
Fibronectin: Molecular Structure, Fibrillar Structure and Mechanochemical Signaling

Abstract
1. Introduction
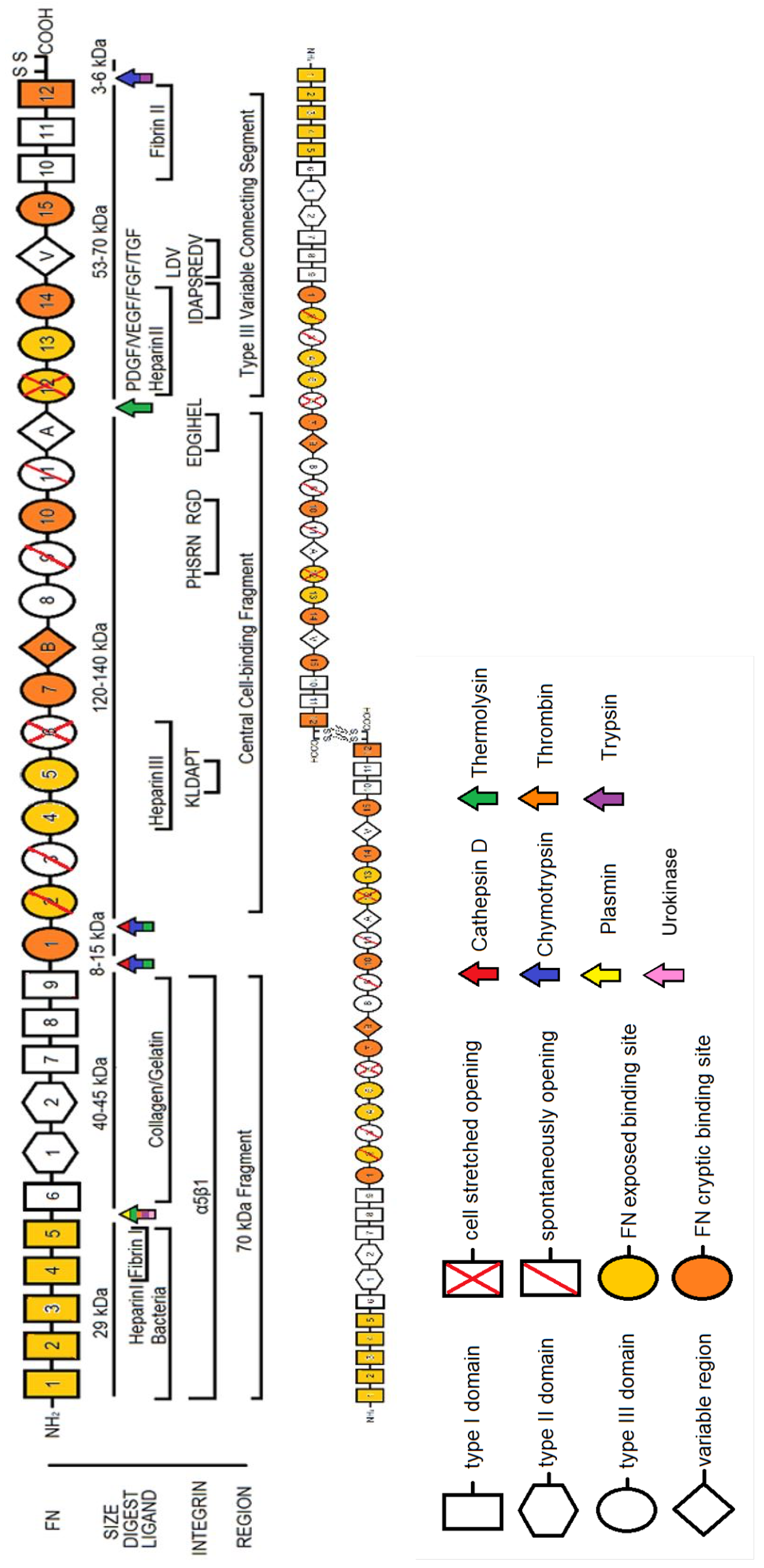
2. Fibronectin: The Molecule
2.1. Plasma FN vs. Cellular FN
2.2. The Role of Alternatively Spliced FN
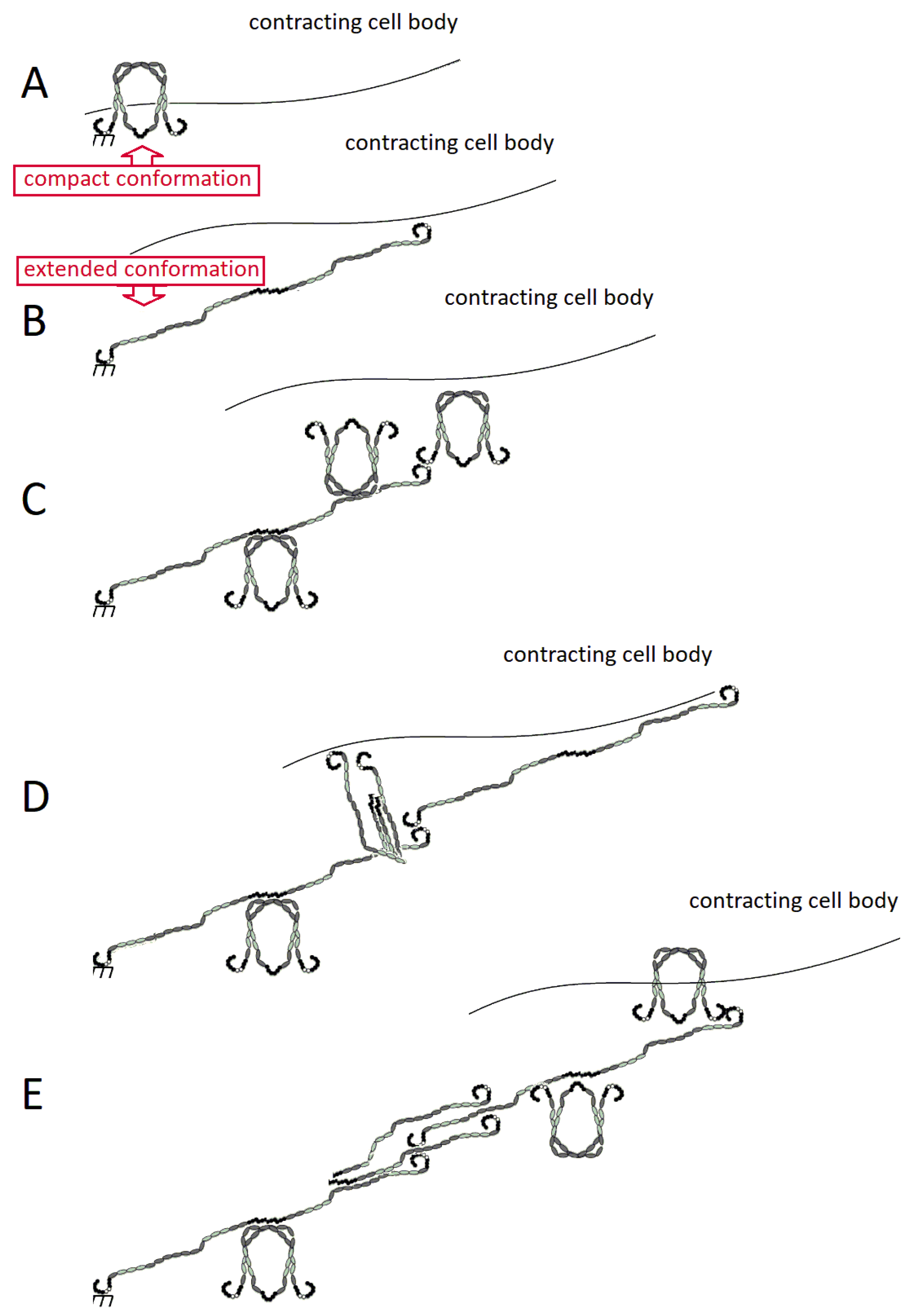
2.3. FN Molecular Conformation
3. Fibronectin: The Fibril
3.1. Assembly of FN Molecules into a Fibril
3.2. Destruction and Turnover of FN Fibrils
3.3. Inhibiting FN Fibrillogenesis
3.4. Artificially-Derived FN Fibers
3.5. Models of FN Fibrillogenesis
4. Interactions of FN with the Extracellular Matrix
5. FN Fibril Biophysics
6. Strategies for Studying FN Biophysical Properties
7. Commentary and Outlook
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Singh, P.; Carraher, C.; Schwarzbauer, J.E. Assembly of fibronectin extracellular matrix. Annu. Rev. Cell Dev. Biol. 2010, 26, 397–419. [Google Scholar] [CrossRef]
- Muncie, J.M.; Weaver, V.M. The Physical and Biochemical Properties of the Extracellular Matrix Regulate Cell Fate. Curr. Top. Dev. Biol. 2018, 130, 1–37. [Google Scholar] [CrossRef] [PubMed]
- Taha, I.N.; Naba, A. Exploring the extracellular matrix in health and disease using proteomics. Essays Biochem. 2019, 63, 417–432. [Google Scholar] [CrossRef] [PubMed]
- Weihermann, A.C.; Lorencini, M.; Brohem, C.A.; de Carvalho, C.M. Elastin structure and its involvement in skin photoageing. Int. J. Cosmet. Sci. 2017, 39, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Ricard-Blum, S. The collagen family. Cold Spring Harb. Perspect. Biol. 2011, 3, a004978. [Google Scholar] [CrossRef]
- Iozzo, R.V.; Schaefer, L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015, 42, 11–55. [Google Scholar] [CrossRef]
- Weinberg, S.H.; Mair, D.B.; Lemmon, C.A. Mechanotransduction Dynamics at the Cell-Matrix Interface. Biophys. J. 2017, 112, 1962–1974. [Google Scholar] [CrossRef]
- Kadler, K.E.; Hill, A.; Canty-Laird, E.G. Collagen fibrillogenesis: Fibronectin, integrins, and minor collagens as organizers and nucleators. Curr. Opin. Cell Biol. 2008, 20, 495–501. [Google Scholar] [CrossRef]
- Musiime, M.; Chang, J.; Hansen, U.; Kadler, K.E.; Zeltz, C.; Gullberg, D. Collagen Assembly at the Cell Surface: Dogmas Revisited. Cells 2021, 10, 662. [Google Scholar] [CrossRef]
- Yao, Y. Laminin: Loss-of-function studies. Cell. Mol. Life Sci. 2017, 74, 1095–1115. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Doyle, A.D.; Shinsato, Y.; Wang, S.; Bodendorfer, M.A.; Zheng, M.; Yamada, K.M. Basement Membrane Regulates Fibronectin Organization Using Sliding Focal Adhesions Driven by a Contractile Winch. Dev. Cell 2020, 52, 631–646.e4. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.G.; Budoff, G.; Prenner, J.L.; Schwarzbauer, J.E. Minireview: Fibronectin in retinal disease. Exp. Biol. Med. 2017, 242, 1–7. [Google Scholar] [CrossRef]
- Pankov, R.; Yamada, K.M. Fibronectin at a glance. J. Cell Sci. 2002, 115, 3861–3863. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Fibronectins; Springer: New York, NY, USA, 1990. [Google Scholar]
- Williams, M.J.; Phan, I.; Harvey, T.S.; Rostagno, A.; Gold, L.I.; Campbell, I.D. Solution structure of a pair of fibronectin type 1 modules with fibrin binding activity. J. Mol. Biol. 1994, 235, 1302–1311. [Google Scholar] [CrossRef]
- Graille, M.; Pagano, M.; Rose, T.; Ravaux, M.R.; van Tilbeurgh, H. Zinc induces structural reorganization of gelatin binding domain from human fibronectin and affects collagen binding. Structure 2010, 18, 710–718. [Google Scholar] [CrossRef]
- Pickford, A.R.; Smith, S.P.; Staunton, D.; Boyd, J.; Campbell, I.D. The hairpin structure of the (6)F1(1)F2(2)F2 fragment from human fibronectin enhances gelatin binding. EMBO J. 2001, 20, 1519–1529. [Google Scholar] [CrossRef]
- Leahy, D.J.; Aukhil, I.; Erickson, H.P. 2.0 A crystal structure of a four-domain segment of human fibronectin encompassing the RGD loop and synergy region. Cell 1996, 84, 155–164. [Google Scholar] [CrossRef]
- Schwarzbauer, J.E.; DeSimone, D.W. Fibronectins, their fibrillogenesis, and in vivo functions. Cold Spring Harb. Perspect. Biol. 2011, 3, a005041. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Craig, D.; Vogel, V.; Schulten, K. Identifying unfolding intermediates of FN-III(10) by steered molecular dynamics. J. Mol. Biol. 2002, 323, 939–950. [Google Scholar] [CrossRef]
- Lemmon, C.A.; Ohashi, T.; Erickson, H.P. Probing the folded state of fibronectin type III domains in stretched fibrils by measuring buried cysteine accessibility. J. Biol. Chem. 2011, 286, 26375–26382. [Google Scholar] [CrossRef]
- Lemmon, C.A.; Chen, C.S.; Romer, L.H. Cell traction forces direct fibronectin matrix assembly. Biophys. J. 2009, 96, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, T.; Augustus, A.M.; Erickson, H.P. Transient opening of fibronectin type III (FNIII) domains: The interaction of the third FNIII domain of FN with anastellin. Biochemistry 2009, 48, 4189–4197. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Abu-Lail, N.I.; Ohashi, T.; Clark, R.L.; Erickson, H.P.; Zauscher, S. Understanding the elasticity of fibronectin fibrils: Unfolding strengths of FN-III and GFP domains measured by single molecule force spectroscopy. Matrix Biol. 2006, 25, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, C.A.; Weinberg, S.H. Multiple Cryptic Binding Sites are Necessary for Robust Fibronectin Assembly: An in Silico Study. Sci. Rep. 2017, 7, 18061. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.M.; Kennedy, D.W. Fibroblast cellular and plasma fibronectins are similar but not identical. J. Cell Biol. 1979, 80, 492–498. [Google Scholar] [CrossRef]
- Cui, C.; Wang, S.; Myneni, V.D.; Hitomi, K.; Kaartinen, M.T. Transglutaminase activity arising from Factor XIIIA is required for stabilization and conversion of plasma fibronectin into matrix in osteoblast cultures. Bone 2014, 59, 127–138. [Google Scholar] [CrossRef]
- Kumra, H.; Sabatier, L.; Hassan, A.; Sakai, T.; Mosher, D.F.; Brinckmann, J.; Reinhardt, D.P. Roles of fibronectin isoforms in neonatal vascular development and matrix integrity. PLoS Biol. 2018, 16, e2004812. [Google Scholar] [CrossRef]
- Patten, J.; Wang, K. Fibronectin in development and wound healing. Adv. Drug Deliv. Rev. 2021, 170. [Google Scholar] [CrossRef]
- Santas, A.J.; Peterson, J.A.; Halbleib, J.L.; Craig, S.E.; Humphries, M.J.; Peters, D.M.P. Alternative splicing of the IIICS domain in fibronectin governs the role of the Heparin II domain in fibrillogenesis and cell spreading. J. Biol. Chem. 2002, 277, 13650–13658. [Google Scholar] [CrossRef]
- Lemańska-Perek, A.; Adamik, B. Fibronectin and its soluble EDA-FN isoform as biomarkers for inflammation and sepsis. Adv. Clin. Exp. Med. 2019, 28, 1561–1567. [Google Scholar] [CrossRef]
- Sens, C.; Huck, K.; Pettera, S.; Uebel, S.; Wabnitz, G.; Moser, M.; Nakchbandi, I.A. Fibronectins containing extradomain A or B enhance osteoblast differentiation via distinct integrins. J. Biol. Chem. 2017, 292, 7745–7760. [Google Scholar] [CrossRef]
- Muro, A.F.; Chauhan, A.K.; Gajovic, S.; Iaconcig, A.; Porro, F.; Stanta, G.; Baralle, F.E. Regulated splicing of the fibronectin EDA exon is essential for proper skin wound healing and normal lifespan. J. Cell Biol. 2003, 162, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Goossens, K.; Soom, A.V.; Zeveren, A.V.; Favoreel, H.; Peelman, L.J. Quantification of Fibronectin 1 (FN1) splice variants, including two novel ones, and analysis of integrins as candidate FN1 receptors in bovine preimplantation embryos. BMC Dev. Biol. 2009, 9, 1. [Google Scholar] [CrossRef]
- Fukuda, T.; Yoshida, N.; Kataoka, Y.; ichiroh Manabe, R.; Mizuno-Horikawa, Y.; Sato, M.; Kuriyama, K.; Yasui, N.; Sekiguchi, K. Mice lacking the EDB segment of fibronectin develop normally but exhibit reduced cell growth and fibronectin matrix assembly in vitro. Cancer Res. 2002, 62, 5603–5610. [Google Scholar] [PubMed]
- Humphries, M.J.; Komoriya, A.; Akiyama, S.K.; Olden, K.; Yamada, K.M. Identification of two distinct regions of the type III connecting segment of human plasma fibronectin that promote cell type-specific adhesion. J. Biol. Chem. 1987, 262, 6886–6892. [Google Scholar] [CrossRef]
- Wayner, E.A.; Garcia-Pardo, A.; Humphries, M.J.; McDonald, J.A.; Carter, W.G. Identification and characterization of the T lymphocyte adhesion receptor for an alternative cell attachment domain (CS-1) in plasma fibronectin. J. Cell Biol. 1989, 109, 1321–1330. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.L.; Hynes, R.O. Lymphoid cells recognize an alternatively spliced segment of fibronectin via the integrin receptor α4β1. Cell 1990, 60, 53–61. [Google Scholar] [CrossRef]
- Mould, A.P.; Wheldon, L.A.; Komoriya, A.; Wayner, E.A.; Yamada, K.M.; Humphries, M.J. Affinity chromatographic isolation of the melanoma adhesion receptor for the IIICS region of fibronectin and its identification as the integrin α4β1. J. Biol. Chem. 1990, 265, 4020–4024. [Google Scholar] [CrossRef]
- Mould, A.P.; Komoriya, A.; Yamada, K.M.; Humphries, M.J. The CS5 peptide is a second site in the IIICS region of fibronectin recognized by the integrin α4β1: Inhibition of α4β1 function by RGD peptide homologues. J. Biol. Chem. 1991, 266. [Google Scholar] [CrossRef]
- Mostafavi-Pour, Z.; Askari, J.A.; Whittard, J.D.; Humphries, M.J. Identification of a novel heparin-binding site in the alternatively spliced IIICS region of fibronectin: Roles of integrins and proteoglycans in cell adhesion to fibronectin splice variants. Matrix Biol. 2001, 20, 63–73. [Google Scholar] [CrossRef]
- Ohashi, T.; Lemmon, C.A.; Erickson, H.P. Fibronectin Conformation and Assembly: Analysis of Fibronectin Deletion Mutants and Fibronectin Glomerulopathy (GFND) Mutants. Biochemistry 2017, 56, 4584–4591. [Google Scholar] [CrossRef]
- Johnson, K.J.; Sage, H.; Briscoe, G.; Erickson, H.P. The compact conformation of fibronectin is determined by intramolecular ionic interactions. J. Biol. Chem. 1999, 274, 15473–15479. [Google Scholar] [CrossRef]
- Dzamba, B.J.; Peters, D.M.P. Arrangement of cellular fibronectin in noncollagenous fibrils in human fibroblast cultures. J. Cell Sci. 1991, 100, 605–612. [Google Scholar] [CrossRef]
- Früh, S.M.; Schoen, I.; Ries, J.; Vogel, V. Molecular architecture of native fibronectin fibrils. Nat. Commun. 2015, 6, 7275. [Google Scholar] [CrossRef]
- Wennerberg, K.; Lohikangas, L.; Gullberg, D.; Pfaff, M.; Johansson, S.; Fässler, R. β1 integrin-dependent and -independent polymerization of fibronectin. J. Cell Biol. 1996, 132. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.E.; Mair, D.B.; Narang, J.D.; Feleke, K.; Lemmon, C.A. Fibronectin fibrillogenesis facilitates mechano-dependent cell spreading, force generation, and nuclear size in human embryonic fibroblasts. Integr. Biol. 2015, 7, 1454–1465. [Google Scholar] [CrossRef]
- Zhong, C.; Chrzanowska-Wodnicka, M.; Brown, J.; Shaub, A.; Belkin, A.M.; Burridge, K. Rho-mediated contractility exposes a cryptic site in fibronectin and induces fibronectin matrix assembly. J. Cell Biol. 1998, 141, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.E.; Weinberg, S.H.; Lemmon, C.A. Mechanochemical Signaling of the Extracellular Matrix in Epithelial-Mesenchymal Transition. Front. Cell Dev. Biol. 2019, 7, 135. [Google Scholar] [CrossRef]
- Schwarzbauer, J.E. Identification of the fibronectin sequences required for assembly of a fibrillar matrix. J. Cell Biol. 1991, 113, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- Sechler, J.L.; Takada, Y.; Schwarzbauer, J.E. Altered rate of fibronectin matrix assembly by deletion of the first type III repeats. J. Cell Biol. 1996, 134, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, T.; Erickson, H.P. Fibronectin aggregation and assembly: The unfolding of the second fibronectin type III domain. J. Biol. Chem. 2011, 286, 39188–39199. [Google Scholar] [CrossRef]
- McKeown-Longo, P.J.; Mosher, D.F. Interaction of the 70,000-mol-wt amino-terminal fragment of fibronectin with the matrix-assembly receptor of fibroblasts. J. Cell Biol. 1985, 100, 364–374. [Google Scholar] [CrossRef]
- Maurer, L.M.; Ma, W.; Eickstaedt, N.L.; Johnson, I.A.; Tomasini-Johansson, B.R.; Annis, D.S.; Mosher, D.F. Ligation of the fibrin-binding domain by β-strand addition is sufficient for expansion of soluble fibronectin. J. Biol. Chem. 2012, 287, 13303–13312. [Google Scholar] [CrossRef]
- Shi, F.; Sottile, J. MT1-MMP regulates the turnover and endocytosis of extracellular matrix fibronectin. J. Cell Sci. 2011, 124, 4039–4050. [Google Scholar] [CrossRef]
- Steffensen, B.; Chen, Z.; Pal, S.; Mikhailova, M.; Su, J.; Wang, Y.; Xu, X. Fragmentation of fibronectin by inherent autolytic and matrix metalloproteinase activities. Matrix Biol. 2011, 30, 34–42. [Google Scholar] [CrossRef][Green Version]
- Maqueda, A.; Moyano, J.V.; Cerro, M.H.D.; Peters, D.M.; Garcia-Pardo, A. The heparin III-binding domain of fibronectin (III4-5 repeats) binds to fibronectin and inhibits fibronectin matrix assembly. Matrix Biol. 2007, 26, 642–651. [Google Scholar] [CrossRef]
- Bultmann, H.; Santas, A.J.; Peters, D.M.P. Fibronectin fibrillogenesis involves the heparin II binding domain of fibronectin. J. Biol. Chem. 1998, 273, 2601–2609. [Google Scholar] [CrossRef]
- Ohashi, T.; Erickson, H.P. Domain unfolding plays a role in superfibronectin formation. J. Biol. Chem. 2005, 280, 39143–39151. [Google Scholar] [CrossRef]
- Yi, M.; Ruoslahti, E. A fibronectin fragment inhibits tumor growth, angiogenesis, and metastasis. Proc. Natl. Acad. Sci. USA 2001, 98, 620–624. [Google Scholar] [CrossRef]
- Ohashi, T.; Kiehart, D.P.; Erickson, H.P. Dynamics and elasticity of the fibronectin matrix in living cell culture visualized by fibronectin–green fluorescent protein. Proc. Natl. Acad. Sci. USA 1999, 96, 2153–2158. [Google Scholar] [CrossRef]
- Ruoslahti, E. Fibronectin in cell adhesion and invasion. Cancer Metastasis Rev. 1984, 3, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Little, W.C.; Smith, M.L.; Ebneter, U.; Vogel, V. Assay to mechanically tune and optically probe fibrillar fibronectin conformations from fully relaxed to breakage. Matrix Biol. 2008, 27, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Klotzsch, E.; Smith, M.L.; Kubow, K.E.; Muntwyler, S.; Little, W.C.; Beyeler, F.; Gourdon, D.; Nelson, B.J.; Vogel, V. Fibronectin forms the most extensible biological fibers displaying switchable force-exposed cryptic binding sites. Proc. Natl. Acad. Sci. USA 2009, 106, 18267–18272. [Google Scholar] [CrossRef] [PubMed]
- Peleg, O.; Savin, T.; Kolmakov, G.V.; Salib, I.G.; Balazs, A.C.; Kröger, M.; Vogel, V. Fibers with integrated mechanochemical switches: Minimalistic design principles derived from fibronectin. Biophys. J. 2012, 103, 1909–1918. [Google Scholar] [CrossRef] [PubMed]
- Chabria, M.; Hertig, S.; Smith, M.L.; Vogel, V. Stretching fibronectin fibres disrupts binding of bacterial adhesins by physically destroying an epitope. Nat. Commun. 2010, 1, 135. [Google Scholar] [CrossRef]
- Bradshaw, M.J.; Cheung, M.C.; Ehrlich, D.J.; Smith, M.L. Using Molecular Mechanics to Predict Bulk Material Properties of Fibronectin Fibers. PLoS Comput. Biol. 2012, 8, e1002845. [Google Scholar] [CrossRef]
- Martino, M.M.; Hubbell, J.A. The 12th–14th type III repeats of fibronectin function as a highly promiscuous growth factor-binding domain. FASEB J. 2010, 24, 4711–4721. [Google Scholar] [CrossRef]
- Griggs, L.A.; Hassan, N.T.; Malik, R.S.; Griffin, B.P.; Martinez, B.A.; Elmore, L.W.; Lemmon, C.A. Fibronectin fibrils regulate TGF-β1-induced Epithelial-Mesenchymal Transition. Matrix Biol. 2017, 60, 157–175. [Google Scholar] [CrossRef]
- Sible, J.C.; Eriksson, E.; Smith, S.P.; Oliver, N. Fibronectin gene expression differs in normal and abnormal human wound healing. Wound Repair Regen. 1994, 2. [Google Scholar] [CrossRef]
- Bae, Y.K.; Kim, A.; Kim, M.K.; Choi, J.E.; Kang, S.H.; Lee, S.J. Fibronectin expression in carcinoma cells correlates with tumor aggressiveness and poor clinical outcome in patients with invasive breast cancer. Hum. Pathol. 2013, 44, 2028–2037. [Google Scholar] [CrossRef]
- Zollinger, A.J.; Smith, M.L. Fibronectin, the extracellular glue. Matrix Biol. 2017, 60-61, 27–37. [Google Scholar] [CrossRef]
- Hynes, R.O.; Naba, A.; Lu, P.; Takai, K.; Weaver, V.M.; Huttenlocher, A.; Horwitz, A.R.; Geiger, B.; Yamada, K.M.; Adams, J.C.; et al. Overview of the Matrisome—An Inventory of. CSH Perspect. 2012, 4, 4. [Google Scholar] [CrossRef]
- Shimizu, M.; Minakuchi, K.; Moon, M.; Koga, J. Difference in interaction of fibronectin with type I collagen and type IV collagen. Biochim. Biophys. Acta 1997, 1339, 53–61. [Google Scholar] [CrossRef]
- Sevilla, C.A.; Dalecki, D.; Hocking, D.C. Regional Fibronectin and Collagen Fibril Co-Assembly Directs Cell Proliferation and Microtissue Morphology. PLoS ONE 2013, 8, e77316. [Google Scholar] [CrossRef]
- Paten, J.A.; Martin, C.L.; Wanis, J.T.; Siadat, S.M.; Figueroa-Navedo, A.M.; Ruberti, J.W.; Deravi, L.F. Molecular Interactions between Collagen and Fibronectin: A Reciprocal Relationship that Regulates De Novo Fibrillogenesis. Chem 2019, 5, 2126–2145. [Google Scholar] [CrossRef]
- Shi, F.; Harman, J.; Fujiwara, K.; Sottile, J. Collagen I matrix turnover is regulated by fibronectin polymerization. Am. J. Physiol. Cell Physiol. 2010, 298, C1265–C1275. [Google Scholar] [CrossRef] [PubMed]
- Velling, T.; Risteli, J.; Wennerberg, K.; Mosher, D.F.; Johansson, S. Polymerization of type I and III collagens is dependent on fibronectin and enhanced by integrins α11β1 and α2β1. J. Biol. Chem. 2002, 277. [Google Scholar] [CrossRef] [PubMed]
- Dzamba, B.J.; Wu, H.; Jaenisch, R.; Peters, D.M. Fibronectin binding site in type I collagen regulates fibronectin fibril formation. J. Cell Biol. 1993, 121, 1165–1172. [Google Scholar] [CrossRef]
- Sottile, J.; Shi, F.; Rublyevska, I.; Chiang, H.Y.; Lust, J.; Chandler, J. Fibronectin-dependent collagen I deposition modulates the cell response to fibronectin. Am. J. Physiol. Cell Physiol. 2007, 293, C1934–C1946. [Google Scholar] [CrossRef]
- Calabro, N.E.; Kristofik, N.J.; Kyriakides, T.R. Thrombospondin-2 and extracellular matrix assembly. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 2396–2402. [Google Scholar] [CrossRef] [PubMed]
- Kudo, A.; Kii, I. Periostin function in communication with extracellular matrices. J. Cell Commun. Signal. 2018, 12, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Fogelgren, B.; Polgár, N.; Szauter, K.M.; Ujfaludi, Z.; Laczkó, R.; Fong, K.S.K.; Csiszar, K. Cellular fibronectin binds to lysyl oxidase with high affinity and is critical for its proteolytic activation. J. Biol. Chem. 2005, 280, 24690–24697. [Google Scholar] [CrossRef] [PubMed]
- Midwood, K.S.; Chiquet, M.; Tucker, R.P.; Orend, G. Tenascin-C at a glance. J. Cell Sci. 2016, 129, 4321–4327. [Google Scholar] [CrossRef] [PubMed]
- Tremble, P.; Chiquet-Ehrismann, R.; Werb, Z. The extracellular matrix ligands fibronectin and tenascin collaborate in regulating collagenase gene expression in fibroblasts. Mol. Biol. Cell 1994, 5, 439–453. [Google Scholar] [CrossRef]
- Weisel, J.W.; Litvinov, R.I. Fibrin formation, structure and properties. Sub-Cell. Biochem. 2017, 82, 405–456. [Google Scholar] [CrossRef]
- Kinsey, R.; Williamson, M.R.; Chaudhry, S.; Mellody, K.T.; McGovern, A.; Takahashi, S.; Shuttleworth, C.A.; Kielty, C.M. Fibrillin-1 microfibril deposition is dependent on fibronectin assembly. J. Cell Sci. 2008, 121, 2696–2704. [Google Scholar] [CrossRef]
- Sabatier, L.; Chen, D.; Fagotto-Kaufmann, C.; Hubmacher, D.; McKee, M.D.; Annis, D.S.; Osher, D.F.; Reinhardt, D.P. Fibrillin assembly requires fibronectin. Mol. Biol. Cell 2009, 20, 846–858. [Google Scholar] [CrossRef]
- Sawicka, K.M.; Seeliger, M.; Musaev, T.; Macri, L.K.; Clark, R.A.F. Fibronectin Interaction and Enhancement of Growth Factors: Importance for Wound Healing. Adv. Wound Care 2015, 4, 469–478. [Google Scholar] [CrossRef]
- Vehviläinen, P.; Hyytiäinen, M.; Keski-Oja, J. Matrix association of latent TGF-beta binding protein-2 (LTBP-2) is dependent on fibrillin-1. J. Cell. Physiol. 2009, 221, 586–593. [Google Scholar] [CrossRef]
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N.K. Extracellular matrix structure. Adv. Drug Deliv. Rev. 2016, 97, 4–27. [Google Scholar] [CrossRef]
- Lundmark, K.; Tran, P.K.; Kinsella, M.G.; Clowes, A.W.; Wight, T.N.; Hedin, U. Perlecan inhibits smooth muscle cell adhesion to fibronectin: Role of heparan sulfate. J. Cell. Physiol. 2001, 188, 67–74. [Google Scholar] [CrossRef]
- Tomasini-Johansson, B.R.; Kaufman, N.R.; Ensenberger, M.G.; Ozeri, V.; Hanski, E.; Mosher, D.F. A 49-Residue Peptide from Adhesin F1 of Streptococcus pyogenes Inhibits Fibronectin Matrix Assembly. J. Biol. Chem. 2001, 276, 23430–23439. [Google Scholar] [CrossRef]
- Schwarz-Linek, U.; Werner, J.M.; Pickford, A.R.; Gurusiddappa, S.; Ewa, J.H.K.; Pilka, S.; Briggs, J.A.; Gough, T.S.; Höök, M.; Campbell, I.D.; et al. Pathogenic bacteria attach to human fibronectin through a tandem β-zipper. Nature 2003, 423, 177–181. [Google Scholar] [CrossRef]
- Schwarz-Linek, U.; Höök, M.; Potts, J.R. The molecular basis of fibronectin-mediated bacterial adherence to host cells. Mol. Microbiol. 2004, 52, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Marjenberg, Z.R.; Ellis, I.R.; Hagan, R.M.; Prabhakaran, S.; Höök, M.; Talay, S.R.; Potts, J.R.; Staunton, D.; Schwarz-Linek, U. Cooperative binding and activation of fibronectin by a bacterial surface protein. J. Biol. Chem. 2011, 286, 1884–1894. [Google Scholar] [CrossRef] [PubMed]
- Obara, M.; Kang, M.S.; Yamada, K.M. Site-directed mutagenesis of the cell-binding domain of human fibronectin: Separable, synergistic sites mediate adhesive function. Cell 1988, 53, 649–657. [Google Scholar] [CrossRef]
- Bowditch, R.D.; Hariharan, M.; Tominna, E.F.; Smith, J.W.; Yamada, K.M.; Getzoff, E.D.; Ginsberg, M.H. Identification of a novel integrin binding site in fibronectin. Differential utilization by β3 integrins. J. Biol. Chem. 1994, 269. [Google Scholar] [CrossRef]
- Moretti, L.; Strohmeyer, N.; Jones, M.; Barker, T. Differential Engagement of The Integrin Binding Domain of Fibronectin Impacts Fibroblast Behavior. FASEB J. 2021, 35. [Google Scholar] [CrossRef]
- Carter, S.B. Haptotaxis and the mechanism of cell motility. Nature 1967, 213, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.M.; Wang, H.B.; Dembo, M.; Wang, Y.L. Cell movement is guided by the rigidity of the substrate. Biophys. J. 2000, 79, 144–152. [Google Scholar] [CrossRef]
- Pieuchot, L.; Marteau, J.; Guignandon, A.; Santos, T.D.; Brigaud, I.; Chauvy, P.F.; Cloatre, T.; Ponche, A.; Petithory, T.; Rougerie, P.; et al. Curvotaxis directs cell migration through cell-scale curvature landscapes. Nat. Commun. 2018, 9, 3995. [Google Scholar] [CrossRef]
- Roca-Cusachs, P.; Sunyer, R.; Trepat, X. Mechanical guidance of cell migration: Lessons from chemotaxis. Curr. Opin. Cell Biol. 2013, 25, 543–549. [Google Scholar] [CrossRef]
- Chen, C.S. Mechanotransduction—A field pulling together? J. Cell Sci. 2008, 121, 3285–3292. [Google Scholar] [CrossRef] [PubMed]
- Kubow, K.E.; Vukmirovic, R.; Zhe, L.; Klotzsch, E.; Smith, M.L.; Gourdon, D.; Luna, S.; Vogel, V. Mechanical forces regulate the interactions of fibronectin and collagen i in extracellular matrix. Nat. Commun. 2015, 6, 8026. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, M.J.; Hoffmann, G.A.; Wong, J.Y.; Smith, M.L. Fibronectin fiber creep under constant force loading. Acta Biomater. 2019, 88, 78–85. [Google Scholar] [CrossRef]
- DeMali, K.A.; Sun, X.; Bui, G.A. Force transmission at cell-cell and cell-matrix adhesions. Biochemistry 2014, 53, 7706–7717. [Google Scholar] [CrossRef] [PubMed]
- Manibog, K.; Li, H.; Rakshit, S.; Sivasankar, S. Resolving the molecular mechanism of cadherin catch bond formation. Nat. Commun. 2014, 5, 3941. [Google Scholar] [CrossRef] [PubMed]
- Baneyx, G.; Baugh, L.; Vogel, V. Fibronectin extension and unfolding within cell matrix fibrils controlled by cytoskeletal tension. Proc. Natl. Acad. Sci. USA 2002, 99, 5139–5143. [Google Scholar] [CrossRef] [PubMed]
- Erickson, H.P. Protein unfolding under isometric tension—What force can integrins generate, and can it unfold FNIII domains? Curr. Opin. Struct. Biol. 2017, 42, 98–105. [Google Scholar] [CrossRef]
- Li, D.; Colin-York, H.; Barbieri, L.; Javanmardi, Y.; Guo, Y.; Korobchevskaya, K.; Moeendarbary, E.; Li, D.; Fritzsche, M. Astigmatic traction force microscopy (aTFM). Nat. Commun. 2021, 12, 2168. [Google Scholar] [CrossRef] [PubMed]
- Kron, S.J.; Spudich, J.A. Fluorescent actin filaments move on myosin fixed to a glass surface. Proc. Natl. Acad. Sci. USA 1986, 83, 6272–6276. [Google Scholar] [CrossRef] [PubMed]
- Longo, P.J.M.; Mosher, D.F. Binding of plasma fibronectin to cell layers of human skin fibroblasts. J. Cell Biol. 1983, 97, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Wheatley, B.B. Investigating Passive Muscle Mechanics With Biaxial Stretch. Front. Physiol. 2020, 11, 1021. [Google Scholar] [CrossRef]
- Laurence, D.W.; Homburg, H.; Yan, F.; Tang, Q.; Fung, K.M.; Bohnstedt, B.N.; Holzapfel, G.A.; Lee, C.H. A pilot study on biaxial mechanical, collagen microstructural, and morphological characterizations of a resected human intracranial aneurysm tissue. Sci. Rep. 2021, 11, 3525. [Google Scholar] [CrossRef]
- Deravi, L.F.; Su, T.; Paten, J.A.; Ruberti, J.W.; Bertoldi, K.; Parker, K.K. Differential contributions of conformation extension and domain unfolding to properties of fibronectin nanotextiles. Nano Lett. 2012, 12, 5587–5592. [Google Scholar] [CrossRef]
- Ren, K.; Gao, J.; Han, D. AFM Force Relaxation Curve Reveals that the Decrease of Membrane Tension Is the Essential Reason for the Softening of Cancer Cells. Front. Cell Dev. Biol. 2021, 9, 663021. [Google Scholar] [CrossRef]
- Gaiko-Shcherbak, A.; Eschenbruch, J.; Kronenberg, N.M.; Teske, M.; Wolters, B.; Springer, R.; Gather, M.C.; Merkel, R.; Hoffmann, B.; Noetzel, E. Cell force-driven basement membrane disruption fuels egf-and stiffness-induced invasive cell dissemination from benign breast gland acini. Int. J. Mol. Sci. 2021, 22, 3962. [Google Scholar] [CrossRef]
- Griffin, B.P.; Largaespada, C.J.; Rinaldi, N.A.; Lemmon, C.A. A novel method for quantifying traction forces on hexagonal micropatterned protein features on deformable poly-dimethyl siloxane sheets. MethodsX 2019, 6, 1343–1352. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Cell-Derived | Artificially-Derived | |
|---|---|---|
| Experimental | • 5–1000 nm, heterogeneous diameters [44] | • 2–5 µm, homogeneous diameters |
| • <5 to >50 µm length [61] | • 1–2 cm length [63] | |
| • 3–4-fold stretch [61] | • 5–8-fold stretch [63,64] | |
| • preliminary mechanical data available for elastic, viscoelastic, and cyclical properties (unpublished) | • high, reversible strain; low rupture events [63] | |
| • formed by cell secretion and stretch via self affinity [24,48] | • formed from surface tension/air-liquid interface [63,64] | |
| • isoforms determined by soluble content and alternative splicing | • millions of FN molecules, isoforms determined by solution preparation [63] | |
| • insoluble, typically remain submerged in aqeuous environment [62] | • insoluble, may be dried in preparation [63] | |
| Computational | • 10–50 nm, hexagonally packed cross-section with randomized orientation depending on FN molecule spring configuration [7] | • density considered over cross-section, often organized as small clusters of linearized chains with cylindrical geometries [65] |
| • 1000–2000 nm lengths [7,25] | • lengths measured by bond stretch, limited by force laws [65] | |
| • 2–3-fold stretch [7,25] | • stretch set by stiffness parameter and force applied [65] | |
| • distinct subtype populations predicted [25] | • demonstrates stress relaxation with domain extension with destabilized drops, dependent on number of neighboring bonds [65] | |
| • modeled as a different number of springs in series with unique stiffness values [7,25] | • designed as series of domain repeats [65] | |
| • hundreds of molecules, isoforms set as combinations of springs [7,25] | • less than 100 molecules [65] | |
| • n number of integrin clutch states with reversible binding [7,25] | • random FN–FN domain interaction within the fibril during stretch [65] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dalton, C.J.; Lemmon, C.A. Fibronectin: Molecular Structure, Fibrillar Structure and Mechanochemical Signaling. Cells 2021, 10, 2443. https://doi.org/10.3390/cells10092443
Dalton CJ, Lemmon CA. Fibronectin: Molecular Structure, Fibrillar Structure and Mechanochemical Signaling. Cells. 2021; 10(9):2443. https://doi.org/10.3390/cells10092443
Chicago/Turabian StyleDalton, Caleb J., and Christopher A. Lemmon. 2021. "Fibronectin: Molecular Structure, Fibrillar Structure and Mechanochemical Signaling" Cells 10, no. 9: 2443. https://doi.org/10.3390/cells10092443
APA StyleDalton, C. J., & Lemmon, C. A. (2021). Fibronectin: Molecular Structure, Fibrillar Structure and Mechanochemical Signaling. Cells, 10(9), 2443. https://doi.org/10.3390/cells10092443