
Substrate- and Calcium-Dependent Differential Regulation of Mitochondrial Oxidative Phosphorylation and Energy Production in the Heart and Kidney

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Mitochondrial Isolation
2.1.1. Heart Mitochondria
2.1.2. Kidney Outer Medulla (OM) Mitochondria
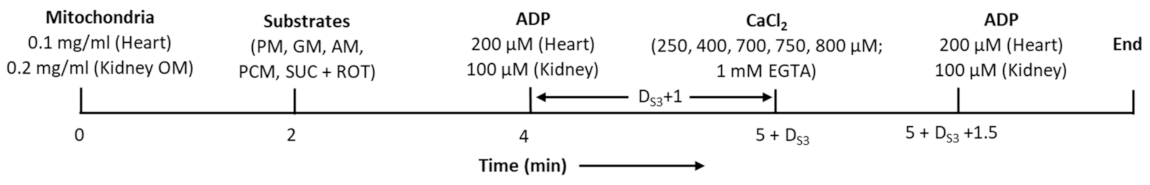
2.2. Mitochondrial O2 Consumption Measurement
2.3. Control of Extra-Mitochondrial free Ca2+ Concentrations
2.4. Data Analysis and Statistics
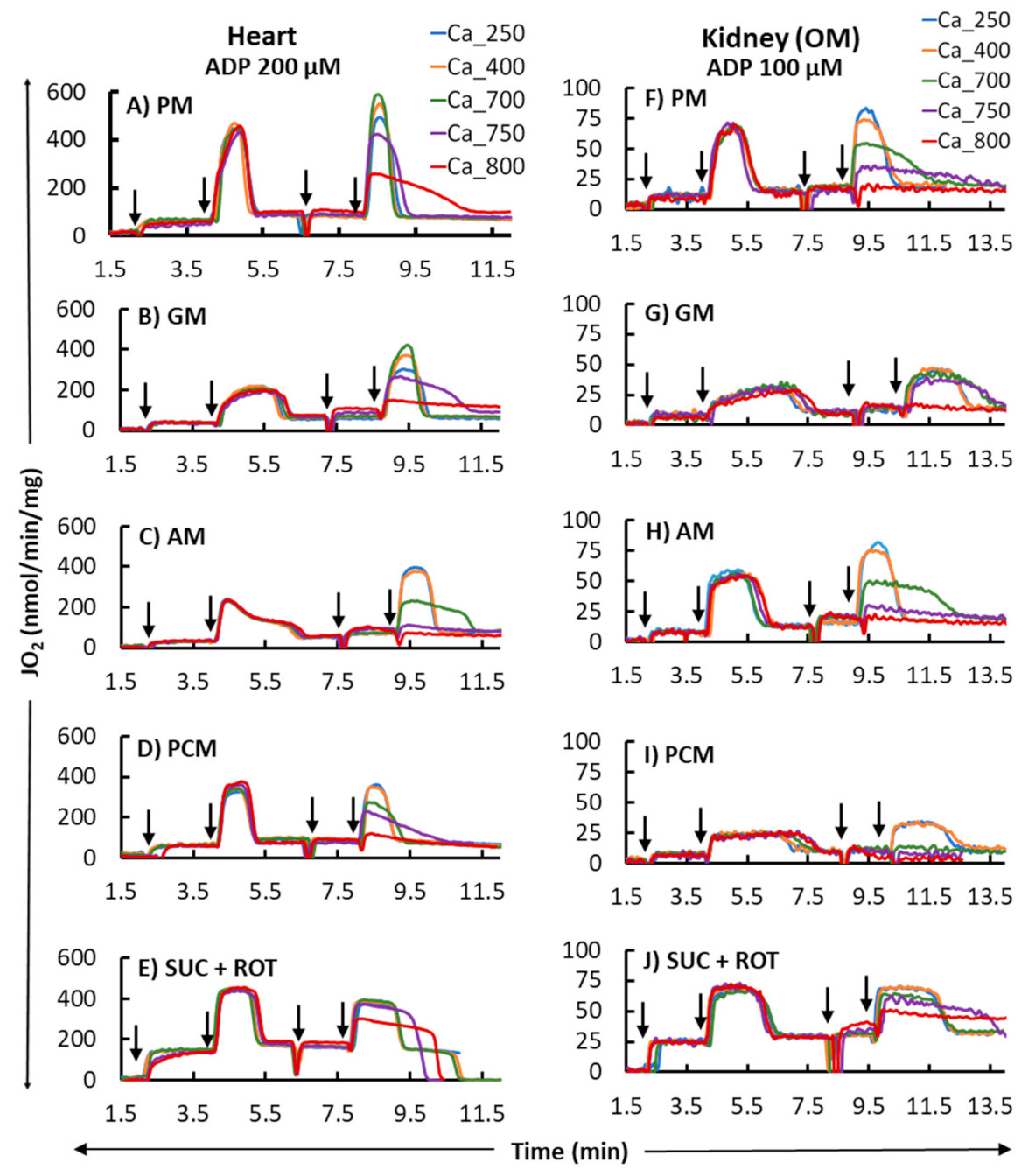
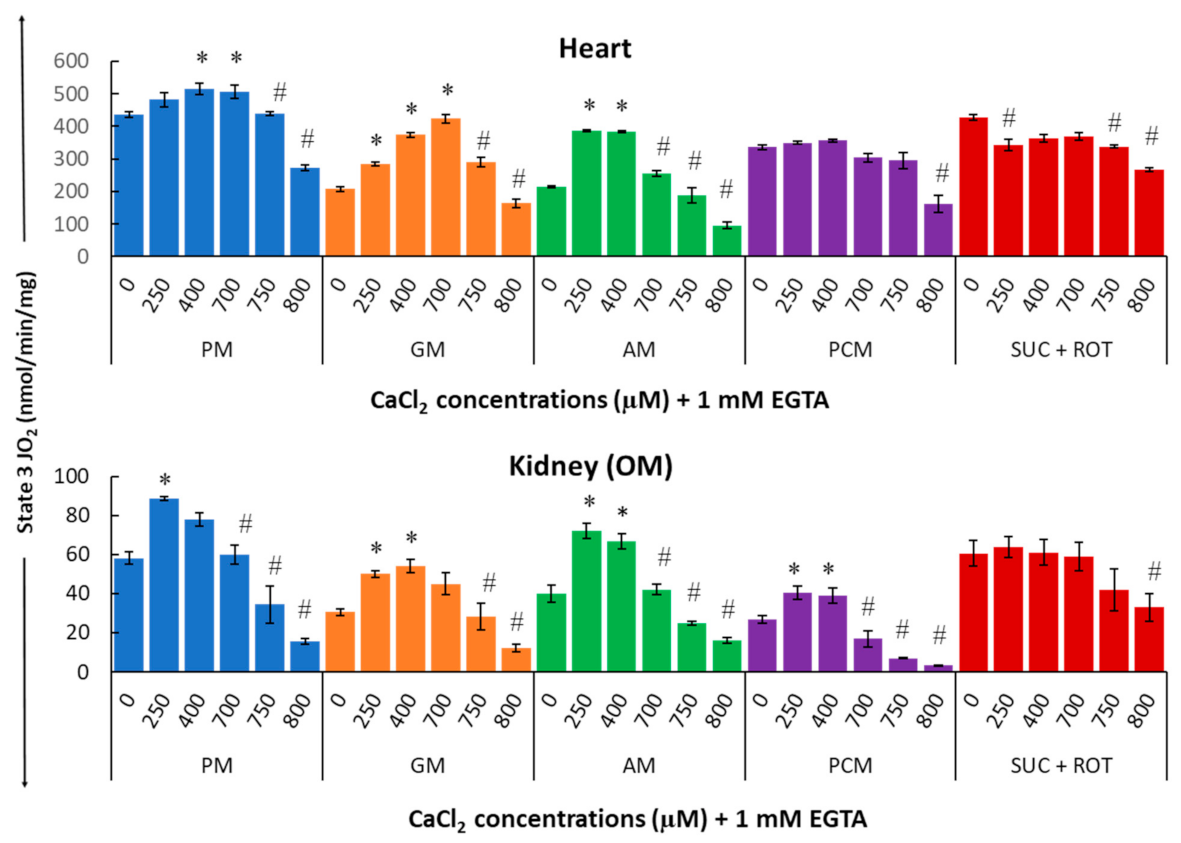
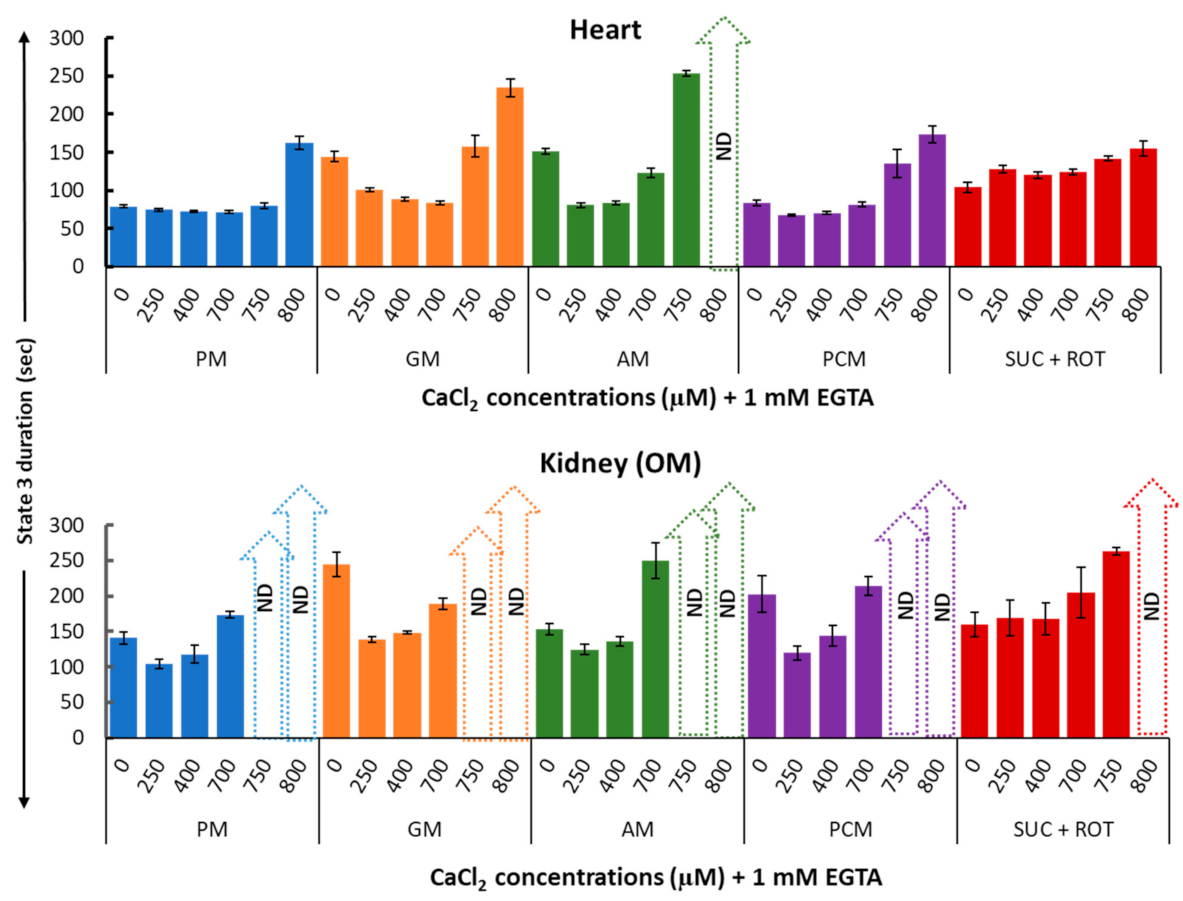
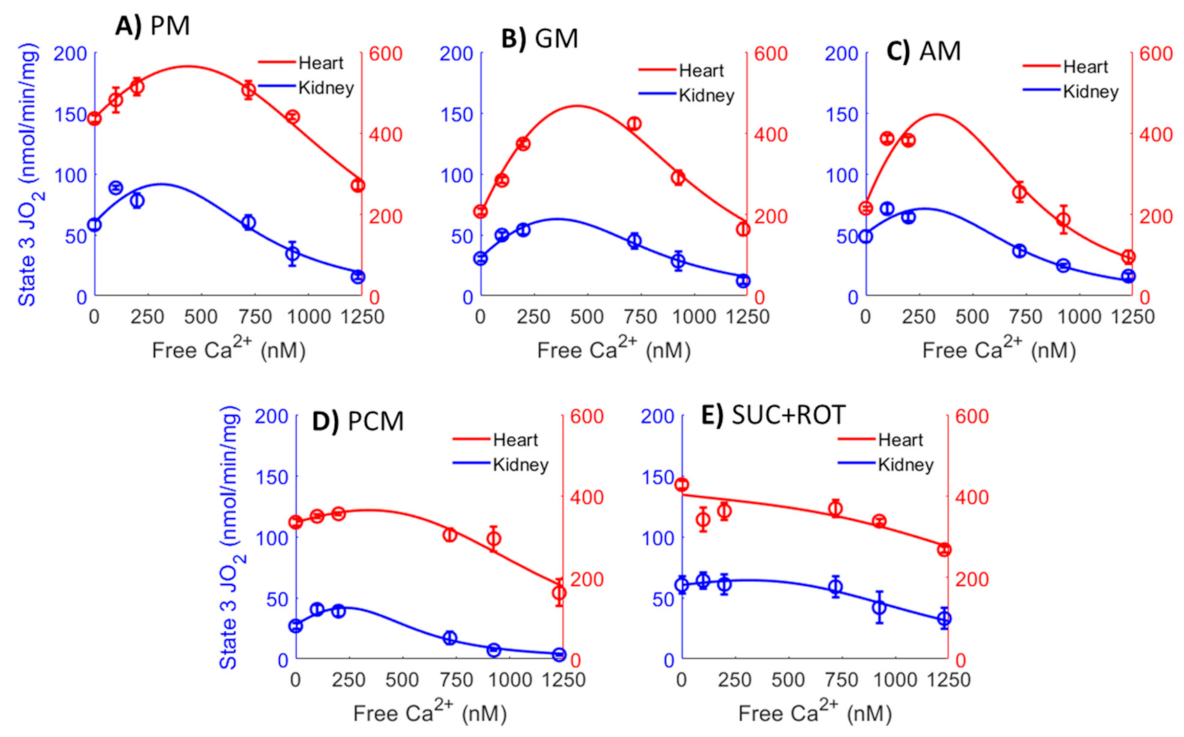
3. Results
4. Discussion
4.1. Intermediate Metabolism and Energy Production: Unique Challenges Due to Different Energy Demands of the Heart and Kidneys
4.2. Substrate-Dependent Mitochondrial Respiratory Rates for the Heart and Kidney OM with Addition of a Fixed ADP Concentration
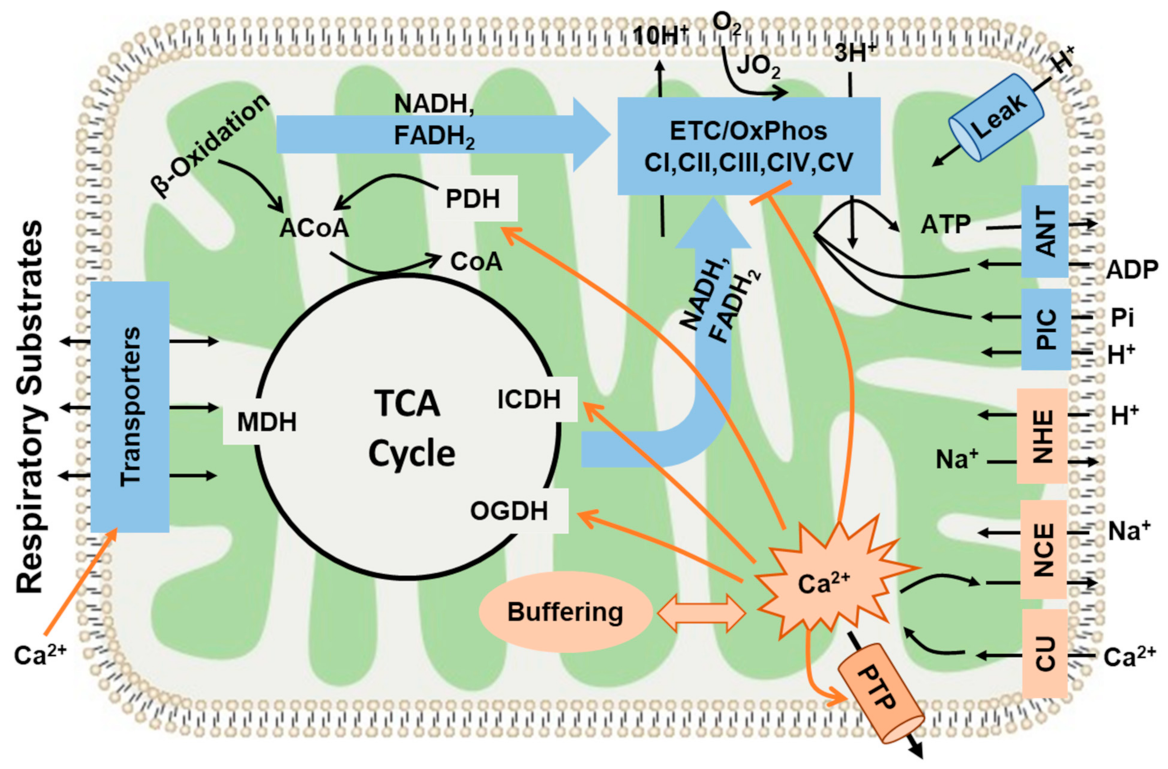
4.3. Current Understanding of Mitochondrial Ca2+ Regulation and Regulation of Mitochondrial Substrate Transport and Energy Metabolism by Ca2+
4.4. Complex Effects of Respiratory Substrates upon Ca2+ Activation of Mitochondrial Respiration and ATP Production in the Heart and Kidney OM
4.5. Mechanisms That May Explain the Seemingly Paradoxical Inhibitory Effects of Higher Ca2+ Concentrations on Mitochondrial State 3 Respiration
4.6. Simple Kinetic Model Comparing the Activation and Inhibition Effects of Free Ca2+ on Mitochondria State 3 Respiration
5. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviation
| AKGDH | Alpha-ketoglutarate dehydrogenase |
| AM | Alpha-ketoglutarate + malate |
| ANT | Adenine nucleotide translocase |
| BSA | Bovine serum albumin |
| ETC | Electron transport chain |
| GM | Glutamate + malate |
| IB | Isolation buffer |
| IMM | Inner mitochondrial membrane |
| ICDH | Isocitrate dehydrogenase |
| JO2 | Oxygen consumption flux |
| MAS | Malate-Aspartate shuttle |
| MCU | Mitochondrial Ca2+ uniporter |
| MDH | Malate dehydrogenase |
| mPTP | Mitochondrial permeability transition pore |
| mTAL | Medullary thick ascending limbs of loop of Henle |
| NCE | Na+/Ca2+ Exchanger |
| NHE | Na+/H+ Exchanger |
| O2k | Oxygraph-2k |
| OCR | Oxygen consumption rate |
| OGDH | 2-oxo glutarate dehydrogenase |
| OM | Outer medulla |
| OxPhos | Oxidative Phosphorylation |
| PCM | Palmitoyl-carnitine + malate |
| PIC | Inorganic phosphate carrier |
| PDH | Pyruvate dehydrogenase |
| PM | Pyruvate + malate |
| RB | Respiration buffer |
| RCR | Respiratory control ratio |
| ROT | Rotenone |
| SD | Sprague-Dawley |
| SUC | Succinate |
| TCA | Tricarboxylic acid cycle |
References
- Tian, Z.; Liang, M. Renal metabolism and hypertension. Nat. Commun. 2021, 12, 963. [Google Scholar] [CrossRef]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef] [PubMed]
- Bertero, E.; Maack, C. Metabolic remodelling in heart failure. Nat. Rev. Cardiol. 2018, 15, 457–470. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G.; Ferguson, S.J. Bioenergetics, 4th ed.; Academic Press: London, UK, 2013. [Google Scholar]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Reviews. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Duchen, M.R. Mitochondria and calcium: From cell signalling to cell death. J. Physiol. 2000, 529 Pt 1, 57–68. [Google Scholar] [CrossRef]
- Bernardi, P. Mitochondrial transport of cations: Channels, exchangers, and permeability transition. Physiol. Rev. 1999, 79, 1127–1155. [Google Scholar] [CrossRef] [PubMed]
- Gunter, T.E.; Gunter, K.K.; Sheu, S.S.; Gavin, C.E. Mitochondrial calcium transport: Physiological and pathological relevance. Am. J. Physiol. 1994, 267, C313–C339. [Google Scholar] [CrossRef]
- Vinnakota, K.C.; Bazil, J.N.; Van den Bergh, F.; Wiseman, R.W.; Beard, D.A. Feedback Regulation and Time Hierarchy of Oxidative Phosphorylation in Cardiac Mitochondria. Biophys. J. 2016, 110, 972–980. [Google Scholar] [CrossRef][Green Version]
- Wu, F.; Zhang, E.Y.; Zhang, J.; Bache, R.J.; Beard, D.A. Phosphate metabolite concentrations and ATP hydrolysis potential in normal and ischaemic hearts. J. Physiol. 2008, 586, 4193–4208. [Google Scholar] [CrossRef]
- Wu, F.; Jeneson, J.A.; Beard, D.A. Oxidative ATP synthesis in skeletal muscle is controlled by substrate feedback. Am. J. Physiol. Cell Physiol. 2007, 292, C115–C124. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.; French, S.; Evans, F.J.; Joubert, F.; Balaban, R.S. Metabolic network control of oxidative phosphorylation: Multiple roles of inorganic phosphate. J. Biol. Chem. 2003, 278, 39155–39165. [Google Scholar] [CrossRef]
- Scholz, T.D.; Laughlin, M.R.; Balaban, R.S.; Kupriyanov, V.V.; Heineman, F.W. Effect of substrate on mitochondrial NADH, cytosolic redox state, and phosphorylated compounds in isolated hearts. Am. J. Physiol. 1995, 268, H82–H91. [Google Scholar] [CrossRef]
- LaNoue, K.F.; Bryla, J.; Williamson, J.R. Feedback interactions in the control of citric acid cycle activity in rat heart mitochondria. J. Biol. Chem. 1972, 247, 667–679. [Google Scholar] [CrossRef]
- LaNoue, K.; Nicklas, W.J.; Williamson, J.R. Control of citric acid cycle activity in rat heart mitochondria. J. Biol. Chem. 1970, 245, 102–111. [Google Scholar] [CrossRef]
- O’Donnell, J.M.; Doumen, C.; LaNoue, K.F.; White, L.T.; Yu, X.; Alpert, N.M.; Lewandowski, E.D. Dehydrogenase regulation of metabolite oxidation and efflux from mitochondria in intact hearts. Am. J. Physiol. 1998, 274, H467–H476. [Google Scholar] [CrossRef]
- Romani, A.M.P. Physiology and Pathology of Mitochondrial Dehydrogenases. In Secondary Metabolites—Sources and Applications; Ramasamy Vijayakumar, S.S.S.R., Ed.; IntechOpen: London, UK, 2018. [Google Scholar]
- Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 2009, 1787, 1309–1316. [Google Scholar] [CrossRef] [PubMed]
- McCormack, J.G.; Halestrap, A.P.; Denton, R.M. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol. Rev. 1990, 70, 391–425. [Google Scholar] [CrossRef]
- McCormack, J.G.; Denton, R.M. The role of Ca2+ in the regulation of intramitochondrial energy production in heart. Biomed. Biochim. Acta 1987, 46, S487–S492. [Google Scholar]
- Denton, R.M.; McCormack, J.G. The calcium sensitive dehydrogenases of vertebrate mitochondria. Cell Calcium 1986, 7, 377–386. [Google Scholar] [CrossRef]
- Denton, R.M.; McCormack, J.G.; Edgell, N.J. Role of calcium ions in the regulation of intramitochondrial metabolism. Effects of Na+, Mg2+ and ruthenium red on the Ca2+-stimulated oxidation of oxoglutarate and on pyruvate dehydrogenase activity in intact rat heart mitochondria. Biochem. J. 1980, 190, 107–117. [Google Scholar] [CrossRef] [PubMed]
- McCormack, J.G.; Denton, R.M. The effects of calcium ions and adenine nucleotides on the activity of pig heart 2-oxoglutarate dehydrogenase complex. Biochem. J. 1979, 180, 533–544. [Google Scholar] [CrossRef]
- Denton, R.M.; Richards, D.A.; Chin, J.G. Calcium ions and the regulation of NAD+-linked isocitrate dehydrogenase from the mitochondria of rat heart and other tissues. Biochem. J. 1978, 176, 899–906. [Google Scholar] [CrossRef]
- Williams, G.S.; Boyman, L.; Lederer, W.J. Mitochondrial calcium and the regulation of metabolism in the heart. J. Mol. Cell Cardiol. 2015, 78, 35–45. [Google Scholar] [CrossRef]
- Rossi, A.; Pizzo, P.; Filadi, R. Calcium, mitochondria and cell metabolism: A functional triangle in bioenergetics. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1068–1078. [Google Scholar] [CrossRef]
- Finkel, T.; Menazza, S.; Holmstrom, K.M.; Parks, R.J.; Liu, J.; Sun, J.; Liu, J.; Pan, X.; Murphy, E. The ins and outs of mitochondrial calcium. Circ. Res. 2015, 116, 1810–1819. [Google Scholar] [CrossRef] [PubMed]
- McCormack, J.G.; Browne, H.M.; Dawes, N.J. Studies on mitochondrial Ca2+-transport and matrix Ca2+ using fura-2-loaded rat heart mitochondria. Biochim. Biophys. Acta 1989, 973, 420–427. [Google Scholar] [CrossRef]
- Boelens, A.D.; Pradhan, R.K.; Blomeyer, C.A.; Camara, A.K.; Dash, R.K.; Stowe, D.F. Extra-matrix Mg2+ limits Ca2+ uptake and modulates Ca2+ uptake-independent respiration and redox state in cardiac isolated mitochondria. J. Bioenerg. Biomembr. 2013, 45, 203–218. [Google Scholar] [CrossRef][Green Version]
- Wan, B.; LaNoue, K.F.; Cheung, J.Y.; Scaduto, R.C., Jr. Regulation of citric acid cycle by calcium. J. Biol. Chem. 1989, 264, 13430–13439. [Google Scholar] [CrossRef]
- Vinnakota, K.C.; Singhal, A.; Van den Bergh, F.; Bagher-Oskouei, M.; Wiseman, R.W.; Beard, D.A. Open-Loop Control of Oxidative Phosphorylation in Skeletal and Cardiac Muscle Mitochondria by Ca2+. Biophys. J. 2016, 110, 954–961. [Google Scholar] [CrossRef]
- Glancy, B.; Balaban, R.S. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 2012, 51, 2959–2973. [Google Scholar] [CrossRef]
- Griffiths, E.J.; Rutter, G.A. Mitochondrial calcium as a key regulator of mitochondrial ATP production in mammalian cells. Biochim. Biophys. Acta 2009, 1787, 1324–1333. [Google Scholar] [CrossRef]
- Balaban, R.S. The role of Ca2+ signaling in the coordination of mitochondrial ATP production with cardiac work. Biochim. Biophys. Acta 2009, 1787, 1334–1341. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Bose, S.; French, S.A.; Territo, P.R. Role of calcium in metabolic signaling between cardiac sarcoplasmic reticulum and mitochondria in vitro. Am. J. Physiol. Cell Physiol. 2003, 284, C285–C293. [Google Scholar] [CrossRef]
- Blomeyer, C.A.; Bazil, J.N.; Stowe, D.F.; Dash, R.K.; Camara, A.K. Mg(2+) differentially regulates two modes of mitochondrial Ca(2+) uptake in isolated cardiac mitochondria: Implications for mitochondrial Ca(2+) sequestration. J. Bioenerg. Biomembr. 2016, 48, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Blomeyer, C.A.; Bazil, J.N.; Stowe, D.F.; Pradhan, R.K.; Dash, R.K.; Camara, A.K. Dynamic buffering of mitochondrial Ca2+ during Ca2+ uptake and Na+-induced Ca2+ release. J. Bioenerg. Biomembr. 2013, 45, 189–202. [Google Scholar] [CrossRef][Green Version]
- Bazil, J.N.; Blomeyer, C.A.; Pradhan, R.K.; Camara, A.K.; Dash, R.K. Modeling the calcium sequestration system in isolated guinea pig cardiac mitochondria. J. Bioenerg. Biomembr. 2013, 45, 177–188. [Google Scholar] [CrossRef]
- Tewari, S.G.; Camara, A.K.; Stowe, D.F.; Dash, R.K. Computational analysis of Ca2+ dynamics in isolated cardiac mitochondria predicts two distinct modes of Ca2+ uptake. J. Physiol. 2014, 592, 1917–1930. [Google Scholar] [CrossRef]
- Dash, R.K.; Beard, D.A. Analysis of cardiac mitochondrial Na+-Ca2+ exchanger kinetics with a biophysical model of mitochondrial Ca2+ handling suggests a 3:1 stoichiometry. J. Physiol. 2008, 586, 3267–3285. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.S.; Boyman, L.; Chikando, A.C.; Khairallah, R.J.; Lederer, W.J. Mitochondrial calcium uptake. Proc. Natl. Acad. Sci. USA 2013, 110, 10479–10486. [Google Scholar] [CrossRef]
- Glancy, B.; Willis, W.T.; Chess, D.J.; Balaban, R.S. Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry 2013, 52, 2793–2809. [Google Scholar] [CrossRef]
- Eisner, D.A.; Caldwell, J.L.; Kistamás, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 2008, 70, 23–49. [Google Scholar] [CrossRef]
- Singh, P.; Thomson, S.C. Metabolic Basis of Solute Transport. In Brenner and Rector’s The Kidney, 11st ed.; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Forbes, J.M. Mitochondria;Power Players in Kidney Function? Trends Endocrinol. Metab. 2016, 27, 441–442. [Google Scholar] [CrossRef]
- Elia, M. Organ and Tissue Contribution to Metabolic Rate; Raven Press: New York, NY, USA, 1992. [Google Scholar]
- Pohjoismaki, J.L.; Goffart, S. The role of mitochondria in cardiac development and protection. Free Radic. Biol. Med. 2017, 106, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Saks, V.; Dzeja, P.; Schlattner, U.; Vendelin, M.; Terzic, A.; Wallimann, T. Cardiac system bioenergetics: Metabolic basis of the Frank-Starling law. J. Physiol. 2006, 571, 253–273. [Google Scholar] [CrossRef]
- Hochachka, P.W.; McClelland, G.B. Cellular metabolic homeostasis during large-scale change in ATP turnover rates in muscles. J. Exp. Biol. 1997, 200, 381–386. [Google Scholar] [CrossRef]
- Soltoff, S.P. ATP and the regulation of renal cell function. Annu. Rev. Physiol. 1986, 48, 9–31. [Google Scholar] [CrossRef]
- Mandel, L.J. Metabolic substrates, cellular energy production, and the regulation of proximal tubular transport. Annu. Rev. Physiol. 1985, 47, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Kiil, F. Renal energy metabolism and regulation of sodium reabsorption. Kidney Int. 1977, 11, 153–160. [Google Scholar] [CrossRef]
- Bankir, L.; Figueres, L.; Prot-Bertoye, C.; Bouby, N.; Crambert, G.; Pratt, J.H.; Houillier, P. Medullary and cortical thick ascending limb: Similarities and differences. Am. J. Physiol. Renal Physiol. 2020, 318, F422–F442. [Google Scholar] [CrossRef] [PubMed]
- Evans, L.C.; Cowley, A.W., Jr. Renal Medullary Circulation, 1st ed.; Morgan & Claypool Publishers: San Rafael, CA, USA, 1990; p. 104. [Google Scholar]
- Cheng, Y.; Song, H.; Pan, X.; Xue, H.; Wan, Y.; Wang, T.; Tian, Z.; Hou, E.; Lanza, I.R.; Liu, P.; et al. Urinary Metabolites Associated with Blood Pressure on a Low- or High-Sodium Diet. Theranostics 2018, 8, 1468–1480. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.L.; Adaniya, S.M.; Cypress, M.W.; Suzuki, Y.; Kusakari, Y.; Jhun, B.S.; O-Uchi, J. Role of mitochondrial Ca(2+) homeostasis in cardiac muscles. Arch. Biochem. Biophys. 2019, 663, 276–287. [Google Scholar] [CrossRef]
- Lopaschuk, G.D. Targets for modulation of fatty acid oxidation in the heart. Curr. Opin. Investig. Drugs 2004, 5, 290–294. [Google Scholar] [PubMed]
- Fink, B.D.; Bai, F.; Yu, L.; Sivitz, W.I. Regulation of ATP production: Dependence on calcium concentration and respiratory state. Am. J. Physiol. Cell Physiol. 2017, 313, C146–C153. [Google Scholar] [CrossRef]
- Tarasov, A.I.; Griffiths, E.J.; Rutter, G.A. Regulation of ATP production by mitochondrial Ca(2+). Cell Calcium 2012, 52, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Wollenman, L.C.; Vander Ploeg, M.R.; Miller, M.L.; Zhang, Y.; Bazil, J.N. The effect of respiration buffer composition on mitochondrial metabolism and function. PLoS ONE 2017, 12, e0187523. [Google Scholar] [CrossRef]
- Vinnakota, K.C.; Dash, R.K.; Beard, D.A. Stimulatory effects of calcium on respiration and NAD(P)H synthesis in intact rat heart mitochondria utilizing physiological substrates cannot explain respiratory control in vivo. J. Biol. Chem. 2011, 286, 30816–30822. [Google Scholar] [CrossRef]
- Panov, A.V.; Scaduto, R.C., Jr. Substrate specific effects of calcium on metabolism of rat heart mitochondria. Am. J. Physiol. 1996, 270, H1398–H1406. [Google Scholar] [CrossRef]
- Tomar, N.; Zhang, X.; Kandel, S.M.; Sadri, S.; Yang, C.; Liang, M.; Audi, S.H.; Cowley, A.W., Jr.; Dash, R.K. Substrate-dependent differential regulation of mitochondrial bioenergetics in the heart and kidney cortex and outer medulla. Biochim. Biophys. Acta Bioenerg. 2021, 1863, 148518. [Google Scholar] [CrossRef]
- Agarwal, B.; Camara, A.K.; Stowe, D.F.; Bosnjak, Z.J.; Dash, R.K. Enhanced charge-independent mitochondrial free Ca(2+) and attenuated ADP-induced NADH oxidation by isoflurane: Implications for cardioprotection. Biochim. Biophys. Acta 2012, 1817, 453–465. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Agarwal, B.; Dash, R.K.; Stowe, D.F.; Bosnjak, Z.J.; Camara, A.K. Isoflurane modulates cardiac mitochondrial bioenergetics by selectively attenuating respiratory complexes. Biochim. Biophys. Acta 2014, 1837, 354–365. [Google Scholar] [CrossRef]
- Schoenmakers, T.J.; Visser, G.J.; Flik, G.; Theuvenet, A.P. CHELATOR: An improved method for computing metal ion concentrations in physiological solutions. Biotechniques 1992, 12, 870–874, 876–879. [Google Scholar]
- McKnight, S.L. On getting there from here. Science 2010, 330, 1338–1339. [Google Scholar] [CrossRef]
- Langman, C.B.; Cannata-Andía, J.B. Calcium in chronic kidney disease: Myths and realities. Introduction. Clin. J. Am. Soc. Nephrol. 2010, 5 (Suppl. 1), S1–S2. [Google Scholar] [CrossRef]
- Chiodini, I.; Bolland, M.J. Calcium supplementation in osteoporosis: Useful or harmful? Eur. J. Endocrinol. 2018, 178, D13–D25. [Google Scholar] [CrossRef]
- Forner, F.; Foster, L.J.; Campanaro, S.; Valle, G.; Mann, M. Quantitative proteomic comparison of rat mitochondria from muscle, heart, and liver. Mol. Cell Proteom. 2006, 5, 608–619. [Google Scholar] [CrossRef]
- Rossignol, R.; Letellier, T.; Malgat, M.; Rocher, C.; Mazat, J.P. Tissue variation in the control of oxidative phosphorylation: Implication for mitochondrial diseases. Biochem. J. 2000, 347 Pt 1, 45–53. [Google Scholar] [CrossRef]
- Shirley, M.K.; Arthurs, O.J.; Seunarine, K.K.; Cole, T.J.; Eaton, S.; Williams, J.E.; Clark, C.A.; Wells, J.C.K. Metabolic rate of major organs and tissues in young adult South Asian women. Eur. J. Clin. Nutr. 2019, 73, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Goffart, S.; von Kleist-Retzow, J.-C.; Wiesner, R.J. Regulation of mitochondrial proliferation in the heart: Power-plant failure contributes to cardiac failure in hypertrophy. Cardiovasc. Res. 2004, 64, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Boyman, L.; Chikando, A.C.; Williams, G.S.; Khairallah, R.J.; Kettlewell, S.; Ward, C.W.; Smith, G.L.; Kao, J.P.; Lederer, W.J. Calcium movement in cardiac mitochondria. Biophys. J. 2014, 107, 1289–1301. [Google Scholar] [CrossRef] [PubMed]
- Rossitto, G.; Maiolino, G.; Lerco, S.; Ceolotto, G.; Blackburn, G.; Mary, S.; Antonelli, G.; Berton, C.; Bisogni, V.; Cesari, M.; et al. High sodium intake, glomerular hyperfiltration, and protein catabolism in patients with essential hypertension. Cardiovasc. Res. 2021, 117, 1372–1381. [Google Scholar] [CrossRef]
- Stillman, I.E.; Brezis, M.; Heyman, S.N.; Epstein, F.H.; Spokes, K.; Rosen, S. Effects of salt depletion on the kidney: Changes in medullary oxygenation and thick ascending limb size. J. Am. Soc. Nephrol. 1994, 4, 1538–1545. [Google Scholar] [CrossRef] [PubMed]
- Friederich-Persson, M.; Thorn, E.; Hansell, P.; Nangaku, M.; Levin, M.; Palm, F. Kidney hypoxia, attributable to increased oxygen consumption, induces nephropathy independently of hyperglycemia and oxidative stress. Hypertension 2013, 62, 914–919. [Google Scholar] [CrossRef] [PubMed]
- Kunz, W.S. Different metabolic properties of mitochondrial oxidative phosphorylation in different cell types--important implications for mitochondrial cytopathies. Exp. Physiol. 2003, 88, 149–154. [Google Scholar] [CrossRef]
- Cortassa, S.; Aon, M.A.; Sollott, S.J. Control and Regulation of Substrate Selection in Cytoplasmic and Mitochondrial Catabolic Networks. A Systems Biology Analysis. Front. Physiol. 2019, 10, 201. [Google Scholar] [CrossRef]
- Hinkle, P.C.; Yu, M.L. The phosphorus/oxygen ratio of mitochondrial oxidative phosphorylation. J. Biol. Chem. 1979, 254, 2450–2455. [Google Scholar] [CrossRef]
- Hinkle, P.C. P/O ratios of mitochondrial oxidative phosphorylation. Biochim. Biophys. Acta 2005, 1706, 1–11. [Google Scholar] [CrossRef]
- Hinkle, P.C.; Kumar, M.A.; Resetar, A.; Harris, D.L. Mechanistic stoichiometry of mitochondrial oxidative phosphorylation. Biochemistry 1991, 30, 3576–3582. [Google Scholar] [CrossRef]
- Santo-Domingo, J.; Demaurex, N. Calcium uptake mechanisms of mitochondria. Biochim. Biophys. Acta 2010, 1797, 907–912. [Google Scholar] [CrossRef]
- Dash, R.K.; Qi, F.; Beard, D.A. A biophysically based mathematical model for the kinetics of mitochondrial calcium uniporter. Biophys. J. 2009, 96, 1318–1332. [Google Scholar] [CrossRef]
- Lehninger, A.L.; Reynafarje, B.; Vercesi, A.; Tew, W.P. Transport and accumulation of calcium in mitochondria. Ann. N. Y. Acad. Sci. 1978, 307, 160–176. [Google Scholar] [CrossRef]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, R.K.; Qi, F.; Beard, D.A.; Dash, R.K. Characterization of Mg2+ inhibition of mitochondrial Ca2+ uptake by a mechanistic model of mitochondrial Ca2+ uniporter. Biophys. J. 2011, 101, 2071–2081. [Google Scholar] [CrossRef] [PubMed]
- Crompton, M.; Kunzi, M.; Carafoli, E. The calcium-induced and sodium-induced effluxes of calcium from heart mitochondria. Evidence for a sodium-calcium carrier. Eur. J. Biochem. 1977, 79, 549–558. [Google Scholar] [CrossRef]
- Gunter, T.E.; Sheu, S.S. Characteristics and possible functions of mitochondrial Ca(2+) transport mechanisms. Biochim. Biophys. Acta 2009, 1787, 1291–1308. [Google Scholar] [CrossRef]
- Puskin, J.S.; Gunter, T.E.; Gunter, K.K.; Russell, P.R. Evidence for more than one Ca2+ transport mechanism in mitochondria. Biochemistry 1976, 15, 3834–3842. [Google Scholar] [CrossRef]
- Fiskum, G.; Lehninger, A.L. Regulated release of Ca2+ from respiring mitochondria by Ca2+/2H+ antiport. J. Biol. Chem. 1979, 254, 6236–6239. [Google Scholar] [CrossRef]
- Chalmers, S.; Nicholls, D.G. The relationship between free and total calcium concentrations in the matrix of liver and brain mitochondria. J. Biol. Chem. 2003, 278, 19062–19070. [Google Scholar] [CrossRef]
- Korge, P.; Yang, L.; Yang, J.H.; Wang, Y.; Qu, Z.; Weiss, J.N. Protective role of transient pore openings in calcium handling by cardiac mitochondria. J. Biol. Chem. 2011, 286, 34851–34857. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G. The regulation of extramitochondrial free calcium ion concentration by rat liver mitochondria. Biochem. J. 1978, 176, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.K.; Coll, K.E.; Cooper, R.H.; Marks, J.S.; Williamson, J.R. Mechanisms underlying calcium homeostasis in isolated hepatocytes. J. Biol. Chem. 1983, 258, 731–741. [Google Scholar] [CrossRef]
- Gherardi, G.; Monticelli, H.; Rizzuto, R.; Mammucari, C. The Mitochondrial Ca(2+) Uptake and the Fine-Tuning of Aerobic Metabolism. Front. Physiol. 2020, 11, 554904. [Google Scholar] [CrossRef]
- Szibor, M.; Gizatullina, Z.; Gainutdinov, T.; Endres, T.; Debska-Vielhaber, G.; Kunz, M.; Karavasili, N.; Hallmann, K.; Schreiber, F.; Bamberger, A.; et al. Cytosolic, but not matrix, calcium is essential for adjustment of mitochondrial pyruvate supply. J. Biol. Chem. 2020, 295, 4383–4397. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Liu, J.; Nguyen, T.; Liu, C.; Sun, J.; Teng, Y.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 2013, 15, 1464–1472. [Google Scholar] [CrossRef]
- Holmstrom, K.M.; Pan, X.; Liu, J.C.; Menazza, S.; Liu, J.; Nguyen, T.T.; Pan, H.; Parks, R.J.; Anderson, S.; Noguchi, A.; et al. Assessment of cardiac function in mice lacking the mitochondrial calcium uniporter. J. Mol. Cell Cardiol. 2015, 85, 178–182. [Google Scholar] [CrossRef]
- Rasmussen, T.P.; Wu, Y.; Joiner, M.L.; Koval, O.M.; Wilson, N.R.; Luczak, E.D.; Wang, Q.; Chen, B.; Gao, Z.; Zhu, Z.; et al. Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart. Proc. Natl. Acad. Sci. USA 2015, 112, 9129–9134. [Google Scholar] [CrossRef]
- Hamilton, J.; Brustovetsky, T.; Rysted, J.E.; Lin, Z.; Usachev, Y.M.; Brustovetsky, N. Deletion of mitochondrial calcium uniporter incompletely inhibits calcium uptake and induction of the permeability transition pore in brain mitochondria. J. Biol. Chem. 2018, 293, 15652–15663. [Google Scholar] [CrossRef]
- Satrustegui, J.; Pardo, B.; Del Arco, A. Mitochondrial transporters as novel targets for intracellular calcium signaling. Physiol. Rev. 2007, 87, 29–67. [Google Scholar] [CrossRef]
- Luongo, T.S.; Lambert, J.P.; Yuan, A.; Zhang, X.; Gross, P.; Song, J.; Shanmughapriya, S.; Gao, E.; Jain, M.; Houser, S.R.; et al. The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell Rep. 2015, 12, 23–34. [Google Scholar] [CrossRef]
- Palmieri, L.; Pardo, B.; Lasorsa, F.M.; del Arco, A.; Kobayashi, K.; Iijima, M.; Runswick, M.J.; Walker, J.E.; Saheki, T.; Satrustegui, J.; et al. Citrin and aralar1 are Ca(2+)-stimulated aspartate/glutamate transporters in mitochondria. EMBO J. 2001, 20, 5060–5069. [Google Scholar] [CrossRef] [PubMed]
- Satrustegui, J.; Contreras, L.; Ramos, M.; Marmol, P.; del Arco, A.; Saheki, T.; Pardo, B. Role of aralar, the mitochondrial transporter of aspartate-glutamate, in brain N-acetylaspartate formation and Ca(2+) signaling in neuronal mitochondria. J. Neurosci. Res. 2007, 85, 3359–3366. [Google Scholar] [CrossRef]
- Llorente-Folch, I.; Rueda, C.B.; Amigo, I.; del Arco, A.; Saheki, T.; Pardo, B.; Satrustegui, J. Calcium-regulation of mitochondrial respiration maintains ATP homeostasis and requires ARALAR/AGC1-malate aspartate shuttle in intact cortical neurons. J. Neurosci. 2013, 33, 13957–13971. [Google Scholar] [CrossRef]
- Gellerich, F.N.; Gizatullina, Z.; Arandarcikaite, O.; Jerzembek, D.; Vielhaber, S.; Seppet, E.; Striggow, F. Extramitochondrial Ca2+ in the nanomolar range regulates glutamate-dependent oxidative phosphorylation on demand. PLoS ONE 2009, 4, e8181. [Google Scholar] [CrossRef]
- LaNoue, K.F.; Williamson, J.R. Interrelationships between malate-aspartate shuttle and citric acid cycle in rat heart mitochondria. Metabolism 1971, 20, 119–140. [Google Scholar] [CrossRef]
- Williamson, J.R.; Safer, B.; LaNoue, K.F.; Smith, C.M.; Walajtys, E. Mitochondrial-cytosolic interactions in cardiac tissue: Role of the malate-aspartate cycle in the removal of glycolytic NADH from the cytosol. Symp. Soc. Exp. Biol. 1973, 27, 241–281. [Google Scholar]
- Vasington, F.D.; Murphy, J.V. Ca ion uptake by rat kidney mitochondria and its dependence on respiration and phosphorylation. J. Biol. Chem. 1962, 237, 2670–2677. [Google Scholar] [CrossRef]
- Carafoli, E.; Lehninger, A.L. A survey of the interaction of calcium ions with mitochondria from different tissues and species. Biochem. J. 1971, 122, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Hansford, R.G.; Moreno-Sanchez, R.; Lewartowski, B. Activation of pyruvate dehydrogenase complex by Ca2+ in intact heart, cardiac myocytes, and cardiac mitochondria. Ann. N. Y. Acad. Sci. 1989, 573, 240–253. [Google Scholar] [CrossRef]
- Randle, P.J.; Denton, R.M.; Pask, H.T.; Severson, D.L. Calcium ions and the regulation of pyruvate dehydrogenase. Biochem. Soc. Symp. 1974, 75–88. [Google Scholar]
- Gellerich, F.N.; Gizatullina, Z.; Trumbeckaite, S.; Nguyen, H.P.; Pallas, T.; Arandarcikaite, O.; Vielhaber, S.; Seppet, E.; Striggow, F. The regulation of OXPHOS by extramitochondrial calcium. Biochim. Biophys. Acta 2010, 1797, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Cole, E.S.; Lepp, C.A.; Holohan, P.D.; Fondy, T.P. Isolation and characterization of flavin-linked glycerol-3-phosphate dehydrogenase from rabbit skeletal muscle mitochondria and comparison with the enzyme from rabbit brain. J. Biol. Chem. 1978, 253, 7952–7959. [Google Scholar] [CrossRef]
- Rutter, G.A.; Denton, R.M. Regulation of NAD+-linked isocitrate dehydrogenase and 2-oxoglutarate dehydrogenase by Ca2+ ions within toluene-permeabilized rat heart mitochondria. Interactions with regulation by adenine nucleotides and NADH/NAD+ ratios. Biochem. J. 1988, 252, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Rutter, G.A.; Denton, R.M. The binding of Ca2+ ions to pig heart NAD+-isocitrate dehydrogenase and the 2-oxoglutarate dehydrogenase complex. Biochem. J. 1989, 263, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Contreras, L.; Gomez-Puertas, P.; Iijima, M.; Kobayashi, K.; Saheki, T.; Satrústegui, J. Ca2+ Activation kinetics of the two aspartate-glutamate mitochondrial carriers, aralar and citrin: Role in the heart malate-aspartate NADH shuttle. J. Biol. Chem. 2007, 282, 7098–7106. [Google Scholar] [CrossRef]
- Qi, F.; Chen, X.; Beard, D.A. Detailed kinetics and regulation of mammalian NAD-linked isocitrate dehydrogenase. Biochim. Biophys. Acta 2008, 1784, 1641–1651. [Google Scholar] [CrossRef]
- Contreras, L.; Drago, I.; Zampese, E.; Pozzan, T. Mitochondria: The calcium connection. Biochim. Biophys. Acta 2010, 1797, 607–618. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Budd, S.L. Mitochondria and neuronal survival. Physiol. Rev. 2000, 80, 315–360. [Google Scholar] [CrossRef]
- Matsuzaki, S.; Szweda, L.I. Inhibition of complex I by Ca2+ reduces electron transport activity and the rate of superoxide anion production in cardiac submitochondrial particles. Biochemistry 2007, 46, 1350–1357. [Google Scholar] [CrossRef]
- Lai, J.C.; DiLorenzo, J.C.; Sheu, K.F. Pyruvate dehydrogenase complex is inhibited in calcium-loaded cerebrocortical mitochondria. Neurochem. Res. 1988, 13, 1043–1048. [Google Scholar] [CrossRef]
- Abou-Khalil, S.; Abou-Khalil, W.H.; Yunis, A.A. Inhibition of Ca2+ of oxidative phosphorylation in myeloid tumor mitochondria. Arch. Biochem. Biophys. 1981, 209, 460–464. [Google Scholar] [CrossRef]
- Malyala, S.; Zhang, Y.; Strubbe, J.O.; Bazil, J.N. Calcium phosphate precipitation inhibits mitochondrial energy metabolism. PLoS Comput. Biol. 2019, 15, e1006719. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate Code | Substrates * | Final Concentrations |
|---|---|---|
| PM | Pyruvate + Malate | 5 mM + 2.5 mM |
| GM | Glutamate + Malate | 5 mM + 2.5 mM |
| AM | Alpha-Ketoglutarate + Malate | 5 mM + 2.5 mM |
| PCM | Palmitoyl-carnitine + Malate | 25 µM + 2.5 mM |
| SUC + ROT | Succinate + Rotenone | 10 mM + 0.5 µM |
| [Ca2+]total (µM) | [Ca2+]free (nM) |
|---|---|
| 0 | 0 |
| 250 | 100 |
| 400 | 200 |
| 700 | 720 |
| 750 | 925 |
| 800 | 1230 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Tomar, N.; Kandel, S.M.; Audi, S.H.; Cowley, A.W., Jr.; Dash, R.K. Substrate- and Calcium-Dependent Differential Regulation of Mitochondrial Oxidative Phosphorylation and Energy Production in the Heart and Kidney. Cells 2022, 11, 131. https://doi.org/10.3390/cells11010131
Zhang X, Tomar N, Kandel SM, Audi SH, Cowley AW Jr., Dash RK. Substrate- and Calcium-Dependent Differential Regulation of Mitochondrial Oxidative Phosphorylation and Energy Production in the Heart and Kidney. Cells. 2022; 11(1):131. https://doi.org/10.3390/cells11010131
Chicago/Turabian StyleZhang, Xiao, Namrata Tomar, Sunil M. Kandel, Said H. Audi, Allen W. Cowley, Jr., and Ranjan K. Dash. 2022. "Substrate- and Calcium-Dependent Differential Regulation of Mitochondrial Oxidative Phosphorylation and Energy Production in the Heart and Kidney" Cells 11, no. 1: 131. https://doi.org/10.3390/cells11010131
APA StyleZhang, X., Tomar, N., Kandel, S. M., Audi, S. H., Cowley, A. W., Jr., & Dash, R. K. (2022). Substrate- and Calcium-Dependent Differential Regulation of Mitochondrial Oxidative Phosphorylation and Energy Production in the Heart and Kidney. Cells, 11(1), 131. https://doi.org/10.3390/cells11010131