
The Presence and Potential Role of ALDH1A2 in the Glioblastoma Microenvironment

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Antibodies
2.3. Polarization of Macrophages
2.4. Glioma Conditioned Media Assay
2.5. Zymograms
2.6. Cell Migration Assay
2.7. Microarray Analysis
2.8. Immune Cell Isolation
2.9. Single-Cell RNA Sequencing
2.10. Western Blot
2.11. Immunostaining
2.12. TCGA Analysis
2.13. Statistical Analysis
3. Results
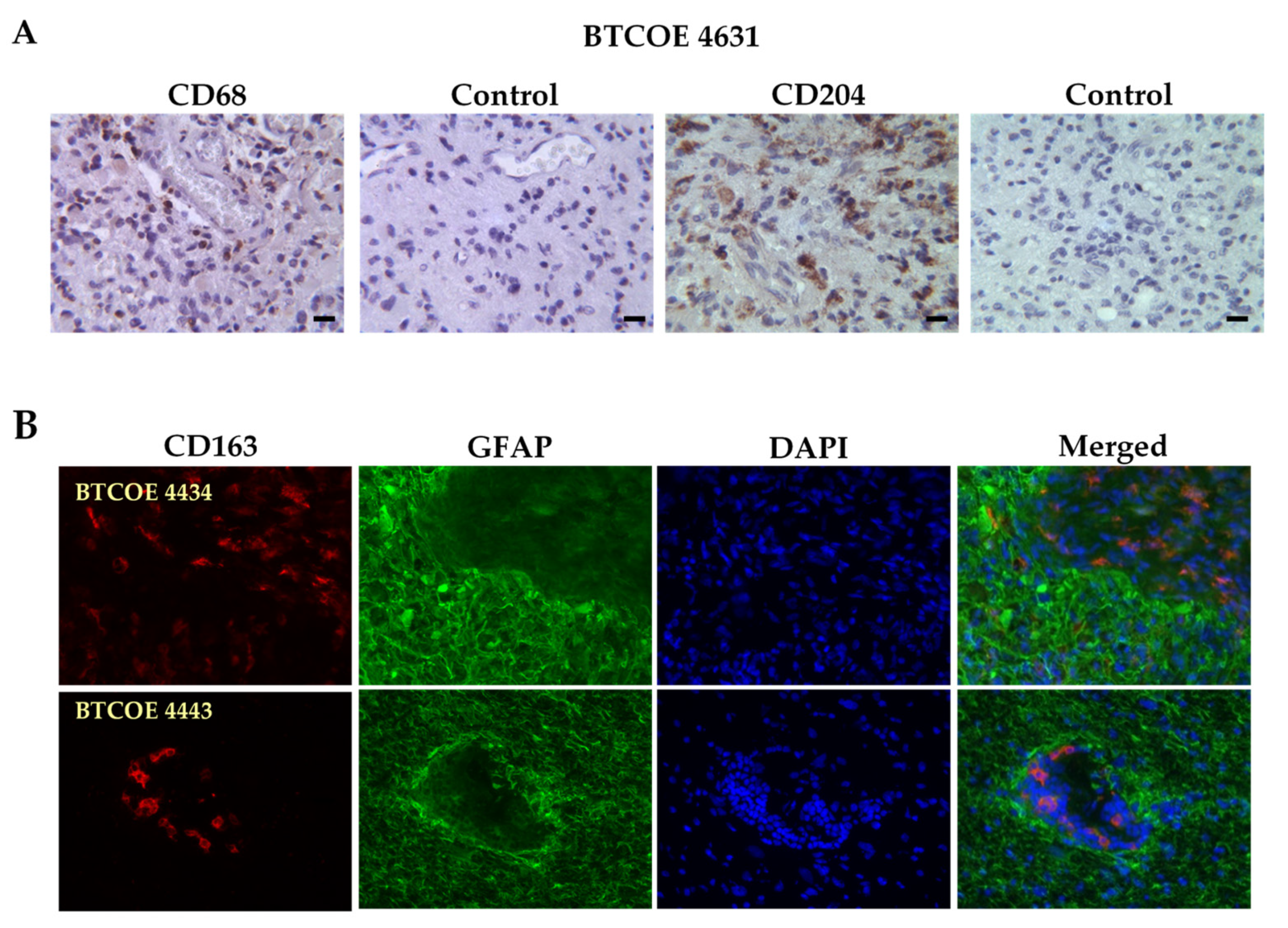
3.1. M2 Macrophages in GBM Specimens
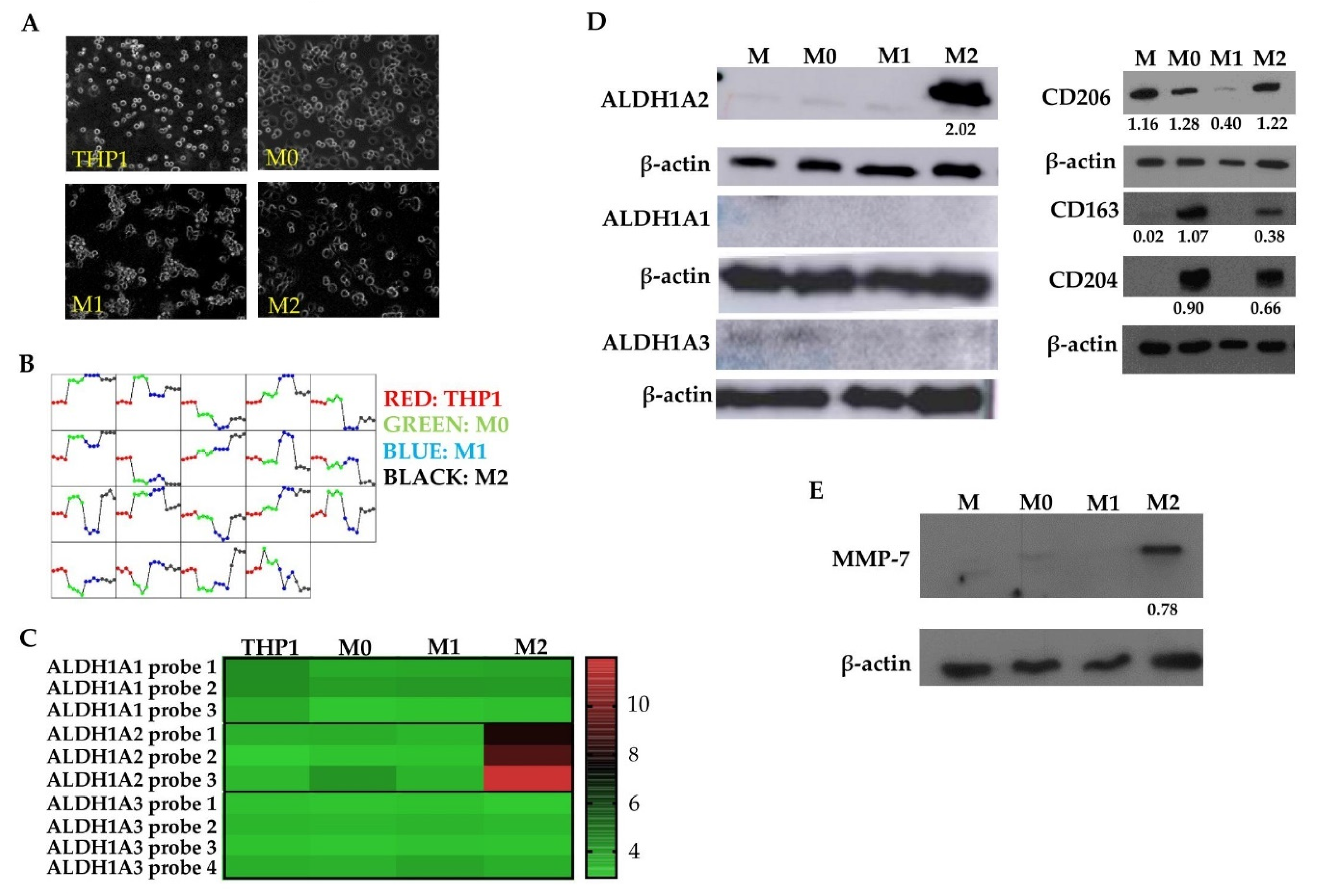
3.2. M2 Macrophages Highly Express ALDH1A2 In Vitro
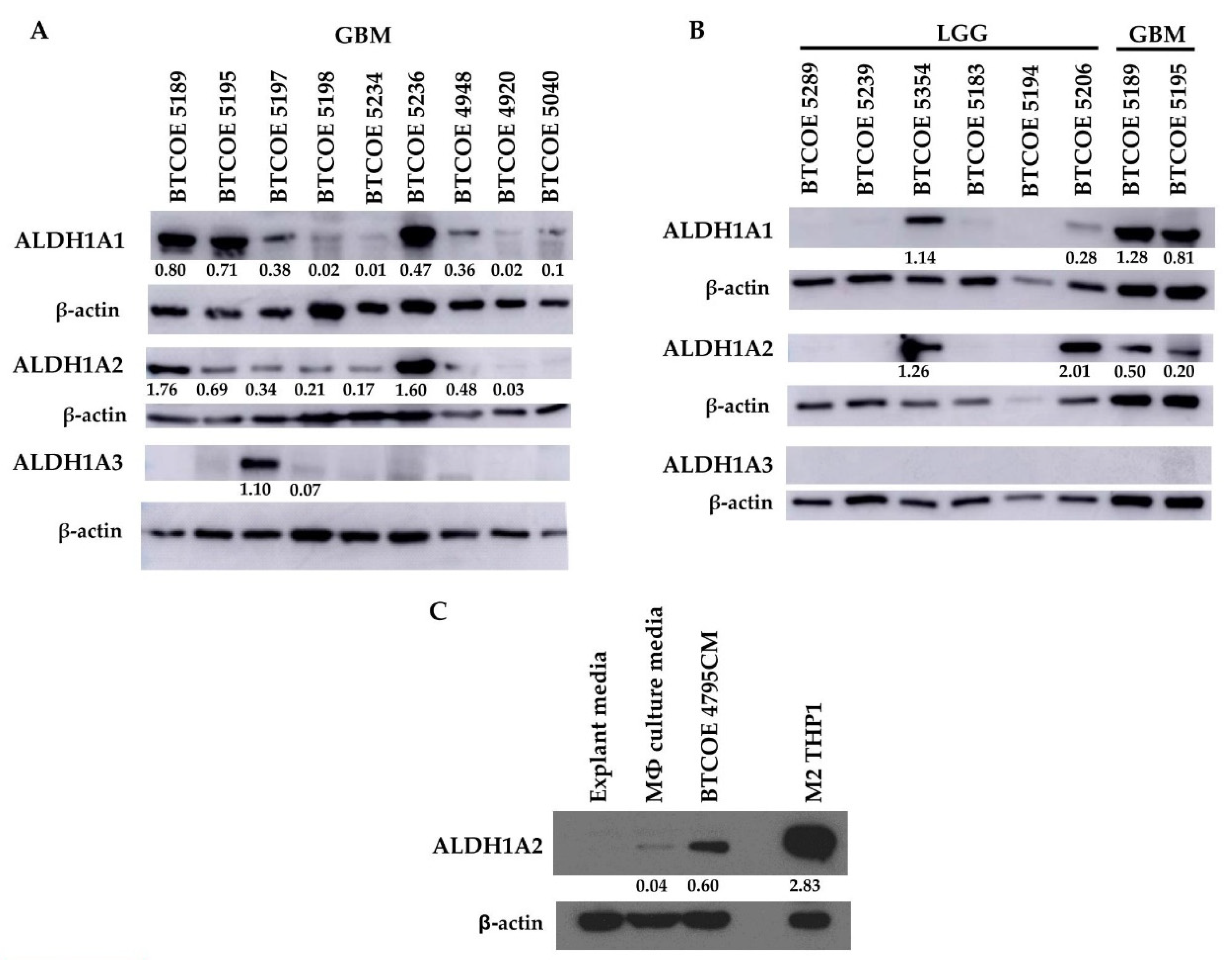
3.3. ALDH1A2 Is Highly Expressed in GBM Compared to LGG and Other ALDH1 Family Proteins
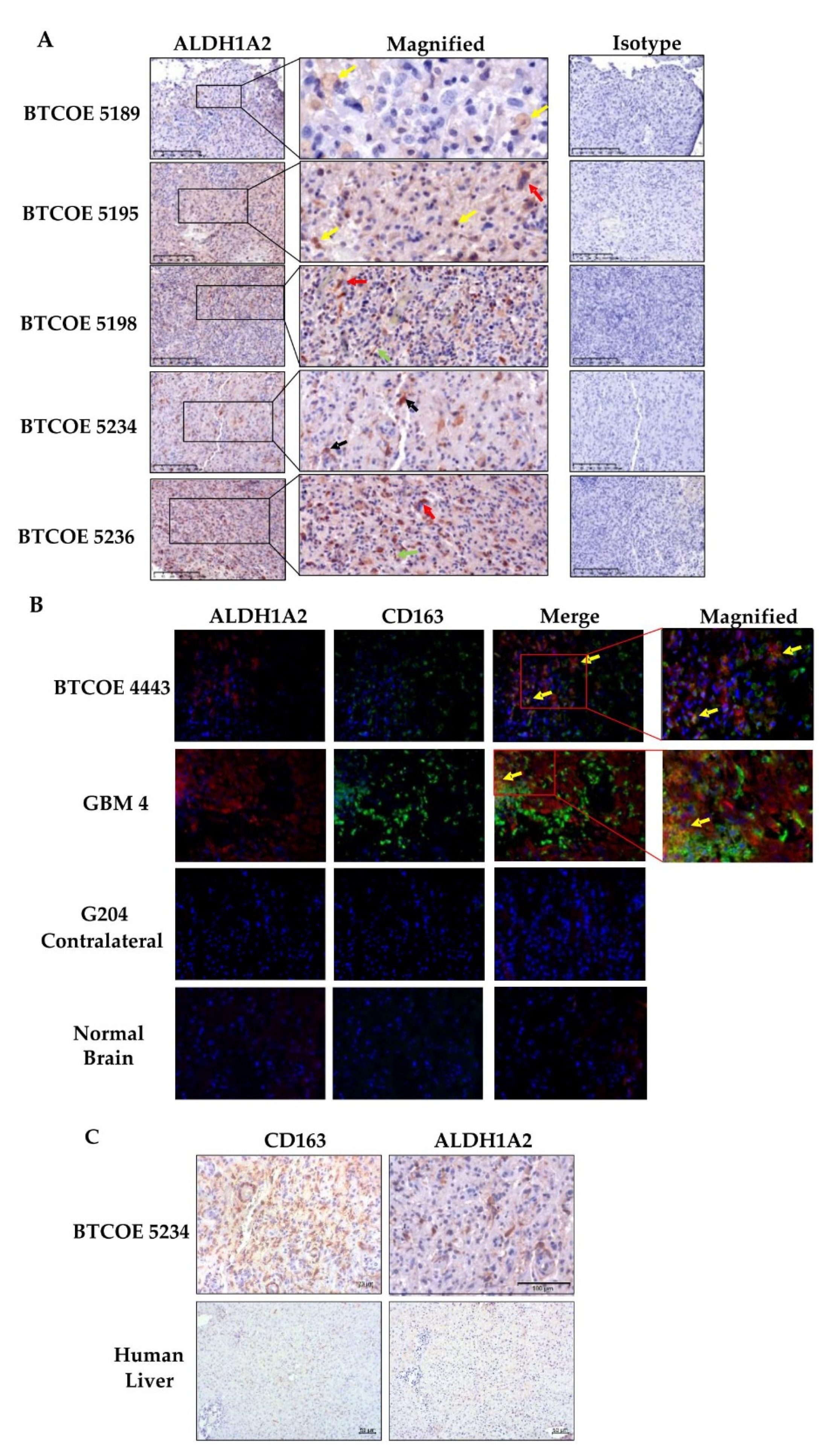
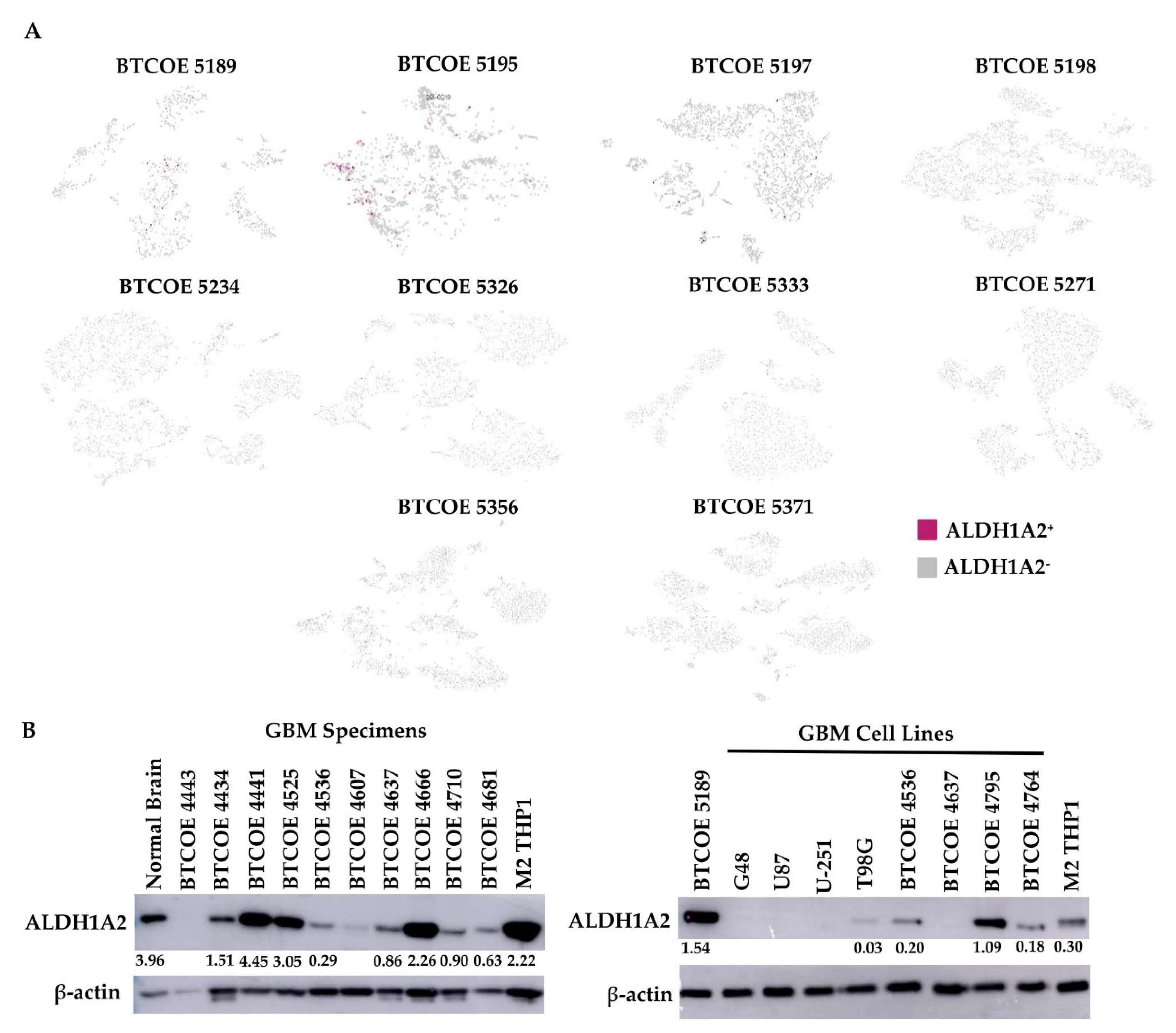
3.4. ALDH1A2 Is Expressed in the Microenvironment of GBM Tumors
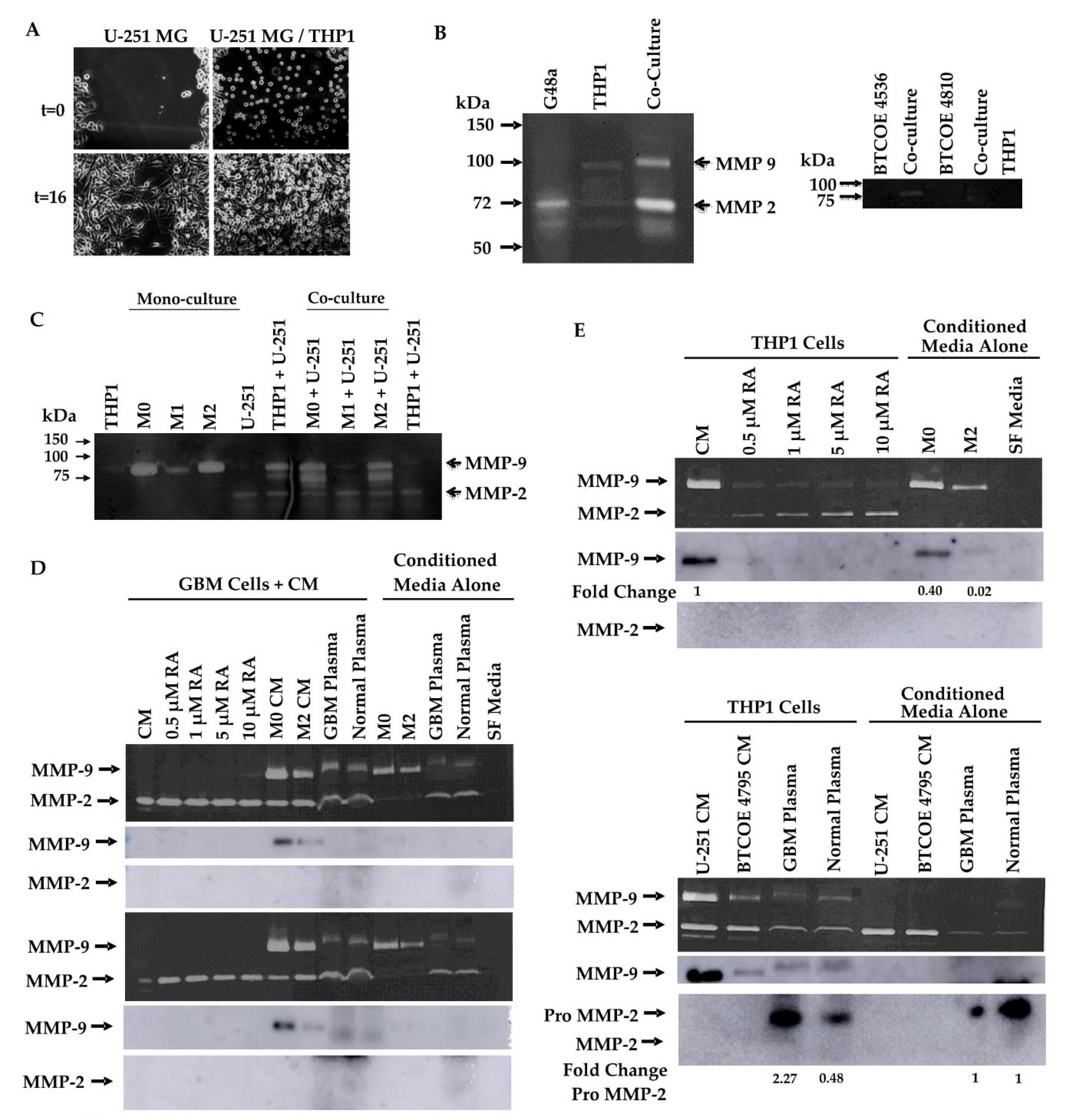
3.5. Retinoic Acid Contributes to the Invasive Potential of GBM Tumor Cells via Modulation of MMP-2 and MMP-9 in Monocytic Cells
4. Discussion
5. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Alexander, B.M.; Cloughesy, T.F. Adult Glioblastoma. J. Clin. Oncol. 2017, 35, 2402–2409. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.; Wakimoto, H.; Cahill, D.; Nahed, B.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20, S2–S8. [Google Scholar] [CrossRef] [Green Version]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849. [Google Scholar] [CrossRef]
- Ferluga, S.; Tomé, C.M.L.; Herpai, D.M.; D’Agostino, R.; Debinski, W. Simultaneous targeting of Eph receptors in glioblastoma. Oncotarget 2016, 7, 59860–59876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferluga, S.; Debinski, W. Ephs and Ephrins in malignant gliomas. Growth Factors 2014, 32, 190–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reardon, D.A.; Wen, P.Y.; Wucherpfennig, K.W.; Sampson, J.H. Immunomodulation for glioblastoma. Curr. Opin. Neurol. 2017, 30, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Dalton, D.; Pitts-Meek, S.; Keshav, S.; Figari, I.; Bradley, A.; Stewart, T. Multiple defects of immune cell function in mice with disrupted interferon-gamma genes. Science 1993, 259, 1739–1742. [Google Scholar] [CrossRef] [PubMed]
- Boldrick, J.C.; Alizadeh, A.A.; Diehn, M.; Dudoit, S.; Liu, C.L.; Belcher, C.E.; Botstein, D.; Staudt, L.M.; Brown, P.O.; Relman, D.A. Stereotyped and specific gene expression programs in human innate immune responses to bacteria. Proc. Natl. Acad. Sci. USA 2002, 99, 972–977. [Google Scholar] [CrossRef] [Green Version]
- Gieryng, A.; Pszczolkowska, D.; Walentynowicz, K.A.; Rajan, W.D.; Kaminska, B. Immune microenvironment of gliomas. Lab. Investig. 2017, 97, 498–518. [Google Scholar] [CrossRef] [Green Version]
- Locati, M.; Curtale, G.; Mantovani, A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 123–147. [Google Scholar] [CrossRef] [Green Version]
- Gabrusiewicz, K.; Rodriguez, B.; Wei, J.; Hashimoto, Y.; Healy, L.M.; Maiti, S.N.; Thomas, G.; Zhou, S.; Wang, Q.; Elakkad, A.; et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight 2016, 1. [Google Scholar] [CrossRef] [PubMed]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS–) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; Decarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, C.E.; Gordon, P.; Barker, R.N.; Wilson, H.M. The activation status of human macrophages presenting antigen determines the efficiency of Th17 responses. Immunobiology 2015, 220, 10–19. [Google Scholar] [CrossRef]
- Domingues, P.; González-Tablas, M.; Otero, Á.; Pascual, D.; Miranda, D.; Ruiz, L.; Sousa, P.; Ciudad, J.; Gonçalves, J.M.; Lopes, M.C.; et al. Tumor infiltrating immune cells in gliomas and meningiomas. Brain Behav. Immun. 2016, 53, 1–15. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Lavin, Y.; Winter, D.; Blecher-Gonen, R.; David, E.; Keren-Shaul, H.; Merad, M.; Jung, S.; Amit, I. Tissue-Resident Macrophage Enhancer Landscapes Are Shaped by the Local Microenvironment. Cell 2014, 159, 1312–1326. [Google Scholar] [CrossRef] [Green Version]
- Hambardzumyan, D.; Gutmann, D.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Markovic, D.S.; Vinnakota, K.; Chirasani, S.; Synowitz, M.; Raguet, H.; Stock, K.; Sliwa, M.; Lehmann, S.; Kalin, R.; van Rooijen, N.; et al. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc. Natl. Acad. Sci. USA 2009, 106, 12530–12535. [Google Scholar] [CrossRef] [Green Version]
- Jackson, B.; Brocker, C.; Thompson, D.C.; Black, W.; Vasiliou, K.; Nebert, D.W.; Vasiliou, V. Update on the aldehyde dehydrogenase gene (ALDH) superfamily. Hum. Genom. 2011, 5, 283–303. [Google Scholar] [CrossRef] [Green Version]
- Tomita, H.; Tanaka, K.; Tanaka, T.; Hara, A. Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget 2016, 7, 11018–11032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, J.A.; Grainger, J.; Spencer, S.; Belkaid, Y. The Role of Retinoic Acid in Tolerance and Immunity. Immunity 2011, 35, 13–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- di Masi, A.; Leboffe, L.; De Marinis, E.; Pagano, F.; Cicconi, L.; Rochette-Egly, C.; Lo-Coco, F.; Ascenzi, P.; Nervi, C. Retinoic acid receptors: From molecular mechanisms to cancer therapy. Mol. Asp. Med. 2015, 41, 1–115. [Google Scholar] [CrossRef]
- Karsy, M.; Albert, L.; Tobias, M.E.; Murali, R.; Jhanwar-Uniyal, M. All-trans retinoic acid modulates cancer stem cells of glioblastoma multiforme in an MAPK-dependent manner. Anticancer Res. 2010, 30, 4915–4920. [Google Scholar] [PubMed]
- Campos, B.; Wan, F.; Farhadi, M.; Ernst, A.; Zeppernick, F.; Tagscherer, K.E.; Ahmadi, R.; Lohr, J.; Dictus, C.; Gdynia, G.; et al. Differentiation Therapy Exerts Antitumor Effects on Stem-like Glioma Cells. Clin. Cancer Res. 2010, 16, 2715–2728. [Google Scholar] [CrossRef] [Green Version]
- Wykosky, J.; Gibo, D.M.; Stanton, C.; Debinski, W. Interleukin-13 Receptor 2, EphA2, and Fos-Related Antigen 1 as Molecular Denominators of High-Grade Astrocytomas and Specific Targets for Combinatorial Therapy. Clin. Cancer Res. 2008, 14, 199–208. [Google Scholar] [CrossRef] [Green Version]
- Debinski, W.; Gibo, D.M. Fos-Related Antigen 1 Modulates Malignant Features of Glioma Cells. Mol. Cancer Res. 2005, 3, 237–249. [Google Scholar] [CrossRef]
- Pandya, H.; Gibo, D.M.; Debinski, W. Molecular Targeting of Intracellular Compartments Specifically in Cancer Cells. Genes Cancer 2010, 1, 421–433. [Google Scholar] [CrossRef]
- Jeong, S.K.; Kim, J.S.; Lee, C.G.; Park, Y.-S.; Kim, S.D.; Yoon, S.O.; Han, D.H.; Lee, K.Y.; Jeong, M.H.; Jo, W.S. Tumor associated macrophages provide the survival resistance of tumor cells to hypoxic microenvironmental condition through IL-6 receptor-mediated signals. Immunobiology 2017, 222, 55–65. [Google Scholar] [CrossRef]
- Beauchamp, A.; Lively, M.O.; Mintz, A.; Gibo, D.; Wykosky, J.; Debinski, W. EphrinA1 Is Released in Three Forms from Cancer Cells by Matrix Metalloproteases. Mol. Cell. Biol. 2012, 32, 3253–3264. [Google Scholar] [CrossRef] [Green Version]
- Song, Q.; Hawkins, G.A.; Wudel, L.; Chou, P.; Forbes, E.; Pullikuth, A.K.; Liu, L.; Jin, G.; Craddock, L.; Topaloglu, U.; et al. Dissecting intratumoral myeloid cell plasticity by single cell RNA-seq. Cancer Med. 2019, 8, 3072–3085. [Google Scholar] [CrossRef] [Green Version]
- Tomé, C.M.L.; Palma, E.; Ferluga, S.; Lowther, W.T.; Hantgan, R.; Wykosky, J.; Debinski, W. Structural and Functional Characterization of Monomeric EphrinA1 Binding Site to EphA2 Receptor. J. Biol. Chem. 2012, 287, 14012–14022. [Google Scholar] [CrossRef] [Green Version]
- Ferluga, S.; Hantgan, R.; Goldgur, Y.; Himanen, J.P.; Nikolov, D.B.; Debinski, W. Biological and Structural Characterization of Glycosylation on Ephrin-A1, a Preferred Ligand for EphA2 Receptor Tyrosine Kinase. J. Biol. Chem. 2013, 288, 18448–18457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chanput, W.; Mes, J.J.; Wichers, H. THP-1 cell line: An in vitro cell model for immune modulation approach. Int. Immunopharmacol. 2014, 23, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Dixon, L.J.; Barnes, M.; Tang, H.; Pritchard, M.T.; Nagy, L.E. Kupffer Cells in the Liver. Compr. Physiol. 2013, 3, 785–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charafe-Jauffret, E.; Ginestier, C.; Iovino, F.; Tarpin, C.; Diebel, M.; Esterni, B.; Houvenaeghel, G.; Extra, J.-M.; Bertucci, F.; Jacquemier, J.; et al. Aldehyde Dehydrogenase 1-Positive Cancer Stem Cells Mediate Metastasis and Poor Clinical Outcome in Inflammatory Breast Cancer. Clin. Cancer Res. 2009, 16, 45–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcato, P.; Dean, C.A.; Giacomantonio, C.A.; Lee, P.W. Aldehyde dehydrogenase: Its role as a cancer stem cell marker comes down to the specific isoform. Cell Cycle 2011, 10, 1378–1384. [Google Scholar] [CrossRef] [PubMed]
- Kaipio, K.; Chen, P.; Roering, P.; Huhtinen, K.; Mikkonen, P.; Östling, P.; Lehtinen, L.; Mansuri, N.; Korpela, T.; Potdar, S.; et al. ALDH1A1-related stemness in high-grade serous ovarian cancer is a negative prognostic indicator but potentially targetable by EGFR/mTOR-PI3K/aurora kinase inhibitors. J. Pathol. 2019, 250, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Tamura, R.; Tanaka, T.; Morimoto, Y.; Kuranari, Y.; Yamamoto, Y.; Takei, J.; Murayama, Y.; Yoshida, K.; Sasaki, H. Alterations of the tumor microenvironment in glioblastoma following radiation and temozolomide with or without bevacizumab. Ann. Transl. Med. 2020, 8, 297. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Burns, T.C. Radiation-Induced Alterations in the Recurrent Glioblastoma Microenvironment: Therapeutic Implications. Front. Oncol. 2018, 8, 503. [Google Scholar] [CrossRef] [Green Version]
- Roomi, M.W.; Kalinovsky, T.; Rath, M.; Niedzwiecki, A. Modulation of MMP-2 and MMP-9 secretion by cytokines, inducers and inhibitors in human glioblastoma T-98G cells. Oncol. Rep. 2017, 37, 1907–1913. [Google Scholar] [CrossRef] [Green Version]
- Vu, H.T.; Hoang, T.X.; Kim, J.Y. All-Trans Retinoic Acid Enhances Matrix Metalloproteinase 2 Expression and Secretion in Human Myeloid Leukemia THP-1 Cells. BioMed Res. Int. 2018, 2018, 5971080. [Google Scholar] [CrossRef] [PubMed]
- Jobani, B.M.; Najafzadeh, N.; Mazani, M.; Arzanlou, M.; Vardin, M.M. Molecular mechanism and cytotoxicity of allicin and all-trans retinoic acid against CD44+ versus CD117+ melanoma cells. Phytomedicine 2018, 48, 161–169. [Google Scholar] [CrossRef]
- Poon, C.C.; Sarkar, S.; Yong, V.W.; Kelly, J.J.P. Glioblastoma-associated microglia and macrophages: Targets for therapies to improve prognosis. Brain 2017, 140, 1548–1560. [Google Scholar] [CrossRef] [Green Version]
- Penas-Prado, M.; Hess, K.R.; Fisch, M.J.; LaGrone, L.W.; Groves, M.D.; Levin, V.A.; De Groot, J.F.; Puduvalli, V.K.; Colman, H.; Volas-Redd, G.; et al. Randomized phase II adjuvant factorial study of dose-dense temozolomide alone and in combination with isotretinoin, celecoxib, and/or thalidomide for glioblastoma. Neuro-Oncology 2014, 17, 266–273. [Google Scholar] [CrossRef]
- Liu, R.-Z.; Li, S.; Garcia, E.; Glubrecht, D.D.; Poon, H.Y.; Easaw, J.C.; Godbout, R. Association between cytoplasmic CRABP2, altered retinoic acid signaling, and poor prognosis in glioblastoma. Glia 2016, 64, 963–976. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanders, S.; Herpai, D.M.; Rodriguez, A.; Huang, Y.; Chou, J.; Hsu, F.-C.; Seals, D.; Mott, R.; Miller, L.D.; Debinski, W. The Presence and Potential Role of ALDH1A2 in the Glioblastoma Microenvironment. Cells 2021, 10, 2485. https://doi.org/10.3390/cells10092485
Sanders S, Herpai DM, Rodriguez A, Huang Y, Chou J, Hsu F-C, Seals D, Mott R, Miller LD, Debinski W. The Presence and Potential Role of ALDH1A2 in the Glioblastoma Microenvironment. Cells. 2021; 10(9):2485. https://doi.org/10.3390/cells10092485
Chicago/Turabian StyleSanders, Stephanie, Denise M. Herpai, Analiz Rodriguez, Yue Huang, Jeff Chou, Fang-Chi Hsu, Darren Seals, Ryan Mott, Lance D. Miller, and Waldemar Debinski. 2021. "The Presence and Potential Role of ALDH1A2 in the Glioblastoma Microenvironment" Cells 10, no. 9: 2485. https://doi.org/10.3390/cells10092485
APA StyleSanders, S., Herpai, D. M., Rodriguez, A., Huang, Y., Chou, J., Hsu, F.-C., Seals, D., Mott, R., Miller, L. D., & Debinski, W. (2021). The Presence and Potential Role of ALDH1A2 in the Glioblastoma Microenvironment. Cells, 10(9), 2485. https://doi.org/10.3390/cells10092485