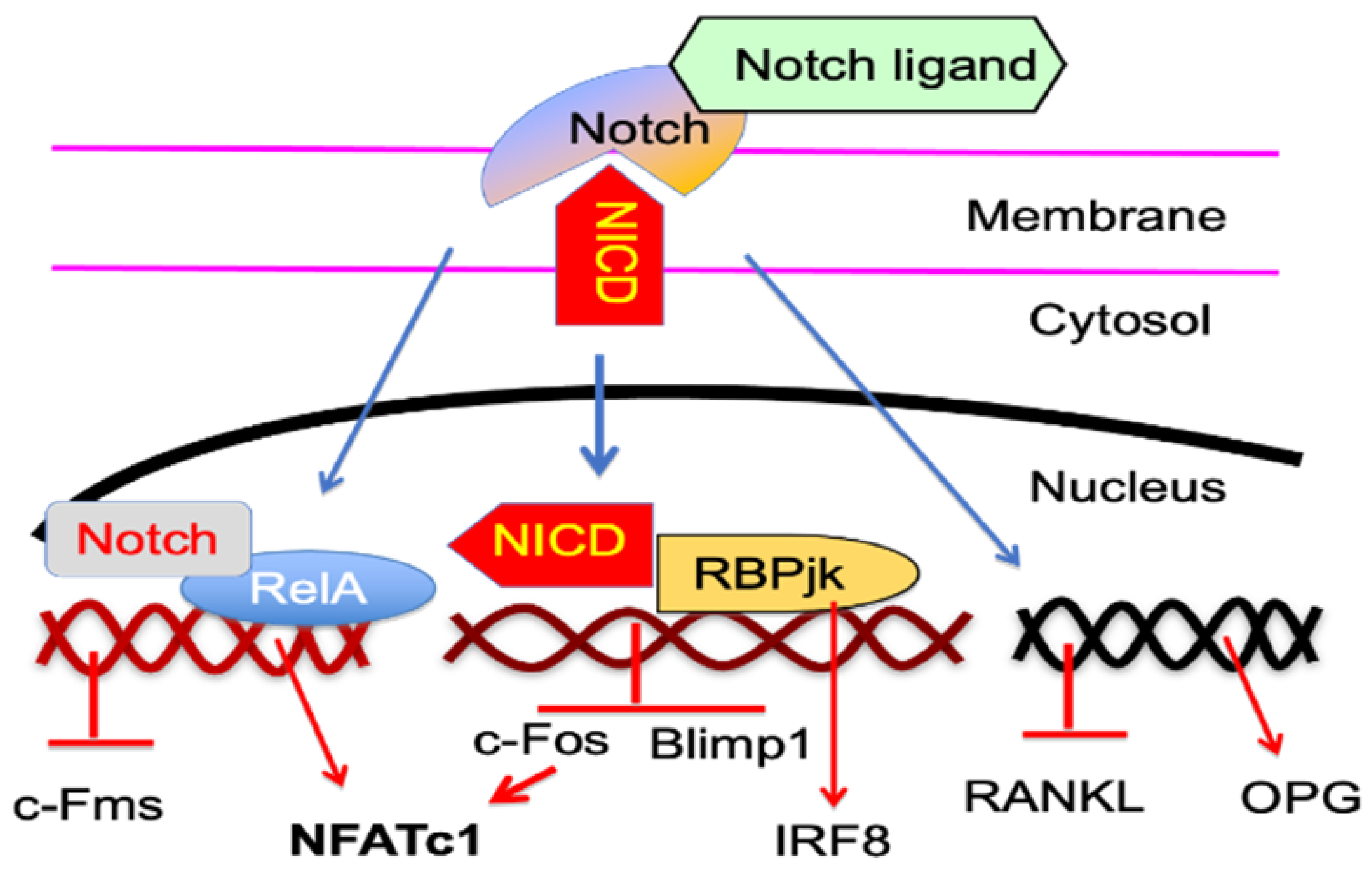
5.2. Notch Signaling Proteins
6. Regulation of OC Forming Potential through Macrophage Polarization
6.1. Macrophage Generation
6.2. Polarized M1 Macrophages with Altered OC Forming Potential
6.3. Polarized M2 Macrophages with Reduced OC Forming Potential
6.4. OC Forming Potential from Unclassified Polarized Macrophages
7. Cytokines That Enhance TNFα Induction of OC Differentiation
7.1. IL-1β
7.2. TGFβ1
8. Limitations of Anti-TNF Therapy to Improve Osteoporosis in RA
9. Prospective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Garnero, P.; Shih, W.J.; Gineyts, E.; Karpf, D.B.; Delmas, P.D. Comparison of new biochemical markers of bone turnover in late postmenopausal osteoporotic women in response to alendronate treatment. J. Clin. Endocrinol. Metab. 1994, 79, 1693–1700. [Google Scholar] [CrossRef]
- Garnero, P.; Sornay-Rendu, E.; Chapuy, M.C.; Delmas, P.D. Increased bone turnover in late postmenopausal women is a major determinant of osteoporosis. J. Bone Miner. Res. 1996, 11, 337–349. [Google Scholar] [CrossRef]
- Eriksen, E.F.; Hodgson, S.F.; Eastell, R.; Riggs, B.L.; Cedel, S.L.; O’Fallon, W.M. Cancellous bone remodeling in type i (postmenopausal) osteoporosis: Quantitative assessment of rates of formation, resorption, and bone loss at tissue and cellular levels. J. Bone Miner. Res. 1990, 5, 311–319. [Google Scholar] [CrossRef]
- Redlich, K.; Hayer, S.; Maier, A.; Dunstan, C.R.; Tohidast-Akrad, M.; Lang, S.; Türk, B.; Pietschmann, P.; Woloszczuk, W.; Haralambous, S.; et al. Tumor necrosis factor α-mediated joint destruction is inhibited by targeting osteoclasts with osteoprotegerin. Arthritis Rheum. 2002, 46, 785–792. [Google Scholar] [CrossRef]
- Redlich, K.; Hayer, S.; Ricci, R.; David, J.-P.; Tohidast-Akrad, M.; Kollias, G.; Steiner, G.; Smolen, J.S.; Wagner, E.F.; Schett, G. Osteoclasts are essential for TNF-α–mediated joint destruction. J. Clin. Investig. 2002, 110, 1419–1427. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.J.; Pollock, C.B.; Kelly, K. Mechanisms of cancer metastasis to the bone. Cell Res. 2005, 15, 57–62. [Google Scholar] [CrossRef]
- Guise, T.A.; Mohammad, K.S.; Clines, G.; Stebbins, E.G.; Wong, D.H.; Higgins, L.S.; Vessella, R.; Corey, E.; Padalecki, S.; Suva, L.; et al. Basic Mechanisms Responsible for Osteolytic and Osteoblastic Bone Metastases. Clin. Cancer Res. 2006, 12, 6213s–6216s. [Google Scholar] [CrossRef] [Green Version]
- Abu-Amer, Y.; Darwech, I.; Clohisy, J.C. Aseptic loosening of total joint replacements: Mechanisms underlying osteolysis and potential therapies. Arthritis Res. Ther. 2007, 9 (Suppl. 1), S6. [Google Scholar] [CrossRef] [Green Version]
- Hienz, S.A.; Paliwal, S.; Ivanovski, S. Mechanisms of Bone Resorption in Periodontitis. J. Immunol. Res. 2015, 2015, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Marie, P.J.; Kassem, M. Osteoblasts in osteoporosis: Past, emerging, and future anabolic targets. Eur. J. Endocrinol. 2011, 165, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Stafford, R.S.; Drieling, R.L.; Hersh, A.L. National Trends in Osteoporosis Visits and Osteoporosis Treatment, 1988–2003. Arch. Intern. Med. 2004, 164, 1525–1530. [Google Scholar] [CrossRef] [Green Version]
- Wasnich, R.D.; Miller, P.D. Antifracture Efficacy of Antiresorptive Agents Are Related to Changes in Bone Density. J. Clin. Endocrinol. Metab. 2000, 85, 231–236. [Google Scholar] [CrossRef]
- Cooper, C.; The IOF CSA Working Group on Fracture Epidemiology; Cole, Z.A.; Holroyd, C.R.; Earl, S.C.; Harvey, N.C.; Dennison, E.M.; Melton, L.J.; Cummings, S.R.; Kanis, J.A. Secular trends in the incidence of hip and other osteoporotic fractures. Osteoporos. Int. 2011, 22, 1277–1288. [Google Scholar] [CrossRef] [Green Version]
- Melton, L.J., 3rd; Kearns, A.E.; Atkinson, E.J.; Bolander, M.E.; Achenbach, S.J.; Huddleston, J.M.; Therneau, T.M.; Leibson, C.L. Secular trends in hip fracture incidence and recurrence. Osteoporos. Int. 2009, 20, 687–694. [Google Scholar] [CrossRef] [Green Version]
- Lewiecki, E.M.; Chastek, B.; Sundquist, K.; Williams, S.A.; Weiss, R.J.; Wang, Y.; Fitzpatrick, L.A.; Curtis, J.R. Osteoporotic fracture trends in a population of US managed care enrollees from 2007 to 2017. Osteoporos. Int. 2020, 31, 1299–1304. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, A.A.; Gludovatz, B.; Riedel, C.; Luengo, E.A.; Saiyed, R.; Marty, E.; Lorich, D.G.; Lane, J.M.; Ritchie, R.O.; Busse, B.; et al. Atypical fracture with long-term bisphosphonate therapy is associated with altered cortical composition and reduced fracture resistance. Proc. Natl. Acad. Sci. USA 2017, 114, 8722–8727. [Google Scholar] [CrossRef] [Green Version]
- Aspenberg, P. Denosumab and atypical femoral fractures. Acta Orthop. 2014, 85, 1. [Google Scholar] [CrossRef]
- Al Elq, A.H. Symptomatic Hypocalcemia Associated with Zoledronic Acid Treatment for Osteoporosis: A Case Report. Oman Med. J. 2013, 28, e043. [Google Scholar] [CrossRef]
- Sedghizadeh, P.P.; Stanley, K.; Caligiuri, M.; Hofkes, S.; Lowry, B.; Shuler, C.F. Oral bisphosphonate use and the prevalence of osteonecrosis of the jaw: An institutional inquiry. J. Am. Dent. Assoc. 2009, 140, 61–66. [Google Scholar] [CrossRef] [Green Version]
- Zavras, A.I. The impact of bisphosphonates on oral health: Lessons from the past and opportunities for the future. Ann. N. Y. Acad. Sci. 2011, 1218, 55–61. [Google Scholar] [CrossRef]
- Benlidayi, I.C.; Guzel, R. Oral Bisphosphonate Related Osteonecrosis of the Jaw: A Challenging Adverse Effect. ISRN Rheumatol. 2013, 2013, 215034. [Google Scholar] [CrossRef] [Green Version]
- Ruggiero, S.L.; Mehrotra, B.; Rosenberg, T.J.; Engroff, S.L. Osteonecrosis of the jaws associated with the use of bisphosphonates: A review of 63 cases. J. Oral Maxillofac. Surg. 2004, 62, 527–534. [Google Scholar] [CrossRef]
- Aghaloo, T.L.; Felsenfeld, A.L.; Tetradis, S. Osteonecrosis of the Jaw in a Patient on Denosumab. J. Oral Maxillofac. Surg. 2010, 68, 959–963. [Google Scholar] [CrossRef] [Green Version]
- Cummings, S.R.; Ferrari, S.; Eastell, R.; Gilchrist, N.; Jensen, J.-E.B.; McClung, M.; Roux, C.; Törring, O.; Valter, I.; Wang, A.T.; et al. Vertebral Fractures After Discontinuation of Denosumab: A Post Hoc Analysis of the Randomized Placebo-Controlled FREEDOM Trial and Its Extension. J. Bone Miner. Res. 2017, 33, 190–198. [Google Scholar] [CrossRef] [Green Version]
- Anastasilakis, A.D.; Makras, P.; Yavropoulou, M.P.; Tabacco, G.; Naciu, A.M.; Palermo, A. Denosumab Discontinuation and the Rebound Phenomenon: A Narrative Review. J. Clin. Med. 2021, 10, 152. [Google Scholar] [CrossRef]
- Breuil, V.; Euller-Ziegler, L. Bisphosphonate therapy in rheumatoid arthritis. Jt. Bone Spine 2006, 73, 349–354. [Google Scholar] [CrossRef]
- Cohen, S.B.; Dore, R.K.; Lane, N.E.; Ory, P.A.; Peterfy, C.G.; Sharp, J.T.; Van Der Heijde, D.; Zhou, L.; Tsuji, W.; Newmark, R.; et al. Denosumab treatment effects on structural damage, bone mineral density, and bone turnover in rheumatoid arthritis: A twelve-month, multicenter, randomized, double-blind, placebo-controlled, phase II clinical trial. Arthritis Rheum. 2008, 58, 1299–1309. [Google Scholar] [CrossRef]
- Sølling, A.S.K.; Harsløf, T.; Langdahl, B. Current Status of Bone-Forming Therapies for the Management of Osteoporosis. Drugs Aging 2019, 36, 625–638. [Google Scholar] [CrossRef]
- Leder, B.Z.; Tsai, J.N.; Jiang, L.A.; Lee, H. Importance of prompt antiresorptive therapy in postmenopausal women discontinuing teriparatide or denosumab: The Denosumab and Teriparatide Follow-up study (DATA-Follow-up). Bone 2017, 98, 54–58. [Google Scholar] [CrossRef]
- Delmas, P.D.; Vergnaud, P.; Arlot, M.E.; Pastoureau, P.; Meunier, P.J.; Nilssen, M.H. The anabolic effect of human PTH (1–34) on bone formation is blunted when bone resorption is inhibited by the bisphosphonate tiludronate—is activated resorption a prerequisite for the in vivo effect of PTH on formation in a remodeling system? Bone 1995, 16, 603–610. [Google Scholar] [CrossRef]
- Black, D.M.; Greenspan, S.L.; Ensrud, K.E.; Palermo, L.; McGowan, J.A.; Lang, T.F.; Garnero, P.; Bouxsein, M.L.; Bilezikian, J.P.; Rosen, C.J.; et al. The Effects of Parathyroid Hormone and Alendronate Alone or in Combination in Postmenopausal Osteoporosis. N. Engl. J. Med. 2003, 349, 1207–1215. [Google Scholar] [CrossRef] [Green Version]
- Finkelstein, J.S.; Hayes, A.; Hunzelman, J.L.; Wyland, J.J.; Lee, H.; Neer, R.M. The Effects of Parathyroid Hormone, Alendronate, or Both in Men with Osteoporosis. N. Engl. J. Med. 2003, 349, 1216–1226. [Google Scholar] [CrossRef]
- Cosman, F.; Nieves, J.W.; Dempster, D.W. Treatment Sequence Matters: Anabolic and Antiresorptive Therapy for Osteoporosis. J. Bone Miner. Res. 2017, 32, 198–202. [Google Scholar] [CrossRef] [Green Version]
- Ponnapakkam, T.; Katikaneni, R.; Sakon, J.; Stratford, R.; Gensure, R.C. Treating osteoporosis by targeting parathyroid hormone to bone. Drug Discov. Today 2014, 19, 204–208. [Google Scholar] [CrossRef] [Green Version]
- Capriani, C.; Irani, D.; Bilezikian, J.P. Safety of osteoanabolic therapy: A decade of experience. J. Bone Miner. Res. 2012, 27, 2419–2428. [Google Scholar] [CrossRef]
- Cosman, F.; Crittenden, D.B.; Adachi, J.D.; Binkley, N.; Czerwinski, E.; Ferrari, S.; Hofbauer, L.C.; Lau, E.; Lewiecki, E.M.; Miyauchi, A.; et al. Romosozumab Treatment in Postmenopausal Women with Osteoporosis. N. Engl. J. Med. 2016, 375, 1532–1543. [Google Scholar] [CrossRef]
- Sleeman, A.; Clements, J.N. Abaloparatide: A new pharmacological option for osteoporosis. Am. J. Health Pharm. 2019, 76, 130–135. [Google Scholar] [CrossRef]
- Chavassieux, P.; Chapurlat, R.; Portero-Muzy, N.; Roux, J.P.; Garcia, P.; Brown, J.P.; Libanati, C.; Boyce, R.W.; Wang, A.; Grauer, A. Bone-Forming and Antiresorptive Effects of Romosozumab in Postmenopausal Women With Osteoporosis: Bone Histomorphometry and Microcomputed Tomography Analysis After 2 and 12 Months of Treatment. J. Bone Miner. Res. 2019, 34, 1597–1608. [Google Scholar] [CrossRef] [Green Version]
- Shoback, D.; Rosen, C.J.; Black, D.M.; Cheung, A.M.; Murad, M.H.; Eastell, R. Pharmacological Management of Osteoporosis in Postmenopausal Women: An Endocrine Society Guideline Update. J. Clin. Endocrinol. Metab. 2020, 105, 587–594. [Google Scholar] [CrossRef]
- Rasmusson, L.; Abtahi, J. Bisphosphonate Associated Osteonecrosis of the Jaw: An Update on Pathophysiology, Risk Factors, and Treatment. Int. J. Dent. 2014, 2014, 471035. [Google Scholar] [CrossRef] [Green Version]
- Kennel, K.A.; Drake, M.T. Adverse Effects of Bisphosphonates: Implications for Osteoporosis Management. Mayo Clin. Proc. 2009, 84, 632–637. [Google Scholar] [CrossRef] [Green Version]
- Boyce, B.F.; Yoneda, T.; Lowe, C.; Soriano, P.; Mundy, G.R. Requirement of pp60c-src expression for osteoclasts to form ruffled borders and resorb bone in mice. J. Clin. Investig. 1992, 90, 1622–1627. [Google Scholar] [CrossRef] [Green Version]
- Stenbeck, G. Formation and function of the ruffled border in osteoclasts. Semin. Cell Dev. Biol. 2002, 13, 285–292. [Google Scholar] [CrossRef]
- Schwartzberg, P.L.; Xing, L.; Hoffmann, O.; Lowell, C.A.; Garrett, L.; Boyce, B.F.; Varmus, H.E. Rescue of osteoclast function by transgenic expression of kinase-deficient Src in src−/− mutant mice. Genes Dev. 1997, 11, 2835–2844. [Google Scholar] [CrossRef] [Green Version]
- McHugh, K.P.; Hodivala-Dilke, K.; Zheng, M.-H.; Namba, N.; Lam, J.; Novack, D.; Feng, X.; Ross, F.P.; Hynes, R.O.; Teitelbaum, S.L. Mice lacking β3 integrins are osteosclerotic because of dysfunctional osteoclasts. J. Clin. Investig. 2000, 105, 433–440. [Google Scholar] [CrossRef] [Green Version]
- Meyerson, G.; Dahl, N.; Pahlman, S. Malignant Osteopetrosis: C-src kinase is not reduced in fibroblasts. Calcif. Tissue Int. 1993, 53, 69–70. [Google Scholar] [CrossRef]
- Bernard, F.; Casanova, J.-L.; Cournot, G.; Jabado, N.; Peake, J.; Jauliac, S.; Fischer, A.; Hivroz, C. The protein tyrosine kinase p60c-Src is not implicated in the pathogenesis of the human autosomal recessive form of osteopetrosis: A study of 13 children. J. Pediatr. 1998, 133, 537–543. [Google Scholar] [CrossRef]
- Botero, J.P.; Lee, K.; Branchford, B.R.; Bray, P.F.; Freson, K.; Lambert, M.P.; Luo, M.; Mohan, S.; Ross, J.E.; Bergmeier, W.; et al. Glanzmann thrombasthenia: Genetic basis and clinical correlates. Haematologica 2020, 105, 888–894. [Google Scholar] [CrossRef] [Green Version]
- Gruber, H.E.; Ivey, J.L.; Thompson, E.R.; Chesnut, C.H., 3rd; Baylink, D.J. Osteoblast and osteoclast cell number and cell activity in postmenopausal osteoporosis. Miner. Electrolyte Metab. 1986, 12, 246–254. [Google Scholar]
- Saag, K.G. Glucocorticoid use in rheumatoid arthritis. Curr. Rheumatol. Rep. 2002, 4, 218–225. [Google Scholar] [CrossRef]
- Maricic, M. Update on Glucocorticoid-Induced Osteoporosis. Rheum. Dis. Clin. N. Am. 2011, 37, 415–431. [Google Scholar] [CrossRef]
- Fischman, D.A.; Hay, E.D. Origin of osteoclasts from mononuclear leucocytes in regenerating newt limbs. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 1962, 143, 329–337. [Google Scholar] [CrossRef]
- Jee, W.S.; Nolan, P.D. Origin of Osteoclasts from the Fusion of Phagocytes. Nature 1963, 200, 225–226. [Google Scholar] [CrossRef]
- Kahn, A.J.; Simmons, D.J. Investigation of cell lineage in bone using a chimaera of chick and quial embryonic tissue. Nature 1975, 258, 325–327. [Google Scholar] [CrossRef]
- Coccia, P.F.; Krivit, W.; Cervenka, J.; Clawson, C.; Kersey, J.H.; Kim, T.H.; Nesbit, M.E.; Ramsay, N.K.; Warkentin, P.I.; Teitelbaum, S.L.; et al. Successful Bone-Marrow Transplantation for Infantile Malignant Osteopetrosis. N. Engl. J. Med. 1980, 302, 701–708. [Google Scholar] [CrossRef]
- Geissmann, F.; Manz, M.G.; Jung, S.; Sieweke, M.H.; Merad, M.; Ley, K. Development of Monocytes, Macrophages, and Dendritic Cells. Science 2010, 327, 656–661. [Google Scholar] [CrossRef] [Green Version]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef]
- Kodama, H.; Nose, M.; Niida, S.; Yamasaki, A. Essential role of macrophage colony-stimulating factor in the osteoclast differentiation supported by stromal cells. J. Exp. Med. 1991, 173, 1291–1294. [Google Scholar] [CrossRef]
- Kodama, H.; Yamasaki, A.; Nose, M.; Niida, S.; Ohgame, Y.; Abe, M.; Kumegawa, M.; Suda, T. Congenital osteoclast deficiency in osteopetrotic (op/op) mice is cured by injections of macrophage colony-stimulating factor. J. Exp. Med. 1991, 173, 269–272. [Google Scholar] [CrossRef]
- Yoshida, H.; Hayashi, S.; Kunisada, T.; Ogawa, M.; Nishikawa, S.; Okamura, H.; Sudo, T.; Shultz, L.D.; Nishikawa, S. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature 1990, 345, 442–444. [Google Scholar] [CrossRef]
- Udagawa, N.; Takahashi, N.; Akatsu, T.; Tanaka, H.; Sasaki, T.; Nishihara, T.; Koga, T.; Martin, T.J.; Suda, T. Origin of osteoclasts: Mature monocytes and macrophages are capable of differentiating into osteoclasts under a suitable microenvironment prepared by bone marrow-derived stromal cells. Proc. Natl. Acad. Sci. USA 1990, 87, 7260–7264. [Google Scholar] [CrossRef] [Green Version]
- Speziani, C.; Rivollier, A.; Gallois, A.; Coury, F.; Mazzorana, M.; Azocar, O.; Flacher, M.; Bella, C.; Tebib, J.; Jurdic, P.; et al. Murine dendritic cell transdifferentiation into osteoclasts is differentially regulated by innate and adaptive cytokines. Eur. J. Immunol. 2007, 37, 747–757. [Google Scholar] [CrossRef]
- Sawant, A.; Hensel, J.A.; Chanda, D.; Harris, B.A.; Siegal, G.P.; Maheshwari, A.; Ponnazhagan, S. Depletion of Plasmacytoid Dendritic Cells Inhibits Tumor Growth and Prevents Bone Metastasis of Breast Cancer Cells. J. Immunol. 2012, 189, 4258–4265. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, T.; Ohneda, O.; Arai, F.; Iwamoto, K.; Okada, S.; Takagi, K.; Anderson, D.M.; Suda, T. Bifurcation of osteoclasts and dendritic cells from common progenitors. Blood 2001, 98, 2544–2554. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.S.; Kim, H.S.; Yeon, J.-T.; Choi, S.-W.; Chun, C.H.; Kwak, H.B.; Oh, J. GM-CSF Regulates Fusion of Mononuclear Osteoclasts into Bone-Resorbing Osteoclasts by Activating the Ras/ERK Pathway. J. Immunol. 2009, 183, 3390–3399. [Google Scholar] [CrossRef] [Green Version]
- Rodan, G.A.; Martin, T.J. Role of osteoblasts in hormonal control of bone resorption—A hypothesis. Calcif. Tissue Int. 1981, 33, 349–351. [Google Scholar] [CrossRef]
- Lacey, D.L.; Timms, E.; Tan, H.L.; Kelley, M.J.; Dunstan, C.R.; Burgess, T.; Elliott, R.; Colombero, A.; Elliott, G.; Scully, S.; et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 1998, 93, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, H.; Shima, N.; Nakagawa, N.; Yamaguchi, K.; Kinosaki, M.; Mochizuki, S.; Tomoyasu, A.; Yano, K.; Goto, M.; Murakami, A.; et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc. Natl. Acad. Sci. USA 1998, 95, 3597–3602. [Google Scholar] [CrossRef] [Green Version]
- Anderson, D.M.; Maraskovsky, E.; Billingsley, W.L.; Dougall, W.C.; Tometsko, M.E.; Roux, E.R.; Teepe, M.C.; DuBose, R.F.; Cosman, D.; Galibert, L. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature 1997, 390, 175–179. [Google Scholar] [CrossRef]
- Dougall, W.C.; Glaccum, M.; Charrier, K.; Rohrbach, K.; Brasel, K.; De Smedt, T.; Daro, E.; Smith, J.; Tometsko, M.E.; Maliszewski, C.R.; et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999, 13, 2412–2424. [Google Scholar] [CrossRef]
- Darnay, B.G.; Besse, A.; Poblenz, A.T.; Lamothe, B.; Jacoby, J.J. TRAFs in RANK Signaling. Adv. Exp. Med. Biol. 2007, 597, 152–159. [Google Scholar] [CrossRef]
- Yamashita, T.; Yao, Z.; Li, F.; Zhang, Q.; Badell, I.R.; Schwarz, E.M.; Takeshita, S.; Wagner, E.F.; Noda, M.; Matsuo, K.; et al. NF-κB p50 and p52 Regulate Receptor Activator of NF-κB Ligand (RANKL) and Tumor Necrosis Factor-induced Osteoclast Precursor Differentiation by Activating c-Fos and NFATc1. J. Biol. Chem. 2007, 282, 18245–18253. [Google Scholar] [CrossRef] [Green Version]
- Takayanagi, H. The Role of NFAT in Osteoclast Formation. Ann. N. Y. Acad. Sci. 2007, 1116, 227–237. [Google Scholar] [CrossRef]
- Takayanagi, H.; Kim, S.; Koga, T.; Nishina, H.; Isshiki, M.; Yoshida, H.; Saiura, A.; Isobe, M.; Yokochi, T.; Inoue, J.-I.; et al. Induction and Activation of the Transcription Factor NFATc1 (NFAT2) Integrate RANKL Signaling in Terminal Differentiation of Osteoclasts. Dev. Cell 2002, 3, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, K.; Galson, D.L.; Zhao, C.; Peng, L.; Laplace, C.; Wang, K.Z.; Bachler, M.A.; Amano, H.; Aburatani, H.; Ishikawa, H.; et al. Nuclear Factor of Activated T-cells (NFAT) Rescues Osteoclastogenesis in Precursors Lacking c-Fos. J. Biol. Chem. 2004, 279, 26475–26480. [Google Scholar] [CrossRef] [Green Version]
- Boyce, B.F.; Xing, L. Biology of RANK, RANKL, and osteoprotegerin. Arthritis Res. Ther. 2007, 9 (Suppl. 1), S1. [Google Scholar] [CrossRef] [Green Version]
- Bucay, N.; Sarosi, I.; Dunstan, C.R.; Morony, S.; Tarpley, J.; Capparelli, C.; Scully, S.; Tan, H.L.; Xu, W.; Lacey, D.L.; et al. Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998, 12, 1260–1268. [Google Scholar] [CrossRef]
- Simonet, W.; Lacey, D.; Dunstan, C.; Kelley, M.; Chang, M.-S.; Lüthy, R.; Nguyen, H.; Wooden, S.; Bennett, L.; Boone, T.; et al. Osteoprotegerin: A Novel Secreted Protein Involved in the Regulation of Bone Density. Cell 1997, 89, 309–319. [Google Scholar] [CrossRef] [Green Version]
- Whyte, M.P.; Obrecht, S.E.; Finnegan, P.M.; Jones, J.L.; Podgornik, M.N.; McAlister, W.H.; Mumm, S. Osteoprotegerin Deficiency and Juvenile Paget’s Disease. N. Engl. J. Med. 2002, 347, 175–184. [Google Scholar] [CrossRef]
- Hofbauer, L.C. Clinical Implications of the Osteoprotegerin/RANKL/RANK System for Bone and Vascular Diseases. JAMA 2004, 292, 490–495. [Google Scholar] [CrossRef]
- Kerschan-Schindl, K.; Wendlova, J.; Kudlacek, S.; Gleiss, A.; Woloszczuk, W.; Pietschmann, P. Serum Levels of Receptor Activator of Nuclear Factor κB Ligand (RANKL) in Healthy Women and Men. Exp. Clin. Endocrinol. Diabetes 2007, 116, 491–495. [Google Scholar] [CrossRef]
- Uemura, H.; Yasui, T.; Miyatani, Y.; Yamada, M.; Hiyoshi, M.; Arisawa, K.; Irahara, M. Circulating profiles of osteoprotegerin and soluble receptor activator of nuclear factor kappaB ligand in post-menopausal women. J. Endocrinol. Investig. 2008, 31, 163–168. [Google Scholar]
- Fichna, M.; Zurawek, M.; Fichna, P.; Gryczynska, M.; Nowak, J.; Ruchala, M. Increased serum osteoprotegerin in patients with primary adrenal insufficiency receiving conventional hydrocortisone substitution. J. Physiol. Pharmacol. 2012, 63, 677–682. [Google Scholar]
- Abrahamsen, B.; Hjelmborg, J.V.; Kostenuik, P.; Stilgren, L.S.; Kyvik, K.; Adamu, S.; Brixen, K.; Langdahl, B.L. Circulating amounts of osteoprotegerin and RANK ligand: Genetic influence and relationship with BMD assessed in female twins. Bone 2005, 36, 727–735. [Google Scholar] [CrossRef]
- Ferrari-Lacraz, S.; Ferrari, S.L. Do RANKL inhibitors (denosumab) affect inflammation and immunity? Osteoporos. Int. 2010, 22, 435–446. [Google Scholar] [CrossRef]
- Kobayashi, K.; Takahashi, N.; Jimi, E.; Udagawa, N.; Takami, M.; Kotake, S.; Nakagawa, N.; Kinosaki, M.; Yamaguchi, K.; Shima, N.; et al. Tumor Necrosis Factor α Stimulates Osteoclast Differentiation by a Mechanism Independent of the Odf/Rankl–Rank Interaction. J. Exp. Med. 2000, 191, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Kadono, Y.; Takami, M.; Lee, J.; Lee, S.-H.; Okada, F.; Kim, J.H.; Kobayashi, T.; Odgren, P.R.; Nakano, H.; et al. Osteoclast differentiation independent of the TRANCE–RANK–TRAF6 axis. J. Exp. Med. 2005, 202, 589–595. [Google Scholar] [CrossRef]
- Yao, Z.; Xing, L.; Qin, C.; Schwarz, E.M.; Boyce, B.F. Osteoclast Precursor Interaction with Bone Matrix Induces Osteoclast Formation Directly by an Interleukin-1-mediated Autocrine Mechanism. J. Biol. Chem. 2008, 283, 9917–9924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Sarosi, I.; Yan, X.-Q.; Morony, S.; Capparelli, C.; Tan, H.-L.; McCabe, S.; Elliott, R.; Scully, S.; Van, G.; et al. RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc. Natl. Acad. Sci. USA 2000, 97, 1566–1571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, J.; Takeshita, S.; Barker, J.E.; Kanagawa, O.; Ross, F.P.; Teitelbaum, S.L. TNF-α induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J. Clin. Investig. 2000, 106, 1481–1488. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Xing, L.; Boyce, B.F. NF-κB p100 limits TNF-induced bone resorption in mice by a TRAF3-dependent mechanism. J. Clin. Investig. 2009, 119, 3024–3034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiu, Y.; Xu, H.; Zhao, C.; Li, J.; Morita, Y.; Yao, Z.; Xing, L.; Boyce, B.F. Chloroquine reduces osteoclastogenesis in murine osteoporosis by preventing TRAF3 degradation. J. Clin. Investig. 2013, 124, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Lei, W.; Duan, R.; Li, Y.; Luo, L.; Boyce, B.F. RANKL cytokine enhances TNF-induced osteoclastogenesis independently of TNF receptor associated factor (TRAF) 6 by degrading TRAF3 in osteoclast precursors. J. Biol. Chem. 2017, 292, 10169–10179. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Ayoub, A.; Xiu, Y.; Yin, X.; Sanders, J.O.; Mesfin, A.; Xing, L.; Yao, Z.; Boyce, B.F. TGFβ-induced degradation of TRAF3 in mesenchymal progenitor cells causes age-related osteoporosis. Nat. Commun. 2019, 10, 2795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Grimes, S.N.; Li, S.; Hu, X.; Ivashkiv, L.B. TNF-induced osteoclastogenesis and inflammatory bone resorption are inhibited by transcription factor RBP-J. J. Exp. Med. 2012, 209, 319–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Takami, M.; Yamada, A.; Wang, X.; Koga, T.; Hu, X.; Tamura, T.; Ozato, K.; Choi, Y.; Ivashkiv, L.B.; et al. Interferon regulatory factor-8 regulates bone metabolism by suppressing osteoclastogenesis. Nat. Med. 2009, 15, 1066–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thumbigere-Math, V.; Foster, B.L.; Bachu, M.; Yoshii, H.; Brooks, S.R.; Coulter, A.; Chavez, M.; Togi, S.; Neely, A.L.; Deng, Z.; et al. Inactivating Mutation in IRF8 Promotes Osteoclast Transcriptional Programs and Increases Susceptibility to Tooth Root Resorption. J. Bone Miner. Res. 2019, 34, 1155–1168. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Hou, X.; Yin, X.; Li, Y.; Duan, R.; Boyce, B.F.; Yao, Z. TNF Induction of NF-κB RelB Enhances RANKL-Induced Osteoclastogenesis by Promoting Inflammatory Macrophage Differentiation but also Limits It through Suppression of NFATc1 Expression. PLoS ONE 2015, 10, e0135728. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Yamazaki, H.; Yamane, T.; Yoshino, M.; Okuyama, H.; Tsuneto, M.; Kurino, T.; Hayashi, S.-I.; Sakano, S. Regulation of osteoclast development by Notch signaling directed to osteoclast precursors and through stromal cells. Blood 2003, 101, 2227–2234. [Google Scholar] [CrossRef]
- Canalis, E.; Bridgewater, D.; Schilling, L.; Zanotti, S. Canonical Notch activation in osteocytes causes osteopetrosis. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E171–E182. [Google Scholar] [CrossRef] [Green Version]
- Engin, F.; Yao, Z.; Yang, T.; Zhou, G.; Bertin, T.; Jiang, M.M.; Chen, Y.; Wang, L.; Zheng, H.; E Sutton, R.; et al. Dimorphic effects of Notch signaling in bone homeostasis. Nat. Med. 2008, 14, 299–305. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takayanagi, H.; Ogasawara, K.; Hida, S.; Chiba, T.; Murata, S.; Sato, K.; Takaoka, A.; Yokochi, T.; Oda, H.; Tanaka, K.; et al. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-γ. Nature 2000, 408, 600–605. [Google Scholar] [CrossRef] [PubMed]
- AlQranei, M.S.; Senbanjo, L.T.; Aljohani, H.; Hamza, T.; Chellaiah, M.A. Lipopolysaccharide- TLR-4 Axis regulates Osteoclastogenesis independent of RANKL/RANK signaling. BMC Immunol. 2021, 22, 23. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Bar-Shavit, Z. Dual Modulation of Osteoclast Differentiation by Lipopolysaccharide. J. Bone Miner. Res. 2002, 17, 1211–1218. [Google Scholar] [CrossRef]
- Horwood, N.J.; Elliott, J.; Martin, T.J.; Gillespie, M.T. IL-12 Alone and in Synergy with IL-18 Inhibits Osteoclast Formation In Vitro. J. Immunol. 2001, 166, 4915–4921. [Google Scholar] [CrossRef] [Green Version]
- Nagata, N.; Kitaura, H.; Yoshida, N.; Nakayama, K. Inhibition of RANKL-induced osteoclast formation in mouse bone marrow cells by IL-12: Involvement of IFN-gamma possibly induced from non-T cell population. Bone 2003, 33, 721–732. [Google Scholar] [CrossRef]
- Ha, H.; Lee, J.H.; Kim, H.N.; Kwak, H.B.; Kim, H.M.; Lee, S.E.; Rhee, J.H.; Kim, H.H.; Lee, Z.H. Stimulation by TLR5 modulates osteoclast differentiation through STAT1/IFN-beta. J. Immunol. 2008, 180, 1382–1389. [Google Scholar] [CrossRef] [Green Version]
- Palmqvist, P.; Lundberg, P.; Persson, E.; Johansson, A.; Lundgren, I.; Lie, A.; Conaway, H.H.; Lerner, U. Inhibition of Hormone and Cytokine-stimulated Osteoclastogenesis and Bone Resorption by Interleukin-4 and Interleukin-13 Is Associated with Increased Osteoprotegerin and Decreased RANKL and RANK in a STAT6-dependent Pathway. J. Biol. Chem. 2006, 281, 2414–2429. [Google Scholar] [CrossRef] [Green Version]
- Yamada, A.; Takami, M.; Kawawa, T.; Yasuhara, R.; Zhao, B.; Mochizuki, A.; Miyamoto, Y.; Eto, T.; Yasuda, H.; Nakamichi, Y.; et al. Interleukin-4 inhibition of osteoclast differentiation is stronger than that of interleukin-13 and they are equivalent for induction of osteoprotegerin production from osteoblasts. Immunology 2007, 120, 573–579. [Google Scholar] [CrossRef]
- E Evans, K.; Fox, S.W. Interleukin-10 inhibits osteoclastogenesis by reducing NFATc1 expression and preventing its translocation to the nucleus. BMC Cell Biol. 2007, 8, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, S.G.-K.; Sugiyama, E.; Shinoda, K.; Taki, H.; Hounoki, H.; Abdel-Aziz, H.O.; Maruyama, M.; Kobayashi, M.; Ogawa, H.; Miyahara, T. Interleukin-10 inhibits RANKL-mediated expression of NFATc1 in part via suppression of c-Fos and c-Jun in RAW264.7 cells and mouse bone marrow cells. Bone 2007, 41, 592–602. [Google Scholar] [CrossRef]
- Ghosh, G.; Wang, V.Y.-F.; Huang, D.-B.; Fusco, A. NF-κB regulation: Lessons from structures. Immunol. Rev. 2012, 246, 36–58. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.-C. The noncanonical NF-κB pathway. Immunol. Rev. 2012, 246, 125–140. [Google Scholar] [CrossRef] [Green Version]
- Hayden, M.S.; Ghosh, S. Shared Principles in NF-κB Signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [Green Version]
- Vallabhapurapu, S.; Karin, M. Regulation and Function of NF-κB Transcription Factors in the Immune System. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef] [PubMed]
- Shibata, W.; Maeda, S.; Hikiba, Y.; Yanai, A.; Ohmae, T.; Sakamoto, K.; Nakagawa, H.; Ogura, K.; Omata, M. Cutting edge: The IkappaB kinase (IKK) inhibitor, NEMO-binding domain peptide, blocks inflammatory injury in murine colitis. J. Immunol. 2007, 179, 2681–2685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarnegar, B.J.; Wang, Y.; Mahoney, D.J.; Dempsey, P.W.; Cheung, H.H.; He, J.; Shiba, T.; Yang, X.; Yeh, W.-C.; Mak, T.W.; et al. Noncanonical NF-κB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat. Immunol. 2008, 9, 1371–1378. [Google Scholar] [CrossRef] [Green Version]
- Bonizzi, G.; Karin, M. The two NF-κB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004, 25, 280–288. [Google Scholar] [CrossRef]
- Senftleben, U.; Cao, Y.; Xiao, G.; Greten, F.R.; Krähn, G.; Bonizzi, G.; Chen, Y.; Hu, Y.; Fong, A.; Sun, S.-C.; et al. Activation by IKKα of a Second, Evolutionary Conserved, NF-κB Signaling Pathway. Science 2001, 293, 1495–1499. [Google Scholar] [CrossRef] [PubMed]
- Franzoso, G.; Carlson, L.; Xing, L.; Poljak, L.; Shores, E.W.; Brown, K.D.; Leonardi, A.; Tran, T.; Boyce, B.F.; Siebenlist, U. Requirement for NF-kappaB in osteoclast and B-cell development. Genes Dev. 1997, 11, 3482–3496. [Google Scholar] [CrossRef] [Green Version]
- Iotsova, V.; Caamano, J.; Loy, J.; Yang, Y.; Lewin, A.; Bravo, R. Osteopetrosis in mice lacking NF-kappaB1 and NF-kappaB2. Nat. Med. 1997, 3, 1285–1289. [Google Scholar] [CrossRef]
- Xing, L.; Carlson, L.; Story, B.; Tai, Z.; Keng, P.; Siebenlist, U.; Boyce, B.F. Expression of Either NF-κB p50 or p52 in Osteoclast Precursors Is Required for IL-1-Induced Bone Resorption. J. Bone Miner. Res. 2003, 18, 260–269. [Google Scholar] [CrossRef]
- Veis, D.; Yin, L.; Hagen-Stapleton, A.; Schreiber, R.D.; Goeddel, D.V.; Ross, F.P.; Teitelbaum, S. The IκB Function of NF-κB2 p100 Controls Stimulated Osteoclastogenesis. J. Exp. Med. 2003, 198, 771–781. [Google Scholar] [CrossRef]
- He, J.Q.; Saha, S.K.; Kang, J.R.; Zarnegar, B.; Cheng, G. Specificity of TRAF3 in Its Negative Regulation of the Noncanonical NF-κB Pathway. J. Biol. Chem. 2007, 282, 3688–3694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.Q.; Zarnegar, B.; Oganesyan, G.; Saha, S.K.; Yamazaki, S.; Doyle, S.E.; Dempsey, P.W.; Cheng, G. Rescue of TRAF3-null mice by p100 NF-κB deficiency. J. Exp. Med. 2006, 203, 2413–2418. [Google Scholar] [CrossRef]
- Vallabhapurapu, S.; Matsuzawa, A.; Zhang, W.; Tseng, P.-H.; Keats, J.J.; Wang, H.; A A Vignali, D.; Bergsagel, P.L.; Karin, M. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-κB signaling. Nat. Immunol. 2008, 9, 1364–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lomaga, M.A.; Yeh, W.-C.; Sarosi, I.; Duncan, G.S.; Furlonger, C.; Ho, A.; Morony, S.; Capparelli, C.; Van, G.; Kaufman, S.; et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 1999, 13, 1015–1024. [Google Scholar] [CrossRef] [Green Version]
- Naito, A.; Azuma, S.; Tanaka, S.; Miyazaki, T.; Takaki, S.; Takatsu, K.; Nakao, K.; Nakamura, K.; Katsuki, M.; Yamamoto, T.; et al. Severe osteopetrosis, defective interleukin-1 signalling and lymph node organogenesis in TRAF6-deficient mice. Genes Cells 1999, 4, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Walsh, P.; Walsh, M.C.; Speirs, K.M.; Chiffoleau, E.; King, C.G.; Hancock, W.W.; Caamano, J.; A Hunter, C.; Scott, P.; et al. TRAF6 Is a Critical Factor for Dendritic Cell Maturation and Development. Immunity 2003, 19, 353–363. [Google Scholar] [CrossRef] [Green Version]
- Vaira, S.; Johnson, T.; Hirbe, A.C.; Alhawagri, M.; Anwisye, I.; Sammut, B.; O’Neal, J.; Zou, W.; Weilbaecher, K.N.; Faccio, R.; et al. RelB is the NF- B subunit downstream of NIK responsible for osteoclast differentiation. Proc. Natl. Acad. Sci. USA 2008, 105, 3897–3902. [Google Scholar] [CrossRef] [Green Version]
- Guruharsha, K.G.; Kankel, M.W.; Artavanis-Tsakonas, S. The Notch signalling system: Recent insights into the complexity of a conserved pathway. Nat. Rev. Genet. 2012, 13, 654–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanotti, S.; Canalis, E. Notch and the Skeleton. Mol. Cell. Biol. 2010, 30, 886–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, S.; Kopan, R.; Zou, W.; Hilton, M.J.; Ong, C.-T.; Long, F.; Ross, F.P.; Teitelbaum, S.L. NOTCH1 Regulates Osteoclastogenesis Directly in Osteoclast Precursors and Indirectly via Osteoblast Lineage Cells. J. Biol. Chem. 2008, 283, 6509–6518. [Google Scholar] [CrossRef] [Green Version]
- Fukushima, H.; Nakao, A.; Okamoto, F.; Shin, M.; Kajiya, H.; Sakano, S.; Bigas, A.; Jimi, E.; Okabe, K. The Association of Notch2 and NF-κB Accelerates RANKL-Induced Osteoclastogenesis. Mol. Cell. Biol. 2008, 28, 6402–6412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, S.; Taylor, P. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Murphy, C.A.; Langrish, C.L.; Chen, Y.; Blumenschein, W.; McClanahan, T.; Kastelein, R.A.; Sedgwick, J.D.; Cua, D.J. Divergent Pro- and Antiinflammatory Roles for IL-23 and IL-12 in Joint Autoimmune Inflammation. J. Exp. Med. 2003, 198, 1951–1957. [Google Scholar] [CrossRef]
- Smith, A.M.; Rahman, F.Z.; Hayee, B.; Graham, S.J.; Marks, D.J.; Sewell, G.W.; Palmer, C.D.; Wilde, J.; Foxwell, B.M.; Gloger, I.S.; et al. Disordered macrophage cytokine secretion underlies impaired acute inflammation and bacterial clearance in Crohn’s disease. J. Exp. Med. 2009, 206, 1883–1897. [Google Scholar] [CrossRef] [Green Version]
- Murai, M.; Turovskaya, O.; Kim, G.; Madan, R.; Karp, C.L.; Cheroutre, H.; Kronenberg, M. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat. Immunol. 2009, 10, 1178–1184. [Google Scholar] [CrossRef]
- Leuschner, F.; Dutta, P.; Gorbatov, R.; I Novobrantseva, T.; Donahoe, J.S.; Courties, G.; Lee, K.M.; I Kim, J.; Markmann, J.F.; Marinelli, B.; et al. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat. Biotechnol. 2011, 29, 1005–1010. [Google Scholar] [CrossRef]
- Li, J.; Hsu, H.-C.; Mountz, J.D. Managing Macrophages in Rheumatoid Arthritis by Reform or Removal. Curr. Rheumatol. Rep. 2012, 14, 445–454. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Hsu, H.-C.; Yang, P.; Wu, Q.; Li, H.; Edgington-Mitchell, L.; Bogyo, M.; Kimberly, R.; Mountz, J.D. Treatment of arthritis by macrophage depletion and immunomodulation: Testing an apoptosis-mediated therapy in a humanized death receptor mouse model. Arthritis Rheum. 2011, 64, 1098–1109. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Liu, X.; Kenney, H.M.; Duan, R.; Lin, X.; Schwarz, E.; Yao, Z. TNF -Polarized Macrophages Produce Insulin-like 6 Peptide to Stimulate Bone Formation in Rheumatoid Arthritis in Mice. J. Bone Miner. Res. 2021, 36, 2426–2439. [Google Scholar] [CrossRef]
- Beek, K.J.; Rusman, T.; Van Der Weijden, M.A.C.; Lems, W.F.; Van Denderen, J.C.; Konsta, M.; Visman, I.; Nurmohamed, M.T.; Van Der Horst-Bruinsma, I.E. Long-Term Treatment With TNF-Alpha Inhibitors Improves Bone Mineral Density But Not Vertebral Fracture Progression in Ankylosing Spondylitis. J. Bone Miner. Res. 2019, 34, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Lim, D.-H.; Oh, J.S.; Kim, Y.-G.; Lee, C.-K.; Yoo, B.; Hong, S. Effect of TNF inhibitors on bone mineral density in rheumatoid arthritis patients receiving bisphosphonate: A retrospective cohort study. Rheumatol. Int. 2020, 40, 481–487. [Google Scholar] [CrossRef]
- Orsolini, G.; Adami, G.; Adami, S.; Viapiana, O.; Idolazzi, L.; Gatti, D.; Rossini, M. Short-Term Effects of TNF Inhibitors on Bone Turnover Markers and Bone Mineral Density in Rheumatoid Arthritis. Calcif. Tissue Int. 2016, 98, 580–585. [Google Scholar] [CrossRef]
- Jackson, S.H.; Alicea, C.; Owens, J.W.; Eigsti, C.L.; Malech, H.L. Characterization of an early dendritic cell precursor derived from murine lineage-negative hematopoietic progenitor cells. Exp. Hematol. 2002, 30, 430–439. [Google Scholar] [CrossRef]
- Schmid, M.A.; Kingston, D.; Boddupalli, S.; Manz, M.G. Instructive cytokine signals in dendritic cell lineage commitment. Immunol. Rev. 2010, 234, 32–44. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.W. Trophic macrophages in development and disease. Nat. Rev. Immunol. 2009, 9, 259–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sierra-Filardi, E.; Vega, M.A.; Sánchez-Mateos, P.; Corbí, A.L.; Puig-Kröger, A. Heme Oxygenase-1 expression in M-CSF-polarized M2 macrophages contributes to LPS-induced IL-10 release. Immunobiology 2010, 215, 788–795. [Google Scholar] [CrossRef]
- Buchacher, T.; Ohradanova-Repic, A.; Stockinger, H.; Fischer, M.B.; Weber, V. M2 Polarization of Human Macrophages Favors Survival of the Intracellular Pathogen Chlamydia pneumoniae. PLoS ONE 2015, 10, e0143593. [Google Scholar] [CrossRef] [Green Version]
- Zajac, E.; Schweighofer, B.; Kupriyanova, T.A.; Juncker-Jensen, A.; Minder, P.; Quigley, J.P.; Deryugina, E.I. Angiogenic capacity of M1- and M2-polarized macrophages is determined by the levels of TIMP-1 complexed with their secreted proMMP-9. Blood 2013, 122, 4054–4067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, H.; Yu, X.; Collin-Osdoby, P.; Osdoby, P. RANKL stimulates inducible nitric-oxide synthase expression and nitric oxide production in developing osteoclasts. An au-tocrine negative feedback mechanism triggered by RANKL-induced interferon-beta via NF-kappaB that restrains osteoclas-togenesis and bone resorption. J. Biol. Chem. 2006, 281, 15809–15820. [Google Scholar] [CrossRef] [Green Version]
- Günthner, R.; Anders, H.-J. Interferon-Regulatory Factors Determine Macrophage Phenotype Polarization. Mediat. Inflamm. 2013, 2013, 731023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, T.; Natoli, G. Transcriptional regulation of macrophage polarization: Enabling diversity with identity. Nat. Rev. Immunol. 2011, 11, 750–761. [Google Scholar] [CrossRef] [PubMed]
- Udagawa, N.; Horwood, N.J.; Elliott, J.; Mackay, A.; Owens, J.; Okamura, H.; Kurimoto, M.; Chambers, T.J.; Martin, T.J.; Gillespie, M.T. Interleukin-18 (interferon-gamma-inducing factor) is produced by osteoblasts and acts via granulocyte/macrophage colony-stimulating factor and not via interferon-gamma to inhibit osteoclast formation. J. Exp. Med. 1997, 185, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi-Matsui, M.; Yano, S.; Matsumoto, N.; Futai, M. Lipopolysaccharide induces multinuclear cell from RAW264.7 line with increased phagocytosis activity. Biochem. Biophys. Res. Commun. 2012, 425, 144–149. [Google Scholar] [CrossRef]
- Jeganathan, S.; Fiorino, C.; Naik, U.; Sun, H.S.; Harrison, R.E. Modulation of Osteoclastogenesis with Macrophage M1- and M2-Inducing Stimuli. PLoS ONE 2014, 9, e104498. [Google Scholar] [CrossRef] [Green Version]
- Takayanagi, H.; Kim, S.; Matsuo, K.; Suzuki, H.; Suzuki, T.; Sato, K.; Yokochi, T.; Oda, H.; Nakamura, K.; Ida, N.; et al. RANKL maintains bone homeostasis through c-Fos-dependent induction of interferon-β. Nature 2002, 416, 744–749. [Google Scholar] [CrossRef]
- Rőszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediat. Inflamm. 2015, 2015, 816460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, S.; Wang, M.W.-H.; Teitelbaum, S.; Ross, F.P. Interleukin-4 Reversibly Inhibits Osteoclastogenesis via Inhibition of NF-κB and Mitogen-activated Protein Kinase Signaling. J. Biol. Chem. 2002, 277, 6622–6630. [Google Scholar] [CrossRef] [Green Version]
- Lewis, D.B.; Liggitt, H.D.; Effmann, E.L.; Motley, S.T.; Teitelbaum, S.; Jepsen, K.J.; Goldstein, S.A.; Bonadio, J.; Carpenter, J.; Perlmutter, R.M. Osteoporosis induced in mice by overproduction of interleukin 4. Proc. Natl. Acad. Sci. USA 1993, 90, 11618–11622. [Google Scholar] [CrossRef] [Green Version]
- Al-Rasheed, A.; Scheerens, H.; Srivastava, A.K.; Rennick, D.M.; Tatakis, D.N. Accelerated alveolar bone loss in mice lacking interleukin-10: Late onset. J. Periodontal Res. 2004, 39, 194–198. [Google Scholar] [CrossRef]
- Sasaki, H.; Hou, L.; Belani, A.; Wang, C.-Y.; Uchiyama, T.; Müller, R.; Stashenko, P. IL-10, But Not IL-4, Suppresses Infection-Stimulated Bone Resorption In Vivo. J. Immunol. 2000, 165, 3626–3630. [Google Scholar] [CrossRef] [Green Version]
- Park-Min, K.-H.; Ji, J.-D.; Antoniv, T.; Reid, A.C.; Silver, R.B.; Humphrey, M.B.; Nakamura, M.; Ivashkiv, L.B. IL-10 Suppresses Calcium-Mediated Costimulation of Receptor Activator NF-κB Signaling during Human Osteoclast Differentiation by Inhibiting TREM-2 Expression. J. Immunol. 2009, 183, 2444–2455. [Google Scholar] [CrossRef] [Green Version]
- Pihusch, V.; Pihusch, M.; Penovici, M.; Kolb, H.J.; Hiller, E.; Pihusch, R. Transforming growth factor beta-1 released from platelets contributes to hypercoagulability in veno-occlusive disease following hematopoetic stem cell transplantation. Thromb. Res. 2005, 116, 233–240. [Google Scholar] [CrossRef]
- Kullberg, M.C.; Hay, V.; Cheever, A.W.; Mamura, M.; Sher, A.; Letterio, J.J.; Shevach, E.M.; Piccirillo, C.A. TGF-β1 production by CD4+CD25+ regulatory T cells is not essential for suppression of intestinal inflammation. Eur. J. Immunol. 2005, 35, 2886–2895. [Google Scholar] [CrossRef]
- Oreffo, R.; Mundy, G.R.; Seyedin, S.M.; Bonewald, L. Activation of the bone-derived latent TGF beta complex by isolated osteoclasts. Biochem. Biophys. Res. Commun. 1989, 158, 817–823. [Google Scholar] [CrossRef]
- Nishikawa, K.; Seo, N.; Torii, M.; Ma, N.; Muraoka, D.; Tawara, I.; Masuya, M.; Tanaka, K.; Takei, Y.; Shiku, H.; et al. Interleukin-17 Induces an Atypical M2-Like Macrophage Subpopulation That Regulates Intestinal Inflammation. PLoS ONE 2014, 9, e108494. [Google Scholar] [CrossRef]
- Lau, Y.S.; Danks, L.; Sun, S.G.; Fox, S.; Sabokbar, A.; Harris, A.; Athanasou, N.A. RANKL-dependent and RANKL-independent mechanisms of macrophage-osteoclast differentiation in breast cancer. Breast Cancer Res. Treat. 2006, 105, 7–16. [Google Scholar] [CrossRef]
- Lau, Y.S.; Adamopoulos, I.; Sabokbar, A.; Giele, H.; Gibbons, C.L.M.H.; A Athanasou, N. Cellular and humoral mechanisms of osteoclast formation in Ewing’s sarcoma. Br. J. Cancer 2007, 96, 1716–1722. [Google Scholar] [CrossRef]
- Boyce, B.F.; Aufdemorte, T.B.; Garrett, I.R.; Yates, A.J.P.; Mundy, G.R. Effects of Interleukin-1 on Bone Turnover in Normal Mice*. Endocrinology 1989, 125, 1142–1150. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Schwarz, E.M.; Boyce, B.F. Osteoclast precursors, RANKL/RANK, and immunology. Immunol. Rev. 2005, 208, 19–29. [Google Scholar] [CrossRef]
- Teitelbaum, S. Osteoclasts; culprits in inflammatory osteolysis. Arthritis Res. Ther. 2005, 8, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofbauer, L.C.; Lacey, D.; Dunstan, C.; Spelsberg, T.; Riggs, B.; Khosla, S. Interleukin-1β and tumor necrosis factor-α, but not interleukin-6, stimulate osteoprotegerin ligand gene expression in human osteoblastic cells. Bone 1999, 25, 255–259. [Google Scholar] [CrossRef]
- Wei, S.; Kitaura, H.; Zhou, P.; Ross, F.P.; Teitelbaum, S. IL-1 mediates TNF-induced osteoclastogenesis. J. Clin. Investig. 2005, 115, 282–290. [Google Scholar] [CrossRef] [Green Version]
- Qin, C.; Baba, O.; Butler, W. Post-translational modifications of sibling proteins and their roles in osteogenesis and dentinogenesis. Crit. Rev. Oral Biol. Med. 2004, 15, 126–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, L.; Torchia, D.; Fohr, B.; Young, M.; Fedarko, N. Flexible Structures of SIBLING Proteins, Bone Sialoprotein, and Osteopontin. Biochem. Biophys. Res. Commun. 2001, 280, 460–465. [Google Scholar] [CrossRef] [Green Version]
- Karst, M.; Gorny, G.; Galvin, R.J.S.; Oursler, M.J. Roles of stromal cell RANKL, OPG, and M-CSF expression in biphasic TGF-β regulation of osteoclast differentiation. J. Cell. Physiol. 2004, 200, 99–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thirunavukkarasu, K.; Miles, R.R.; Halladay, D.L.; Yang, X.; Galvin, R.J.S.; Chandrasekhar, S.; Martin, T.J.; Onyia, J.E. Stimulation of osteoprotegerin (OPG) gene expression by transforming growth factor-beta (TGF-beta). Mapping of the OPG promoter region that mediates TGF-beta effects. J. Biol. Chem. 2001, 276, 36241–36250. [Google Scholar] [CrossRef] [Green Version]
- Fox, S.W.; Lovibond, A.C. Current insights into the role of transforming growth factor-β in bone resorption. Mol. Cell. Endocrinol. 2005, 243, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.M.W.; Itoh, K.; Udagawa, N.; Häusler, K.; Yasuda, H.; Shima, N.; Mizuno, A.; Higashio, K.; Takahashi, N.; Suda, T.; et al. Transforming Growth Factor β Affects Osteoclast Differentiation via Direct and Indirect Actions. J. Bone Miner. Res. 2001, 16, 1787–1794. [Google Scholar] [CrossRef]
- Kaneda, T.; Nojima, T.; Nakagawa, M.; Ogasawara, A.; Kaneko, H.; Sato, T.; Mano, H.; Kumegawa, M.; Hakeda, Y. Endogenous production of TGF-beta is essential for osteoclastogenesis induced by a combination of receptor activator of NF-kappa B ligand and macrophage-colony-stimulating factor. J. Immunol. 2000, 165, 4254–4263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itonaga, I.; Sabokbar, A.; Sun, S.G.; Kudo, O.; Danks, L.; Ferguson, D.; Fujikawa, Y.; Athanasou, N.A. Transforming growth factor-beta induces osteoclast formation in the absence of RANKL. Bone 2004, 34, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Yasui, T.; Kadono, Y.; Nakamura, M.; Oshima, Y.; Matsumoto, T.; Masuda, H.; Hirose, J.; Omata, Y.; Yasuda, H.; Imamura, T.; et al. Regulation of RANKL-induced osteoclastogenesis by TGF-β through molecular interaction between Smad3 and Traf6. J. Bone Miner. Res. 2011, 26, 1447–1456. [Google Scholar] [CrossRef]
- Hafez, E.A.; Mansour, H.E.; Hamza, S.H.; Moftah, S.G.; Younes, T.B.; Ismail, M.A. Bone Mineral Density Changes in Patients with Recent-Onset Rheumatoid Arthritis. Clin. Med. Insights Arthritis Musculoskelet. Disord. 2011, 4, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Goldring, S.R.; Gravallese, E.M. Mechanisms of bone loss in inflammatory arthritis: Diagnosis and therapeutic implications. Arthritis Res. 2000, 2, 33–37. [Google Scholar] [CrossRef] [Green Version]
- Taylor, P.C.; Feldmann, M. Anti-TNF biologic agents: Still the therapy of choice for rheumatoid arthritis. Nat. Rev. Rheumatol. 2009, 5, 578–582. [Google Scholar] [CrossRef]
- Monaco, C.; Nanchahal, J.; Taylor, P.; Feldmann, M. Anti-TNF therapy: Past, present and future. Int. Immunol. 2015, 27, 55–62. [Google Scholar] [CrossRef]
- Kawai, V.K.; Stein, C.M.; Perrien, D.; Griffin, M.R. Effects of anti-tumor necrosis factor α agents on bone. Curr. Opin. Rheumatol. 2012, 24, 576–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, M.S.; Ueki, Y. Does anti-TNF-alpha have a role in the treatment of osteoporosis? Bull. NYU Hosp. Jt. Dis. 2008, 66, 280–281. [Google Scholar] [PubMed]
- Baraliakos, X.; Haibel, H.; Listing, J.; Sieper, J.; Braun, J. Continuous long-term anti-TNF therapy does not lead to an increase in the rate of new bone formation over 8 years in patients with ankylosing spondylitis. Ann. Rheum. Dis. 2013, 73, 710–715. [Google Scholar] [CrossRef]
- Lodder, M.C.; De Jong, Z.; Kostense, P.J.; Molenaar, E.T.H.; Staal, K.; E Voskuyl, A.; Hazes, J.M.W.; Dijkmans, B.A.C.; Lems, W.F. Bone mineral density in patients with rheumatoid arthritis: Relation between disease severity and low bone mineral density. Ann. Rheum. Dis. 2004, 63, 1576–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahata, M.; Maher, J.R.; Juneja, S.C.; Inzana, J.; Xing, L.; Schwarz, E.M.; Berger, A.J.; Awad, H.A. Mechanisms of bone fragility in a mouse model of glucocorticoid-treated rheumatoid arthritis: Implications for insufficiency fracture risk. Arthritis Rheum. 2012, 64, 3649–3659. [Google Scholar] [CrossRef] [Green Version]
- Hill, C.R.; Yuasa, M.; Schoenecker, J.; Goudy, S.L. Jagged1 is essential for osteoblast development during maxillary ossification. Bone 2014, 62, 10–21. [Google Scholar] [CrossRef] [Green Version]
- A Lawal, R.; Zhou, X.; Batey, K.; Hoffman, C.M.; A Georger, M.; Radtke, F.; Hilton, M.J.; Xing, L.; Frisch, B.J.; Calvi, L.M.; et al. The Notch Ligand Jagged1 Regulates the Osteoblastic Lineage by Maintaining the Osteoprogenitor Pool. J. Bone Miner. Res. 2017, 32, 1320–1331. [Google Scholar] [CrossRef]
- E Boyce, B.; Li, P.; Yao, Z.; Zhang, Q.; Badell, I.R.; Schwarz, E.M.; O’Keefe, R.J.; Xing, L. TNFα and pathologic bone resorption. Keio J. Med. 2005, 54, 127–131. [Google Scholar] [CrossRef] [Green Version]
- Hyrich, K.L.; Watson, K.D.; Silman, A.J.; Symmons, D.P.; British Society for Rheumatology Biologics, R. Predictors of response to anti-TNF- therapy among patients with rheumatoid arthritis: Results from the British Society for Rheumatology Biologics Register. Rheumatology 2006, 45, 1558–1565. [Google Scholar] [CrossRef] [Green Version]
- Symmons, D.P.; Silman, A.J. The world of biologics. Lupus 2006, 15, 122–126. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Class | Factors | Functions |
|---|---|---|
| Negative regulators | TRAF3 | Inhibits OC formation directly by preventing p100 processing [91,92,93], and indirectly by limiting RANKL production by MPCs [94]. |
| NF-κB2 p100 | Inhibits OC formation [91] *. | |
| RBP-j | Suppresses NFATc1 to inhibit OC formation [95]. | |
| IRF8 | Inhibits OC formation [96,97]. | |
| Positive regulators | IL-1β | Promotes TNF-pre-activated OCPs expressing c-Fos to form OCs independent of NF-κB p50 and p52 [88]. |
| TGFβ1 | Enhances TNF-induced OC formation independent of RANKL, RANK, and TRAF6 [87,93]. | |
| RANKL | Degrades TRAF3 to enhance TNF-induced OC formation [93]. | |
| NF-κB RelB | Mediates TNF-polarized inflammatory Mφ to enhance OC formation, but inhibits NFATc1 expression to limit terminal OC differentiation [98]. | |
| Untested factors known to regulate RANKL-induced OC formation ** | Notch ligands | Inhibits OC formation directly by suppressing cFms expression by OCPs [99], and indirectly by reducing RANKL production [100]. |
| Notch intracellular domain | Limits OPG production by MPCs [101]. | |
| IFN-γ | Polarizes M-CSF-induced resident to inflammatory Mφ [102], and strongly inhibits OC formation [103]. | |
| GM-CSF | Polarizes inflammatory Mφ [102], induces DCs, and inhibits OC formation [64]. | |
| LPS | Polarizes inflammatory Mφ [102], and inhibits OC formation [104], while promoting OC formation from pre-activated OCPs [105]. | |
| IL-12 | Inflammatory Mφ cytokine, inhibiting OC formation [106,107]. | |
| IL-18 | Inflammatory Mφ cytokine, inhibiting OC formation [106,107]. | |
| STAT1 | Transcription factor that polarizes inflammatory Mφ to mediate IFN-γ, IFN-β, and LPS inhibition of RANKL-induced OC formation [103,108]. | |
| IL-4 | Cytokine that polarizes resident Mφ through STAT6 to inhibit OC formation and activity [109,110]. | |
| IL-13 | Works similarly with IL-4 to inhibit OC formation [109,110]. | |
| IL-10 | Resident Mφ cytokine that inhibits OC formation by suppressing RANKL-induced NFATc1, c-Fos, and c-Jun expression [111,112]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, Z.; Getting, S.J.; Locke, I.C. Regulation of TNF-Induced Osteoclast Differentiation. Cells 2022, 11, 132. https://doi.org/10.3390/cells11010132
Yao Z, Getting SJ, Locke IC. Regulation of TNF-Induced Osteoclast Differentiation. Cells. 2022; 11(1):132. https://doi.org/10.3390/cells11010132
Chicago/Turabian StyleYao, Zhenqiang, Stephen J. Getting, and Ian C. Locke. 2022. "Regulation of TNF-Induced Osteoclast Differentiation" Cells 11, no. 1: 132. https://doi.org/10.3390/cells11010132
APA StyleYao, Z., Getting, S. J., & Locke, I. C. (2022). Regulation of TNF-Induced Osteoclast Differentiation. Cells, 11(1), 132. https://doi.org/10.3390/cells11010132