
Golgi Alpha1,2-Mannosidase IA Promotes Efficient Endoplasmic Reticulum-Associated Degradation of NKCC2


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Yeast Two-Hybrid Assay
2.2. Plasmid Constructions and Site Directed Mutagenesis
2.3. Cell Culture
2.4. Protein Preparation, Immunoblotting and Immunoprecipitation
2.5. Immunocytochemistry
2.6. Cycloheximide-Chase Assays
2.7. siRNA Knockdown
2.8. Statistical Analyses
3. Results
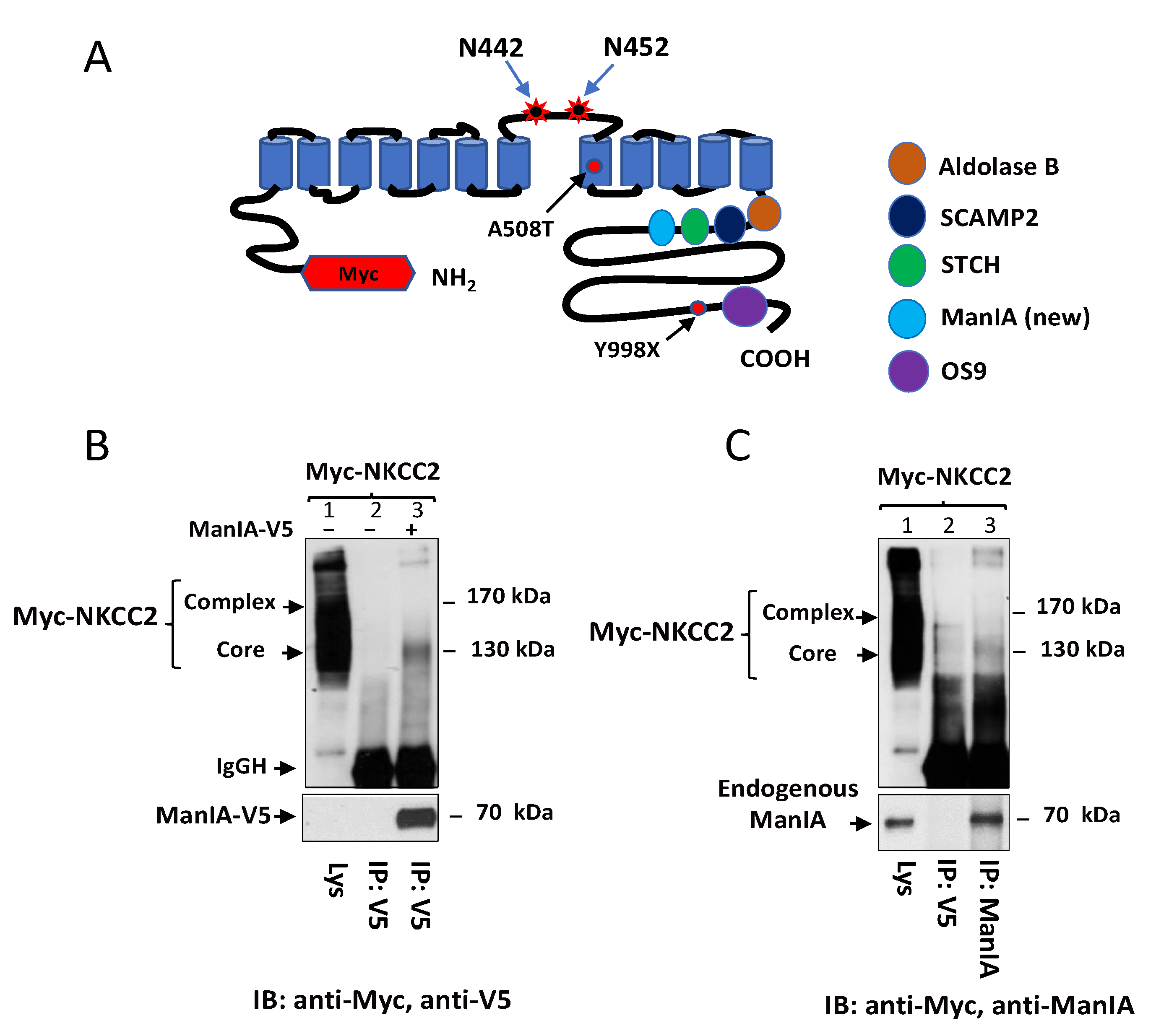
3.1. Identification of Golgi Mannosidase IA as a Novel NKCC2-Interacting Protein
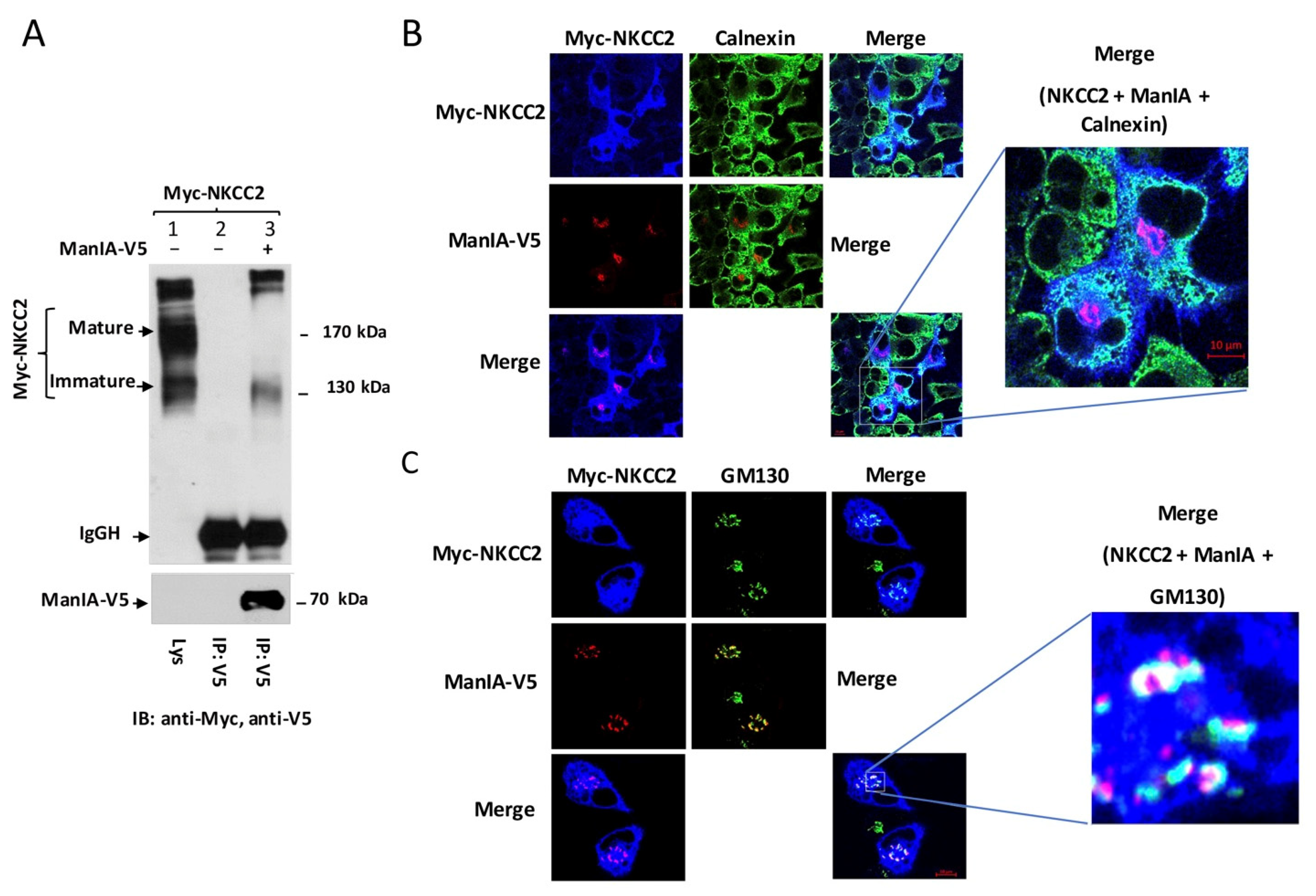
3.2. NKCC2 and ManIA Co-Localize Mainly at the Cis-Golgi Compartment
3.3. ManIA Decreases NKCC2 Stability and Maturation
3.4. ManIA Impairs NKCC2 Maturation Independently of the Expression System
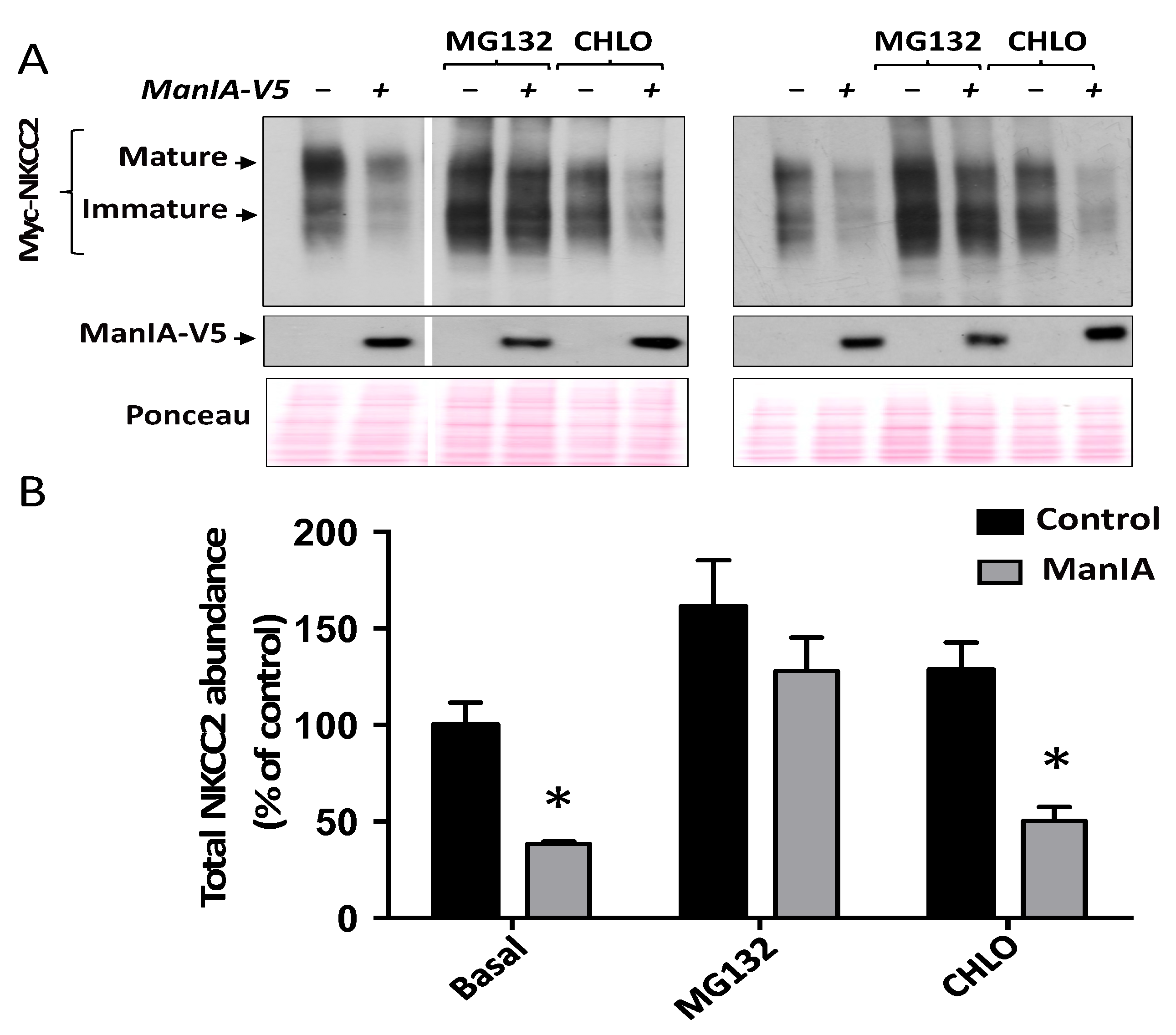
3.5. ManIA Promotes NKCC2 ERAD in a Proteasome-Dependent Manner
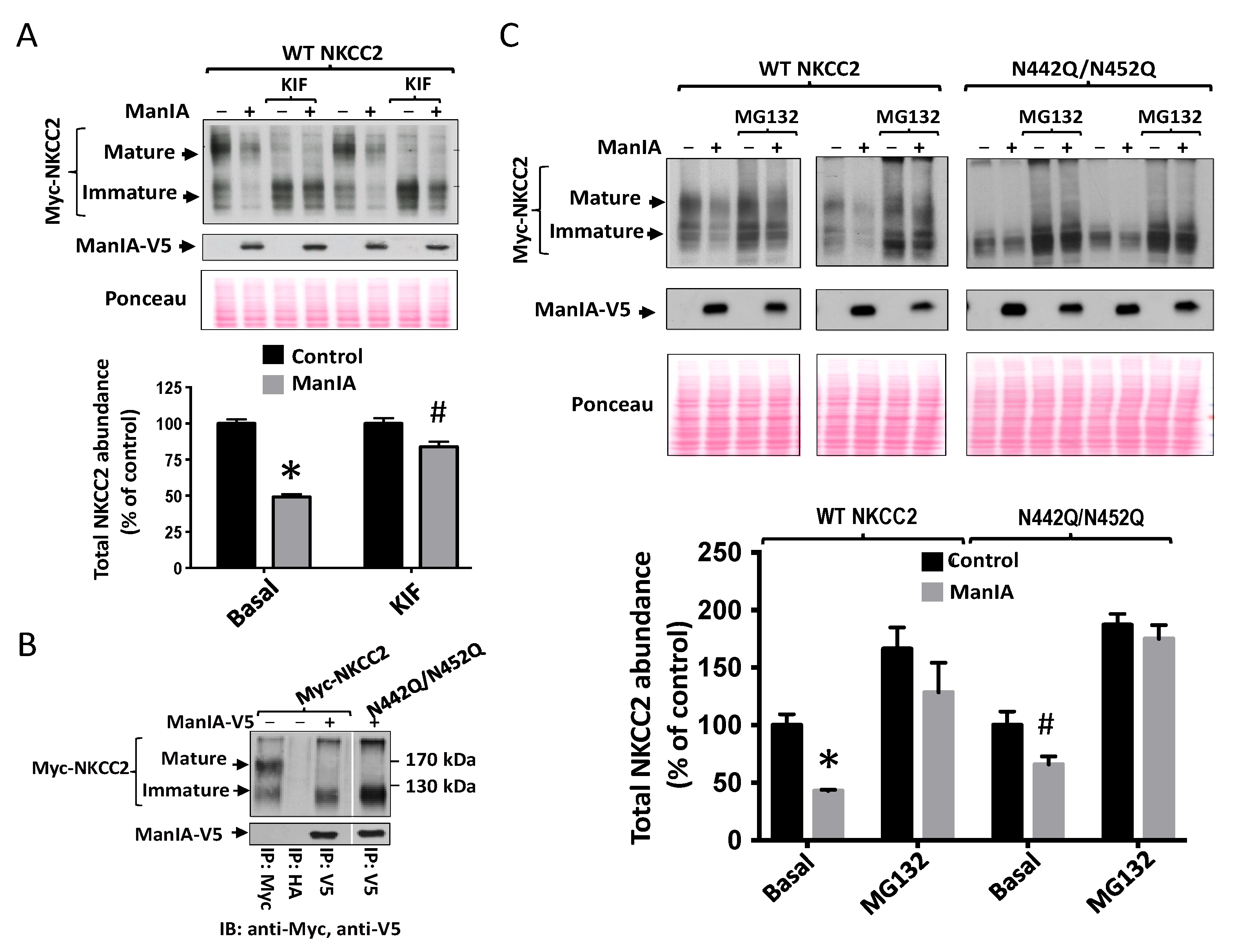
3.6. Effect of Mannose Trimming of NKCC2 on the ManIA-Dependent ERAD Pathway
3.7. Role of the Cytoplasmic Domain of ManIA in its Effect on NKCC2
3.8. Role of the Catalytic Domain of ManIA on its Effect on NKCC2
3.9. ManIA Promotes Efficient Degradation of NKCC2 Folding Mutants
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guyton, A.C. Blood pressure control-special role of the kidneys and body fluids. Science 1991, 252, 1813–1816. [Google Scholar] [CrossRef] [PubMed]
- Lifton, R.P.; Gharavi, A.G.; Geller, D.S. Molecular mechanisms of human hypertension. Cell 2001, 104, 545–556. [Google Scholar] [CrossRef]
- Castrop, H.; Schiessl, I.M. Physiology and pathophysiology of the renal Na-K-2Cl cotransporter (NKCC2). Am. J. Physiol. Ren. Physiol. 2014, 307, F991–F1002. [Google Scholar] [CrossRef]
- Greger, R.; Velazquez, H. The cortical thick ascending limb and early distal convoluted tubule in the urinary concentrating mechanism. Kidney Int. 1987, 31, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Gamba, G. Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol. Rev. 2005, 85, 423–493. [Google Scholar] [CrossRef] [PubMed]
- Ares, G.R.; Caceres, P.S.; Ortiz, P.A. Molecular regulation of NKCC2 in the thick ascending limb. Am. J. Physiol. Ren. Physiol. 2011, 301, F1143–F1159. [Google Scholar] [CrossRef] [PubMed]
- Komhoff, M.; Laghmani, K. Pathophysiology of antenatal Bartter’s syndrome. Curr. Opin. Nephrol. Hypertens. 2017, 26, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Laghmani, K.; Beck, B.B.; Yang, S.S.; Seaayfan, E.; Wenzel, A.; Reusch, B.; Vitzthum, H.; Priem, D.; Demaretz, S.; Bergmann, K.; et al. Polyhydramnios, Transient Antenatal Bartter’s Syndrome, and MAGED2 Mutations. N. Engl. J. Med. 2016, 374, 1853–1863. [Google Scholar] [CrossRef]
- Simon, D.B.; Lifton, R.P. The molecular basis of inherited hypokalemic alkalosis: Bartter’s and Gitelman’s syndromes. Am. J. Physiol. 1996, 271, F961–F966. [Google Scholar] [CrossRef]
- Aviv, A.; Hollenberg, N.K.; Weder, A. Urinary potassium excretion and sodium sensitivity in blacks. Hypertension 2004, 43, 707–713. [Google Scholar] [CrossRef]
- Hoorn, E.J.; Walsh, S.B.; McCormick, J.A.; Furstenberg, A.; Yang, C.L.; Roeschel, T.; Paliege, A.; Howie, A.J.; Conley, J.; Bachmann, S.; et al. The calcineurin inhibitor tacrolimus activates the renal sodium chloride cotransporter to cause hypertension. Nat. Med. 2011, 17, 1304–1309. [Google Scholar] [CrossRef] [PubMed]
- Borschewski, A.; Himmerkus, N.; Boldt, C.; Blankenstein, K.I.; McCormick, J.A.; Lazelle, R.; Willnow, T.E.; Jankowski, V.; Plain, A.; Bleich, M.; et al. Calcineurin and Sorting-Related Receptor with A-Type Repeats Interact to Regulate the Renal Na(+)-K(+)-2Cl(-) Cotransporter. J. Am. Soc. Nephrol. 2016, 27, 107–119. [Google Scholar] [CrossRef]
- Trudu, M.; Janas, S.; Lanzani, C.; Debaix, H.; Schaeffer, C.; Ikehata, M.; Citterio, L.; Demaretz, S.; Trevisani, F.; Ristagno, G.; et al. Common noncoding UMOD gene variants induce salt-sensitive hypertension and kidney damage by increasing uromodulin expression. Nat. Med. 2013, 19, 1655–1660. [Google Scholar] [CrossRef]
- Alvarez-Guerra, M.; Garay, R.P. Renal Na-K-Cl cotransporter NKCC2 in Dahl salt-sensitive rats. J. Hypertens. 2002, 20, 721–727. [Google Scholar] [CrossRef]
- Capasso, G.; Rizzo, M.; Evangelista, C.; Ferrari, P.; Geelen, G.; Lang, F.; Bianchi, G. Altered expression of renal apical plasma membrane Na+ transporters in the early phase of genetic hypertension. Am. J. Physiol. Ren. Physiol. 2005, 288, F1173–F1182. [Google Scholar] [CrossRef]
- Fenton, R.A.; Knepper, M.A. Mouse models and the urinary concentrating mechanism in the new millennium. Physiol. Rev. 2007, 87, 1083–1112. [Google Scholar] [CrossRef] [PubMed]
- Komhoff, M.; Laghmani, K. MAGED2: A novel form of antenatal Bartter’s syndrome. Curr. Opin. Nephrol. Hypertens. 2018, 27, 323–328. [Google Scholar] [CrossRef]
- Sands, J.M. Urinary concentration and dilution in the aging kidney. Semin. Nephrol. 2009, 29, 579–586. [Google Scholar] [CrossRef][Green Version]
- Tian, Y.; Riazi, S.; Khan, O.; Klein, J.D.; Sugimura, Y.; Verbalis, J.G.; Ecelbarger, C.A. Renal ENaC subunit, Na-K-2Cl and Na-Cl cotransporter abundances in aged, water-restricted F344 x Brown Norway rats. Kidney Int. 2006, 69, 304–312. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schiessl, I.M.; Castrop, H. Regulation of NKCC2 splicing and phosphorylation. Curr. Opin. Nephrol. Hypertens. 2015, 24, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.; Fraser, S.A.; Galic, S.; Choy, S.W.; Katerelos, M.; Gleich, K.; Kemp, B.E.; Mount, P.F.; Power, D.A. Novel mechanisms of Na+ retention in obesity: Phosphorylation of NKCC2 and regulation of SPAK/OSR1 by AMPK. Am. J. Physiol. Ren. Physiol. 2014, 307, F96–F106. [Google Scholar] [CrossRef]
- Gamba, G. Regulation of NKCC2 activity by SPAK truncated isoforms. Am. J. Physiol. Ren. Physiol. 2014, 306, F49–F50. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Edwards, A.; Castrop, H.; Laghmani, K.; Vallon, V.; Layton, A.T. Effects of NKCC2 isoform regulation on NaCl transport in thick ascending limb and macula densa: A modeling study. Am. J. Physiol. Ren. Physiol. 2014, 307, F137–F146. [Google Scholar] [CrossRef]
- Vitale, A.; Denecke, J. The endoplasmic reticulum-gateway of the secretory pathway. Plant. Cell 1999, 11, 615–628. [Google Scholar] [PubMed]
- Caplan, M.J. Membrane polarity in epithelial cells: Protein sorting and establishment of polarized domains. Am. J. Physiol. 1997, 272, F425–F429. [Google Scholar] [CrossRef]
- Sun, Z.; Brodsky, J.L. Protein quality control in the secretory pathway. J. Cell Biol 2019, 218, 3171–3187. [Google Scholar] [CrossRef] [PubMed]
- Ruggiano, A.; Foresti, O.; Carvalho, P. Quality control: ER-associated degradation: Protein quality control and beyond. J. Cell Biol. 2014, 204, 869–879. [Google Scholar] [CrossRef] [PubMed]
- Ruddock, L.W.; Molinari, M. N-glycan processing in ER quality control. J. Cell Sci. 2006, 119, 4373–4380. [Google Scholar] [CrossRef]
- Molinari, M. N-glycan structure dictates extension of protein folding or onset of disposal. Nat. Chem. Biol. 2007, 3, 313–320. [Google Scholar] [CrossRef]
- Hebert, D.N.; Garman, S.C.; Molinari, M. The glycan code of the endoplasmic reticulum: Asparagine-linked carbohydrates as protein maturation and quality-control tags. Trends Cell Biol. 2005, 15, 364–370. [Google Scholar] [CrossRef]
- Igdoura, S.A.; Herscovics, A.; Lal, A.; Moremen, K.W.; Morales, C.R.; Hermo, L. Alpha-mannosidases involved in N-glycan processing show cell specificity and distinct subcompartmentalization within the Golgi apparatus of cells in the testis and epididymis. Eur. J. Cell Biol. 1999, 78, 441–452. [Google Scholar] [CrossRef]
- Lederkremer, G.Z.; Glickman, M.H. A window of opportunity: Timing protein degradation by trimming of sugars and ubiquitins. Trends Biochem. Sci. 2005, 30, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Stanley, P.; Taniguchi, N.; Aebi, M. N-Glycans. In Essentials of Glycobiology, 3rd.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., et al., Eds.; Cold Spring Harbor: New York, NY, USA, 2015; pp. 99–111. [Google Scholar]
- Barlowe, C.K.; Miller, E.A. Secretory protein biogenesis and traffic in the early secretory pathway. Genetics 2013, 193, 383–410. [Google Scholar] [CrossRef]
- Wu, X.; Rapoport, T.A. Mechanistic insights into ER-associated protein degradation. Curr. Opin. Cell Biol. 2018, 53, 22–28. [Google Scholar] [CrossRef]
- Sun, Z.; Brodsky, J.L. The degradation pathway of a model misfolded protein is determined by aggregation propensity. Mol. Biol Cell. 2018, 29, 1422–1434. [Google Scholar] [CrossRef]
- Fregno, I.; Molinari, M. Proteasomal and lysosomal clearance of faulty secretory proteins: ER-associated degradation (ERAD) and ER-to-lysosome-associated degradation (ERLAD) pathways. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Briant, K.; Johnson, N.; Swanton, E. Transmembrane domain quality control systems operate at the endoplasmic reticulum and Golgi apparatus. PLoS ONE 2017, 12, e0173924. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.J.; Loo, M.A.; Pind, S.; Williams, D.B.; Goldberg, A.L.; Riordan, J.R. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell 1995, 83, 129–135. [Google Scholar] [CrossRef]
- Ward, C.L.; Omura, S.; Kopito, R.R. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell 1995, 83, 121–127. [Google Scholar] [CrossRef]
- Gong, Q.; Keeney, D.R.; Molinari, M.; Zhou, Z. Degradation of trafficking-defective long QT syndrome type II mutant channels by the ubiquitin-proteasome pathway. J. Biol. Chem. 2005, 280, 19419–19425. [Google Scholar] [CrossRef]
- O’Donnell, B.M.; Mackie, T.D.; Subramanya, A.R.; Brodsky, J.L. Endoplasmic reticulum-associated degradation of the renal potassium channel, ROMK, leads to type II Bartter syndrome. J. Biol. Chem. 2017, 292, 12813–12827. [Google Scholar] [CrossRef] [PubMed]
- Needham, P.G.; Mikoluk, K.; Dhakarwal, P.; Khadem, S.; Snyder, A.C.; Subramanya, A.R.; Brodsky, J.L. The thiazide-sensitive NaCl cotransporter is targeted for chaperone-dependent endoplasmic reticulum-associated degradation. J. Biol. Chem. 2011, 286, 43611–43621. [Google Scholar] [CrossRef] [PubMed]
- Zaarour, N.; Demaretz, S.; Defontaine, N.; Zhu, Y.; Laghmani, K. Multiple evolutionarily conserved Di-leucine like motifs in the carboxyl terminus control the anterograde trafficking of NKCC2. J. Biol. Chem. 2012, 287, 42642–42653. [Google Scholar] [CrossRef]
- Zaarour, N.; Demaretz, S.; Defontaine, N.; Mordasini, D.; Laghmani, K. A highly conserved motif at the COOH terminus dictates endoplasmic reticulum exit and cell surface expression of NKCC2. J. Biol. Chem. 2009, 284, 21752–21764. [Google Scholar] [CrossRef] [PubMed]
- Seaayfan, E.; Defontaine, N.; Demaretz, S.; Zaarour, N.; Laghmani, K. OS9 Protein Interacts with Na-K-2Cl Co-transporter (NKCC2) and Targets Its Immature Form for the Endoplasmic Reticulum-associated Degradation Pathway. J. Biol. Chem. 2016, 291, 4487–4502. [Google Scholar] [CrossRef]
- Brodsky, J.L. The protective and destructive roles played by molecular chaperones during ERAD (endoplasmic-reticulum-associated degradation). Biochem. J. 2007, 404, 353–363. [Google Scholar] [CrossRef]
- Guerriero, C.J.; Brodsky, J.L. The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol. Rev. 2012, 92, 537–576. [Google Scholar] [CrossRef]
- Ahner, A.; Gong, X.; Frizzell, R.A. Cystic fibrosis transmembrane conductance regulator degradation: Cross-talk between the ubiquitylation and SUMOylation pathways. FEBS J. 2013, 280, 4430–4438. [Google Scholar] [CrossRef]
- Donnelly, B.F.; Needham, P.G.; Snyder, A.C.; Roy, A.; Khadem, S.; Brodsky, J.L.; Subramanya, A.R. Hsp70 and Hsp90 multichaperone complexes sequentially regulate thiazide-sensitive cotransporter endoplasmic reticulum-associated degradation and biogenesis. J. Biol. Chem. 2013, 288, 13124–13135. [Google Scholar] [CrossRef]
- Benziane, B.; Demaretz, S.; Defontaine, N.; Zaarour, N.; Cheval, L.; Bourgeois, S.; Klein, C.; Froissart, M.; Blanchard, A.; Paillard, M.; et al. NKCC2 surface expression in mammalian cells: Down-regulation by novel interaction with aldolase B. J. Biol. Chem. 2007, 282, 33817–33830. [Google Scholar] [CrossRef]
- Zaarour, N.; Defontaine, N.; Demaretz, S.; Azroyan, A.; Cheval, L.; Laghmani, K. Secretory carrier membrane protein 2 regulates exocytic insertion of NKCC2 into the cell membrane. J. Biol. Chem. 2011, 286, 9489–9502. [Google Scholar] [CrossRef]
- Bakhos-Douaihy, D.; Seaayfan, E.; Demaretz, S.; Komhoff, M.; Laghmani, K. Differential Effects of STCH and Stress-Inducible Hsp70 on the Stability and Maturation of NKCC2. Int. J. Mol. Sci. 2021, 22, 2207. [Google Scholar] [CrossRef]
- Herscovics, A.; Schneikert, J.; Athanassiadis, A.; Moremen, K.W. Isolation of a mouse Golgi mannosidase cDNA, a member of a gene family conserved from yeast to mammals. J. Biol. Chem. 1994, 269, 9864–9871. [Google Scholar] [CrossRef]
- Tempel, W.; Karaveg, K.; Liu, Z.J.; Rose, J.; Wang, B.C.; Moremen, K.W. Structure of mouse Golgi alpha-mannosidase IA reveals the molecular basis for substrate specificity among class 1 (family 47 glycosylhydrolase) alpha1,2-mannosidases. J. Biol. Chem. 2004, 279, 29774–29786. [Google Scholar] [CrossRef]
- Tulsiani, D.R.; Touster, O. The purification and characterization of mannosidase IA from rat liver Golgi membranes. J. Biol. Chem. 1988, 263, 5408–5417. [Google Scholar] [CrossRef]
- Hosokawa, N.; You, Z.; Tremblay, L.O.; Nagata, K.; Herscovics, A. Stimulation of ERAD of misfolded null Hong Kong alpha1-antitrypsin by Golgi alpha1,2-mannosidases. Biochem. Biophys. Res. Commun. 2007, 362, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Ogen-Shtern, N.; Avezov, E.; Shenkman, M.; Benyair, R.; Lederkremer, G.Z. Mannosidase IA is in Quality Control Vesicles and Participates in Glycoprotein Targeting to ERAD. J. Mol. Biol. 2016, 428, 3194–3205. [Google Scholar] [CrossRef]
- Iannotti, M.J.; Figard, L.; Sokac, A.M.; Sifers, R.N. A Golgi-localized mannosidase (MAN1B1) plays a non-enzymatic gatekeeper role in protein biosynthetic quality control. J. Biol. Chem. 2014, 289, 11844–11858. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Cheng, X.; Sifers, R.N. Golgi-situated endoplasmic reticulum alpha-1, 2-mannosidase contributes to the retrieval of ERAD substrates through a direct interaction with gamma-COP. Mol. Biol. Cell 2013, 24, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Shaukat, I.; Bakhos-Douaihy, D.; Zhu, Y.; Seaayfan, E.; Demaretz, S.; Frachon, N.; Weber, S.; Komhoff, M.; Vargas-Poussou, R.; Laghmani, K. New insights into the role of endoplasmic reticulum-associated degradation in Bartter Syndrome Type 1. Hum. Mutat. 2021, 42, 947–968. [Google Scholar] [CrossRef]
- Fujita, E.; Kouroku, Y.; Isoai, A.; Kumagai, H.; Misutani, A.; Matsuda, C.; Hayashi, Y.K.; Momoi, T. Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: Ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II). Hum. Mol. Genet. 2007, 16, 618–629. [Google Scholar] [CrossRef]
- Bernasconi, R.; Molinari, M. ERAD and ERAD tuning: Disposal of cargo and of ERAD regulators from the mammalian ER. Curr. Opin. Cell. Biol. 2011, 23, 176–183. [Google Scholar] [CrossRef]
- Arvan, P.; Zhao, X.; Ramos-Castaneda, J.; Chang, A. Secretory pathway quality control operating in Golgi, plasmalemmal, and endosomal systems. Traffic 2002, 3, 771–780. [Google Scholar] [CrossRef]
- Stolz, A.; Wolf, D.H. Endoplasmic reticulum associated protein degradation: A chaperone assisted journey to hell. Biochim. Biophys. Acta 2010, 1803, 694–705. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Song, W.; Brancati, G.; Segatori, L. Inhibition of endoplasmic reticulum-associated degradation rescues native folding in loss of function protein misfolding diseases. J. Biol. Chem. 2011, 286, 43454–43464. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, F.; Brostrom, C.; Koide, T.; Arvan, P. Endoplasmic reticulum (ER)-associated degradation of misfolded N-linked glycoproteins is suppressed upon inhibition of ER mannosidase I. J. Biol. Chem. 2000, 275, 40757–40764. [Google Scholar] [CrossRef] [PubMed]
- Groisman, B.; Shenkman, M.; Ron, E.; Lederkremer, G.Z. Mannose trimming is required for delivery of a glycoprotein from EDEM1 to XTP3-B and to late endoplasmic reticulum-associated degradation steps. J. Biol. Chem. 2011, 286, 1292–1300. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Poussou, R.; Feldmann, D.; Vollmer, M.; Konrad, M.; Kelly, L.; van den Heuvel, L.P.; Tebourbi, L.; Brandis, M.; Karolyi, L.; Hebert, S.C.; et al. Novel molecular variants of the Na-K-2Cl cotransporter gene are responsible for antenatal Bartter syndrome. Am. J. Hum. Genet. 1998, 62, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Helenius, A.; Aebi, M. Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 2004, 73, 1019–1049. [Google Scholar] [CrossRef]
- Spiro, R.G. Role of N-linked polymannose oligosaccharides in targeting glycoproteins for endoplasmic reticulum-associated degradation. Cell Mol. Life Sci. 2004, 61, 1025–1041. [Google Scholar] [CrossRef]
- Marcus, N.Y.; Perlmutter, D.H. Glucosidase and mannosidase inhibitors mediate increased secretion of mutant alpha1 antitrypsin Z. J. Biol. Chem. 2000, 275, 1987–1992. [Google Scholar] [CrossRef]
- Liu, Y.; Choudhury, P.; Cabral, C.M.; Sifers, R.N. Intracellular disposal of incompletely folded human alpha1-antitrypsin involves release from calnexin and post-translational trimming of asparagine-linked oligosaccharides. J. Biol. Chem. 1997, 272, 7946–7951. [Google Scholar] [CrossRef] [PubMed]
- Cabral, C.M.; Liu, Y.; Sifers, R.N. Dissecting glycoprotein quality control in the secretory pathway. Trends Biochem. Sci. 2001, 26, 619–624. [Google Scholar] [CrossRef]
- Ermonval, M.; Kitzmuller, C.; Mir, A.M.; Cacan, R.; Ivessa, N.E. N-glycan structure of a short-lived variant of ribophorin I expressed in the MadIA214 glycosylation-defective cell line reveals the role of a mannosidase that is not ER mannosidase I in the process of glycoprotein degradation. Glycobiology 2001, 11, 565–576. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Foulquier, F.; Duvet, S.; Klein, A.; Mir, A.M.; Chirat, F.; Cacan, R. Endoplasmic reticulum-associated degradation of glycoproteins bearing Man5GlcNAc2 and Man9GlcNAc2 species in the MI8-5 CHO cell line. Eur. J. Biochem. 2004, 271, 398–404. [Google Scholar] [CrossRef]
- Avezov, E.; Frenkel, Z.; Ehrlich, M.; Herscovics, A.; Lederkremer, G.Z. Endoplasmic reticulum (ER) mannosidase I is compartmentalized and required for N-glycan trimming to Man5-6GlcNAc2 in glycoprotein ER-associated degradation. Mol. Biol. Cell 2008, 19, 216–225. [Google Scholar] [CrossRef]
- Hosokawa, N.; Tremblay, L.O.; You, Z.; Herscovics, A.; Wada, I.; Nagata, K. Enhancement of endoplasmic reticulum (ER) degradation of misfolded Null Hong Kong alpha1-antitrypsin by human ER mannosidase I. J. Biol. Chem. 2003, 278, 26287–26294. [Google Scholar] [CrossRef]
- Sifers, R.N. Cell biology. Protein degradation unlocked. Science 2003, 299, 1330–1331. [Google Scholar] [CrossRef]
- Wu, Y.; Swulius, M.T.; Moremen, K.W.; Sifers, R.N. Elucidation of the molecular logic by which misfolded alpha 1-antitrypsin is preferentially selected for degradation. Proc. Natl. Acad. Sci. USA 2003, 100, 8229–8234. [Google Scholar] [CrossRef] [PubMed]
- Hirao, K.; Natsuka, Y.; Tamura, T.; Wada, I.; Morito, D.; Natsuka, S.; Romero, P.; Sleno, B.; Tremblay, L.O.; Herscovics, A.; et al. EDEM3, a soluble EDEM homolog, enhances glycoprotein endoplasmic reticulum-associated degradation and mannose trimming. J. Biol. Chem. 2006, 281, 9650–9658. [Google Scholar] [CrossRef]
- Olivari, S.; Cali, T.; Salo, K.E.; Paganetti, P.; Ruddock, L.W.; Molinari, M. EDEM1 regulates ER-associated degradation by accelerating de-mannosylation of folding-defective polypeptides and by inhibiting their covalent aggregation. Biochem. Biophys. Res. Commun. 2006, 349, 1278–1284. [Google Scholar] [CrossRef] [PubMed]
- Bieberich, E.; Bause, E. Man9-mannosidase from human kidney is expressed in COS cells as a Golgi-resident type II transmembrane N-glycoprotein. Eur. J. Biochem. 1995, 233, 644–649. [Google Scholar] [CrossRef] [PubMed]
- Velasco, A.; Hendricks, L.; Moremen, K.W.; Tulsiani, D.R.; Touster, O.; Farquhar, M.G. Cell type-dependent variations in the subcellular distribution of alpha-mannosidase I and II. J. Cell Biol. 1993, 122, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Bieberich, E.; Treml, K.; Volker, C.; Rolfs, A.; Kalz-Fuller, B.; Bause, E. Man9-mannosidase from pig liver is a type-II membrane protein that resides in the endoplasmic reticulum. cDNA cloning and expression of the enzyme in COS 1 cells. Eur. J. Biochem. 1997, 246, 681–689. [Google Scholar] [CrossRef]
- Ron, E.; Shenkman, M.; Groisman, B.; Izenshtein, Y.; Leitman, J.; Lederkremer, G.Z. Bypass of glycan-dependent glycoprotein delivery to ERAD by up-regulated EDEM1. Mol. Biol. Cell 2011, 22, 3945–3954. [Google Scholar] [CrossRef]
- Casagrande, R.; Stern, P.; Diehn, M.; Shamu, C.; Osario, M.; Zuniga, M.; Brown, P.O.; Ploegh, H. Degradation of proteins from the ER of S. cerevisiae requires an intact unfolded protein response pathway. Mol. Cell 2000, 5, 729–735. [Google Scholar] [CrossRef]
- Schroder, M.; Kaufman, R.J. ER stress and the unfolded protein response. Mutat. Res. 2005, 569, 29–63. [Google Scholar] [CrossRef]
- Liang, C.J.; Chang, Y.C.; Chang, H.C.; Wang, C.K.; Hung, Y.C.; Lin, Y.E.; Chan, C.C.; Chen, C.H.; Chang, H.Y.; Sang, T.K. Derlin-1 regulates mutant VCP-linked pathogenesis and endoplasmic reticulum stress-induced apoptosis. PLoS Genet. 2014, 10, e1004675. [Google Scholar] [CrossRef] [PubMed]
- Houck, S.A.; Singh, S.; Cyr, D.M. Cellular responses to misfolded proteins and protein aggregates. Methods Mol. Biol. 2012, 832, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, T.K.; Paul, S. Protein-misfolding diseases and chaperone-based therapeutic approaches. FEBS J. 2006, 273, 1331–1349. [Google Scholar] [CrossRef]
- Yadav, K.; Yadav, A.; Vashistha, P.; Pandey, V.P.; Dwivedi, U.N. Protein Misfolding Diseases and Therapeutic Approaches. Curr. Protein. Pept. Sci. 2019, 20, 1226–1245. [Google Scholar] [CrossRef]
- Fregno, I.; Molinari, M. Endoplasmic reticulum turnover: ER-phagy and other flavors in selective and non-selective ER clearance. F1000Res 2018, 7, 454. [Google Scholar] [CrossRef]
- Okiyoneda, T.; Apaja, P.M.; Lukacs, G.L. Protein quality control at the plasma membrane. Curr. Opin. Cell Biol. 2011, 23, 483–491. [Google Scholar] [CrossRef]
- Caldwell, S.R.; Hill, K.J.; Cooper, A.A. Degradation of endoplasmic reticulum (ER) quality control substrates requires transport between the ER and Golgi. J. Biol. Chem. 2001, 276, 23296–23303. [Google Scholar] [CrossRef] [PubMed]
- Taxis, C.; Vogel, F.; Wolf, D.H. ER-golgi traffic is a prerequisite for efficient ER degradation. Mol. Biol. Cell 2002, 13, 1806–1818. [Google Scholar] [CrossRef] [PubMed]
- Vashist, S.; Ng, D.T. Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J. Cell Biol. 2004, 165, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, O.; Weyer, Y.; Baumann, V.; Widerin, M.A.; Eising, S.; Angelova, M.; Schleiffer, A.; Kremser, L.; Lindner, H.; Peter, M.; et al. Endosome and Golgi-associated degradation (EGAD) of membrane proteins regulates sphingolipid metabolism. EMBO J. 2019, 38, e101433. [Google Scholar] [CrossRef]
- Pan, S.; Wang, S.; Utama, B.; Huang, L.; Blok, N.; Estes, M.K.; Moremen, K.W.; Sifers, R.N. Golgi localization of ERManI defines spatial separation of the mammalian glycoprotein quality control system. Mol. Biol. Cell 2011, 22, 2810–2822. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Demaretz, S.; Seaayfan, E.; Bakhos-Douaihy, D.; Frachon, N.; Kömhoff, M.; Laghmani, K. Golgi Alpha1,2-Mannosidase IA Promotes Efficient Endoplasmic Reticulum-Associated Degradation of NKCC2. Cells 2022, 11, 101. https://doi.org/10.3390/cells11010101
Demaretz S, Seaayfan E, Bakhos-Douaihy D, Frachon N, Kömhoff M, Laghmani K. Golgi Alpha1,2-Mannosidase IA Promotes Efficient Endoplasmic Reticulum-Associated Degradation of NKCC2. Cells. 2022; 11(1):101. https://doi.org/10.3390/cells11010101
Chicago/Turabian StyleDemaretz, Sylvie, Elie Seaayfan, Dalal Bakhos-Douaihy, Nadia Frachon, Martin Kömhoff, and Kamel Laghmani. 2022. "Golgi Alpha1,2-Mannosidase IA Promotes Efficient Endoplasmic Reticulum-Associated Degradation of NKCC2" Cells 11, no. 1: 101. https://doi.org/10.3390/cells11010101
APA StyleDemaretz, S., Seaayfan, E., Bakhos-Douaihy, D., Frachon, N., Kömhoff, M., & Laghmani, K. (2022). Golgi Alpha1,2-Mannosidase IA Promotes Efficient Endoplasmic Reticulum-Associated Degradation of NKCC2. Cells, 11(1), 101. https://doi.org/10.3390/cells11010101