
Epigenetic Modifications Associated with Maternal Anxiety during Pregnancy and Children’s Behavioral Measures

 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design and Participants
2.2. Questionnaires Measuring Maternal Anxiety and Children’s Behavioral Measures
2.3. Covariates
2.4. Buccal Cells Collection, DNA Extraction, and Bisulfite Treatment
2.5. Bisulfite (BS) Specific PCR
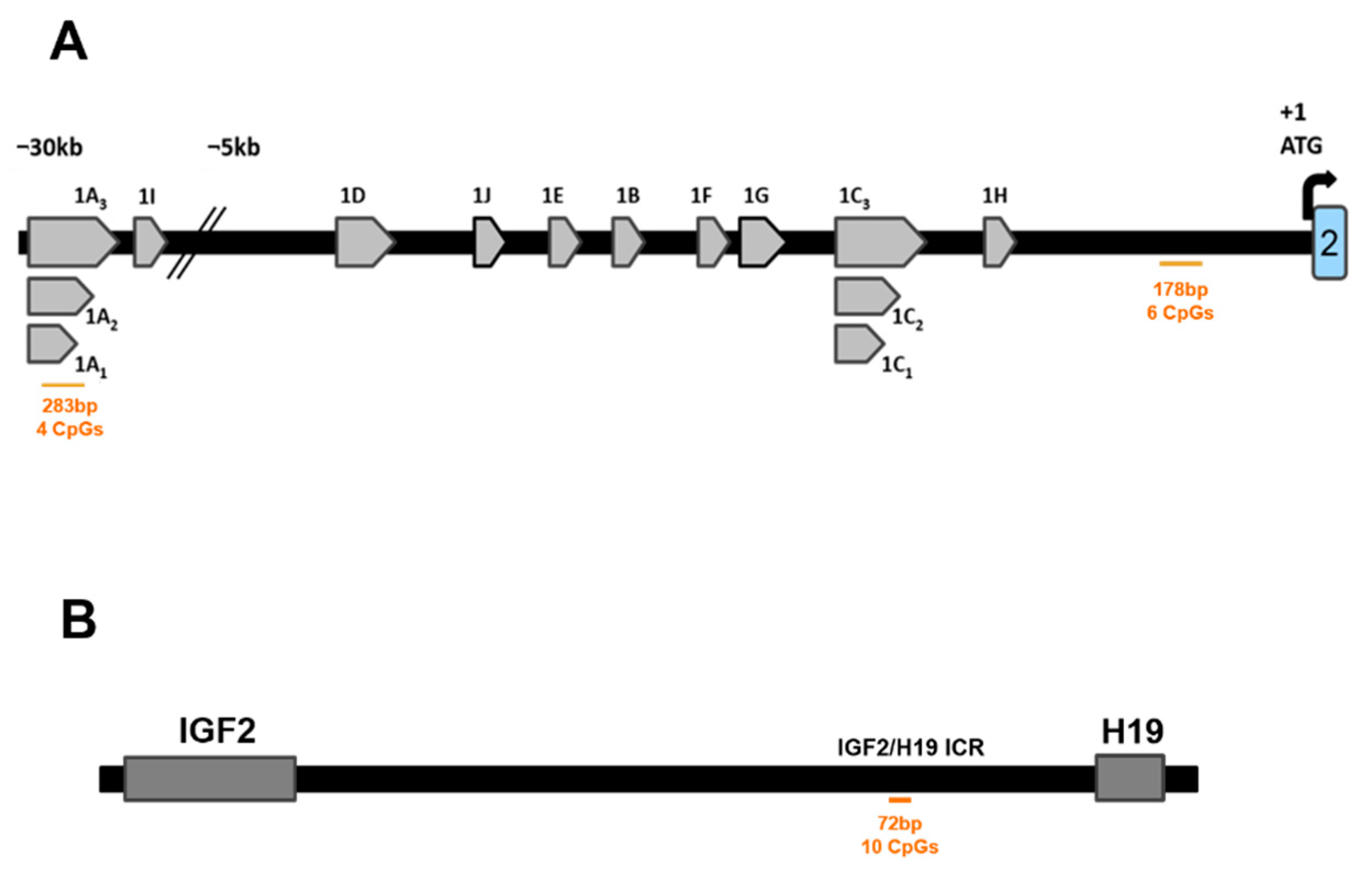
2.5.1. NR3C1
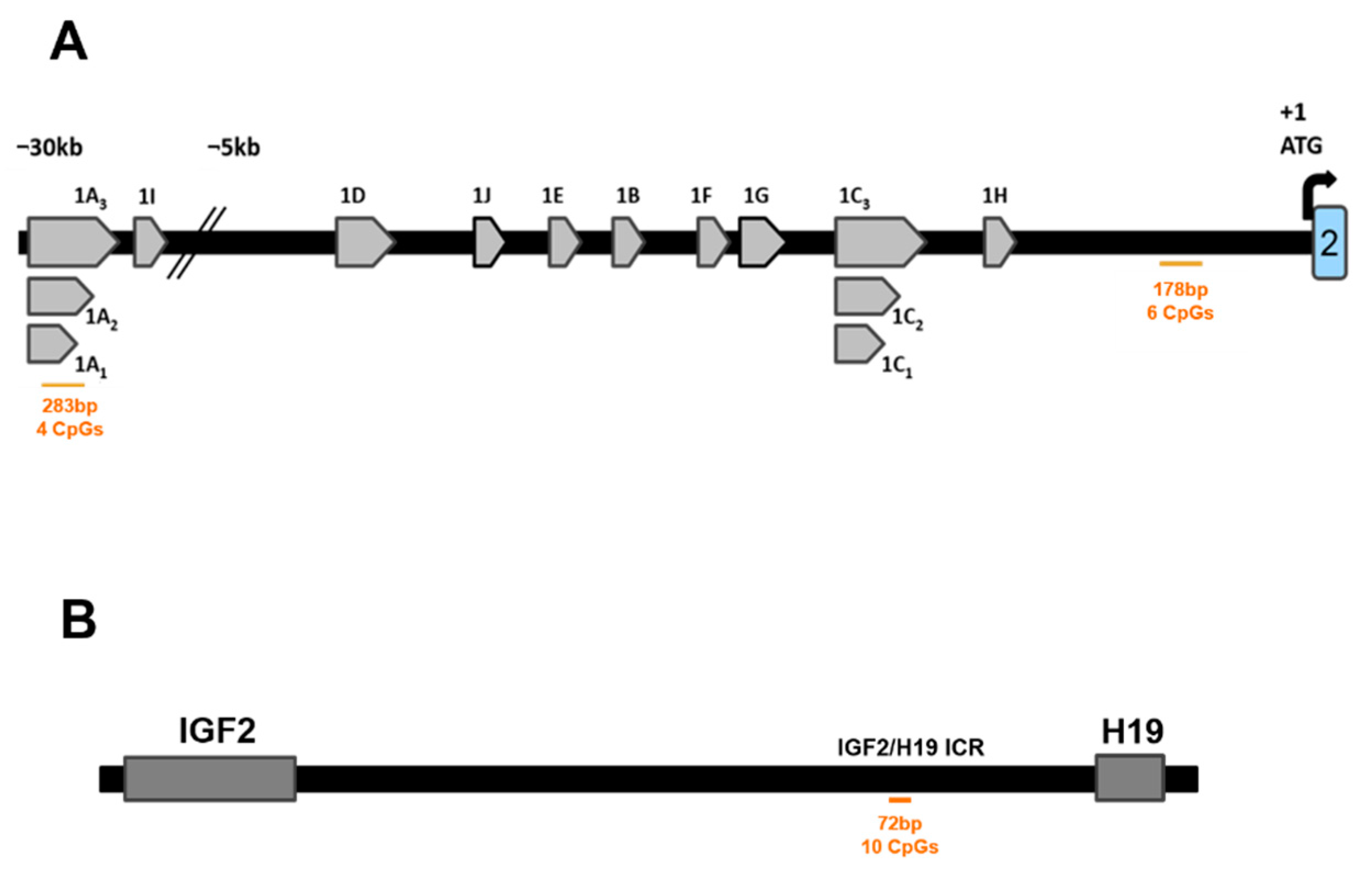
2.5.2. IGF2/H19 Imprinting Control Region (IGF2/H19 ICR)
2.5.3. LINE1 Motifs
2.6. Sequencing and Data Processing
2.7. Statistical Analyses
3. Results
3.1. Participants’ Characteristics; Descriptive Analysis
3.2. Association of Maternal Anxiety with Child’s Buccal DNA Methylation Level
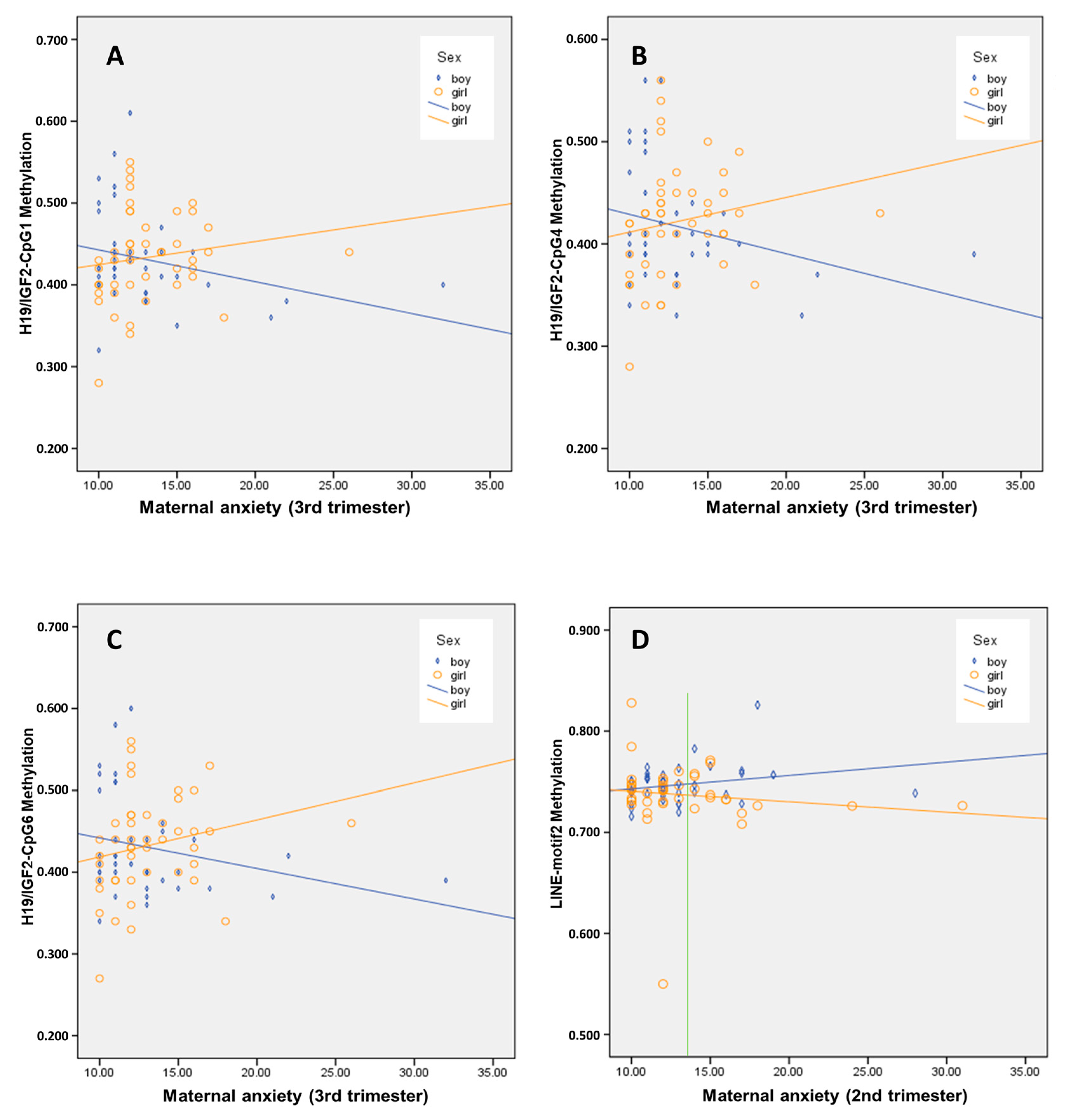
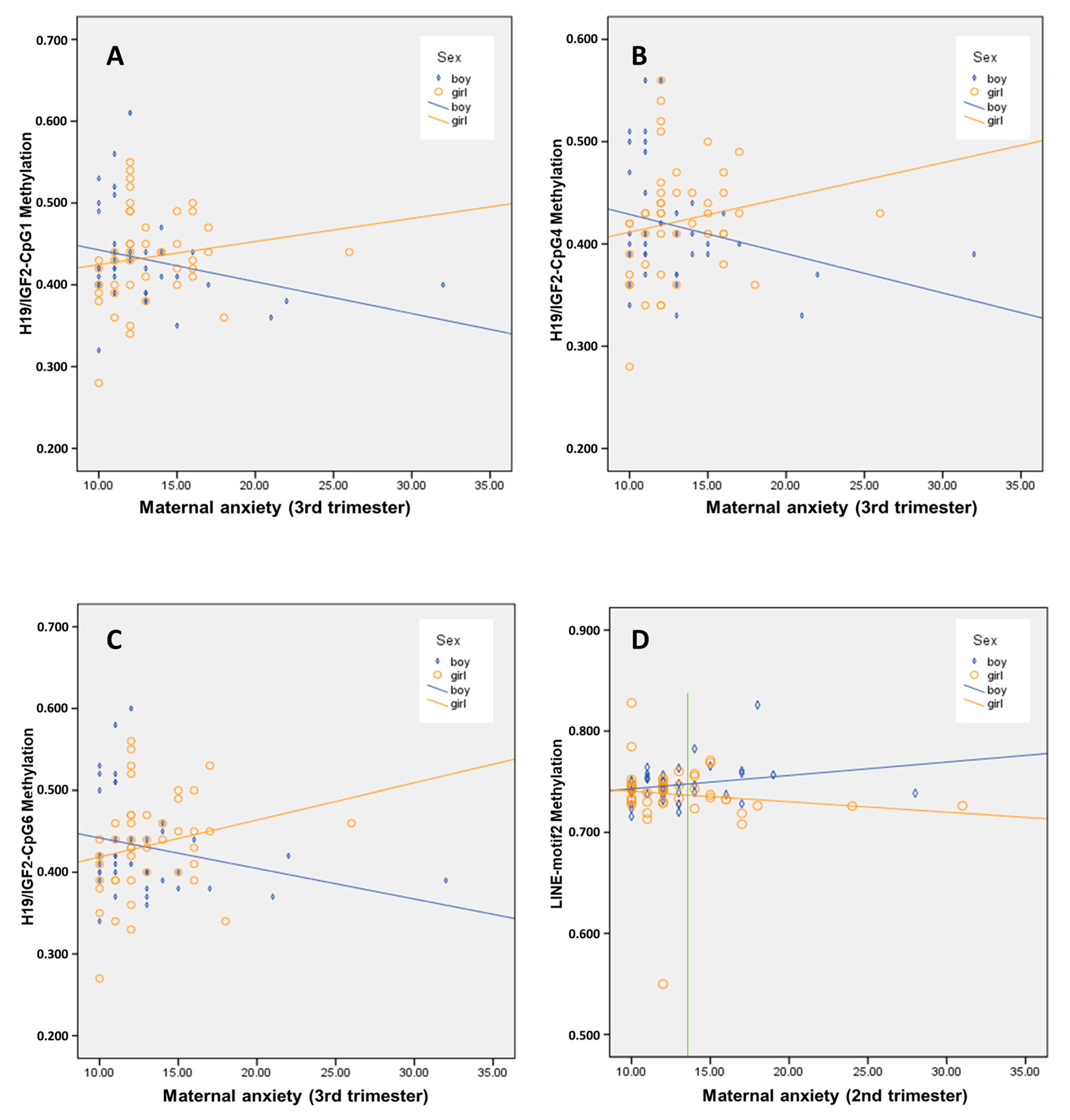
3.3. Interaction Effect of Sex on the Association between Maternal Anxiety and Child’s Buccal DNA Methylation Level
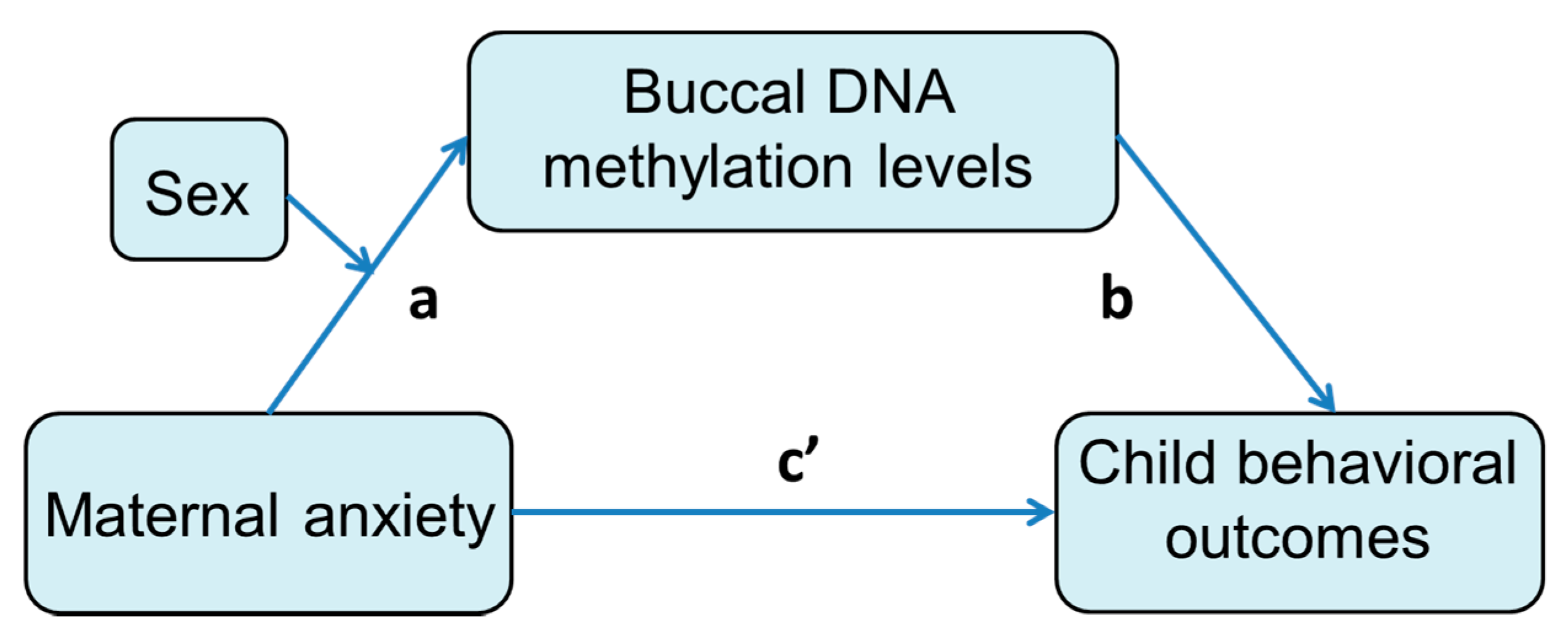
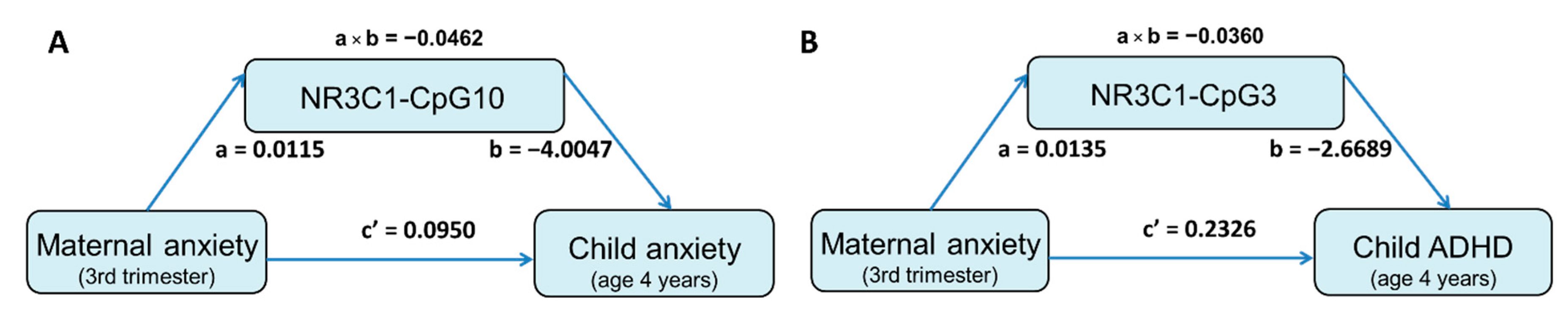
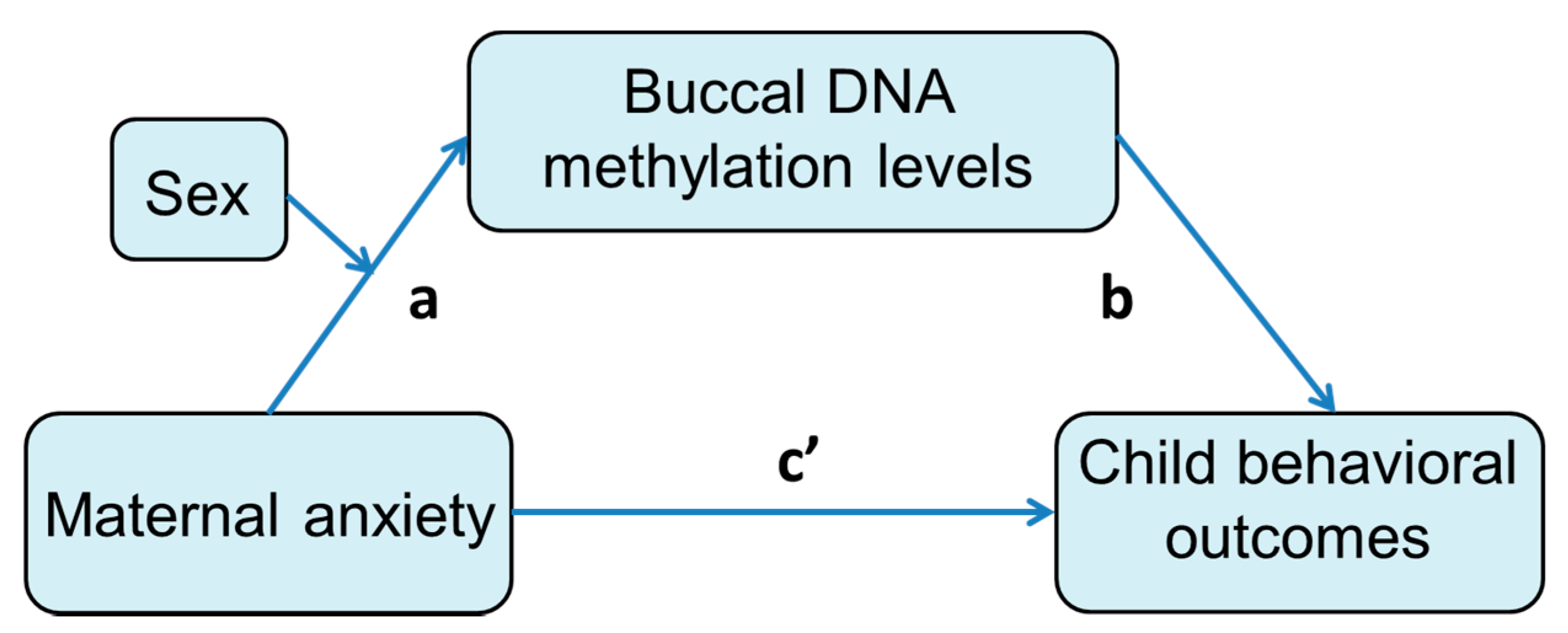
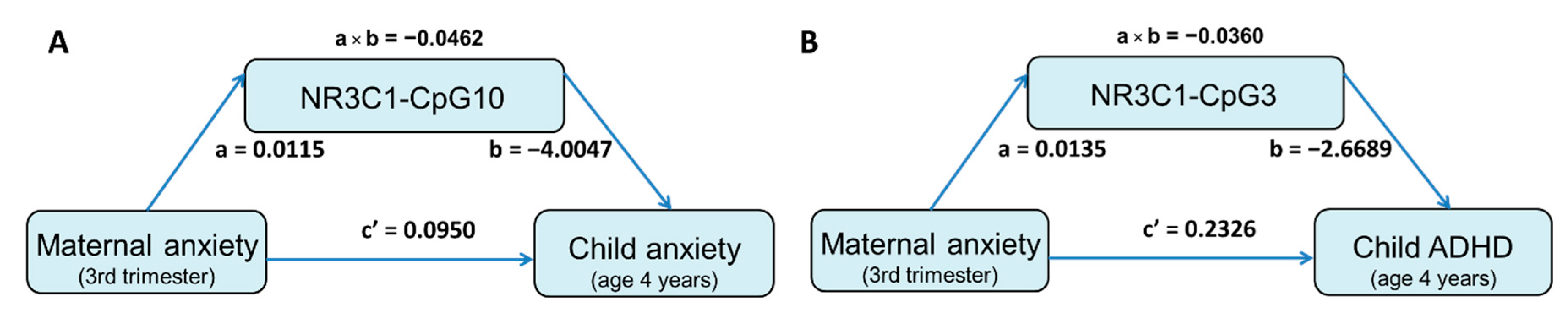
3.4. Mediation Effect of DNA Methylation Level on the Association between Prenatal Maternal Anxiety and Child Behavioral Measures
3.5. Sensitivity Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barter, J.D.; Foster, T.C. Aging in the brain: New roles of epigenetics in cognitive decline. Neuroscientist 2018, 24, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Lardenoije, R.; Iatrou, A.; Kenis, G.; Kompotis, K.; Steinbusch, H.W.; Mastroeni, D.; Coleman, P.; Lemere, C.A.; Hof, P.R.; van den Hove, D.L.; et al. The epigenetics of aging and neurodegeneration. Prog. Neurobiol. 2015, 131, 21–64. [Google Scholar] [CrossRef]
- Daskalakis, N.P.; Yehuda, R. Site-specific methylation changes in the glucocorticoid receptor exon 1f promoter in relation to life adversity: Systematic review of contributing factors. Front. Neurosci. 2014, 8, 369. [Google Scholar] [CrossRef] [Green Version]
- Cao-Lei, L.; de Rooij, S.R.; King, S.; Matthews, S.G.; Metz, G.A.S.; Roseboom, T.J.; Szyf, M. Prenatal stress and epigenetics. Neurosci. Biobehav. Rev. 2020, 117, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Gluckman, P.D.; Hanson, M.A. Living with the past: Evolution, development, and patterns of disease. Science 2004, 305, 1733–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, D.J. The fetal and infant origins of adult disease. BMJ 1990, 301, 1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontein-Kuipers, Y.J.; Nieuwenhuijze, M.J.; Ausems, M.; Budé, L.; de Vries, R. Antenatal interventions to reduce maternal distress: A systematic review and meta-analysis of randomised trials. BJOG 2014, 121, 389–397. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Maternal Mental Health and Child Health and Development in Low and Middle Income Countries: Report of the Meeting 30 January–1 February 2008; WHO: Geneva, Switzerland, 2008. [Google Scholar]
- Dennis, C.L.; Falah-Hassani, K.; Shiri, R. Prevalence of antenatal and postnatal anxiety: Systematic review and meta-analysis. Br. J. Psychiatry 2017, 21, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Loomans, E.M.; van Dijk, A.E.; Vrijkotte, T.G.; van Eijsden, M.; Stronks, K.; Gemke, R.J.; Van den Bergh, B.R. Psychosocial stress during pregnancy is related to adverse birth outcomes: Results from a large multi-ethnic community-based birth cohort. Eur. J. Public Health 2013, 23, 485–491. [Google Scholar] [CrossRef] [Green Version]
- Fairbrother, N.; Janssen, P.; Antony, M.M.; Tucker, E.; Young, A.H. Perinatal anxiety disorder prevalence and incidence. J. Affect. Disord. 2016, 20, 148–155. [Google Scholar] [CrossRef] [Green Version]
- Vigod, S.N.; Wilson, C.A.; Howard, L.M. Depression in pregnancy. BMJ 2016, 352, i1547. [Google Scholar] [CrossRef]
- Bennett, H.A.; Einarson, A.; Taddio, A.; Koren, G.; Einarson, T.R. Prevalence of depression during pregnancy: Systematic review. Obstet. Gynecol. 2004, 103, 698–709. [Google Scholar] [CrossRef] [PubMed]
- Tomfohr-Madsen, L.M.; Racine, N.; Giesbrecht, G.F.; Lebel, C.; Madigan, S. Depression and anxiety in pregnancy during covid-19: A rapid review and meta-analysis. Psychiatry Res. 2021, 30, 113912. [Google Scholar] [CrossRef]
- Boekhorst, M.; Muskens, L.; Hulsbosch, L.P.; Van Deun, K.; Bergink, V.; Pop, V.J.M.; van den Heuvel, M.I. The covid-19 outbreak increases maternal stress during pregnancy, but not the risk for postpartum depression. Arch. Women’s Ment. Health 2021, 8, 1–7. [Google Scholar]
- Gluckman, P.D.; Hanson, M.A.; Beedle, A.S. Early life events and their consequences for later disease: A life history and evolutionary perspective. Am. J. Hum. Biol. 2007, 19, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Champagne, D.L.; Bagot, R.C.; van Hasselt, F.; Ramakers, G.; Meaney, M.J.; de Kloet, E.R.; Joëls, M.; Krugers, H. Maternal care and hippocampal plasticity: Evidence for experience-dependent structural plasticity, altered synaptic functioning, and differential responsiveness to glucocorticoids and stress. J. Neurosci. 2008, 28, 6037–6045. [Google Scholar] [CrossRef] [Green Version]
- Serpeloni, F.; Radtke, K.M.; Hecker, T.; Sill, J.; Vukojevic, V.; de Assis, S.G.; Schauer, M.; Elbert, T.; Nätt, D. Does prenatal stress shape postnatal resilience?—An epigenome-wide study on violence and mental health in humans. Front. Genet. 2019, 1, 269. [Google Scholar] [CrossRef]
- Van den Bergh, B.R.H.; van den Heuvel, M.I.; Lahti, M.; Braeken, M.; de Rooij, S.R.; Entringer, S.; Hoyer, D.; Roseboom, T.; Räikkönen, K.; King, S.; et al. Prenatal developmental origins of behavior and mental health: The influence of maternal stress in pregnancy. Neurosci. Biobehav. Rev. 2020, 117, 26–64. [Google Scholar] [CrossRef] [Green Version]
- Van den Bergh, B.R.; Mulder, E.J.; Mennes, M.; Glover, V. Antenatal maternal anxiety and stress and the neurobehavioural development of the fetus and child: Links and possible mechanisms. A review. Neurosci. Biobehav. Rev. 2005, 29, 237–258. [Google Scholar] [CrossRef]
- Vos, T.; Flaxman, A.D.; Naghavi, M.; Lozano, R.; Michaud, C.; Ezzati, M.; Shibuya, K.; Salomon, J.A.; Abdalla, S.; Aboyans, V.; et al. Years lived with disability (ylds) for 1160 sequelae of 289 diseases and injuries 1990–2010: A systematic analysis for the global burden of disease study 2010. Lancet 2012, 38, 2163–2196. [Google Scholar] [CrossRef]
- Van den Bergh, B.R.H.; Dahnke, R.; Mennes, M. Prenatal stress and the developing brain: Risks for neurodevelopmental disorders. Dev. Psychopathol. 2018, 3, 743–762. [Google Scholar] [CrossRef]
- Bureau, J.F.; Easterbrooks, M.A.; Lyons-Ruth, K. Maternal depressive symptoms in infancy: Unique contribution to children’s depressive symptoms in childhood and adolescence? Dev. Psychopathol. 2009, 21, 519–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grigoriadis, S.; Vonderporten, E.H.; Mamisashvili, L.; Tomlinson, G.; Dennis, C.L.; Koren, G.; Steiner, M.; Mousmanis, P.; Cheung, A.; Ross, L.E. Prenatal exposure to antidepressants and persistent pulmonary hypertension of the newborn: Systematic review and meta-analysis. BMJ 2014, 348, f6932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, H.; Hill, J.; Hellier, J.; Pickles, A. Maternal antenatal anxiety, postnatal stroking and emotional problems in children: Outcomes predicted from pre- and postnatal programming hypotheses. Psychol. Med. 2015, 45, 269–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Lu, Y.C.; Jacobs, M.; Pradhan, S.; Kapse, K.; Zhao, L.; Niforatos-Andescavage, N.; Vezina, G.; du Plessis, A.J.; Limperopoulos, C. Association of prenatal maternal psychological distress with fetal brain growth, metabolism, and cortical maturation. JAMA Netw. Open 2020, 3, e1919940. [Google Scholar] [CrossRef] [PubMed]
- Waterland, R.A.; Michels, K.B. Epigenetic epidemiology of the developmental origins hypothesis. Annu. Rev. Nutr. 2007, 27, 363–388. [Google Scholar] [CrossRef]
- Monk, C.; Spicer, J.; Champagne, F.A. Linking prenatal maternal adversity to developmental outcomes in infants: The role of epigenetic pathways. Dev. Psychopathol. 2012, 24, 1361–1376. [Google Scholar] [CrossRef] [Green Version]
- Conradt, E.; Adkins, D.E.; Crowell, S.E.; Monk, C.; Kobor, M.S. An epigenetic pathway approach to investigating associations between prenatal exposure to maternal mood disorder and newborn neurobehavior. Dev. Psychopathol. 2018, 3, 881–890. [Google Scholar] [CrossRef]
- Palma-Gudiel, H.; Cordova-Palomera, A.; Leza, J.C.; Fananas, L. Glucocorticoid receptor gene (nr3c1) methylation processes as mediators of early adversity in stress-related disorders causality: A critical review. Neurosci. Biobehav. Rev. 2015, 55, 520–535. [Google Scholar] [CrossRef] [Green Version]
- Monk, C.; Feng, T.; Lee, S.; Krupska, I.; Champagne, F.A.; Tycko, B. Distress during pregnancy: Epigenetic regulation of placenta glucocorticoid-related genes and fetal neurobehavior. Am. J. Psychiatry 2016, 173, 705–713. [Google Scholar] [CrossRef] [Green Version]
- Oberlander, T.F.; Weinberg, J.; Papsdorf, M.; Grunau, R.; Misri, S.; Devlin, A.M. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (nr3c1) and infant cortisol stress responses. Epigenetics 2008, 3, 97–106. [Google Scholar] [CrossRef] [Green Version]
- Hompes, T.; Izzi, B.; Gellens, E.; Morreels, M.; Fieuws, S.; Pexsters, A.; Schops, G.; Dom, M.; Van Bree, R.; Freson, K.; et al. Investigating the influence of maternal cortisol and emotional state during pregnancy on the DNA methylation status of the glucocorticoid receptor gene (nr3c1) promoter region in cord blood. J. Psychiatr. Res. 2013, 47, 880–891. [Google Scholar] [CrossRef] [PubMed]
- Agba, O.B.; Lausser, L.; Huse, K.; Bergmeier, C.; Jahn, N.; Groth, M.; Bens, M.; Sahm, A.; Gall, M.; Witte, O.W.; et al. Tissue-, sex-, and age-specific DNA methylation of rat glucocorticoid receptor gene promoter and insulin-like growth factor 2 imprinting control region. Physiol. Genom. 2017, 49, 690–702. [Google Scholar] [CrossRef] [PubMed]
- Ostlund, B.D.; Conradt, E.; Crowell, S.E.; Tyrka, A.R.; Marsit, C.J.; Lester, B.M. Prenatal stress, fearfulness, and the epigenome: Exploratory analysis of sex differences in DNA methylation of the glucocorticoid receptor gene. Front. Behav. Neurosci. 2016, 1, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braithwaite, E.C.; Kundakovic, M.; Ramchandani, P.G.; Murphy, S.E.; Champagne, F.A. Maternal prenatal depressive symptoms predict infant nr3c1 1f and bdnf iv DNA methylation. Epigenetics 2015, 1, 408–417. [Google Scholar] [CrossRef] [Green Version]
- Berretta, E.; Guida, E.; Forni, D.; Provenzi, L. Glucocorticoid receptor gene (nr3c1) methylation during the first thousand days: Environmental exposures and developmental outcomes. Neurosci. Biobehav. Rev. 2021, 125, 493–502. [Google Scholar] [CrossRef]
- Hayes, J.P.; Logue, M.W.; Reagan, A.; Salat, D.; Wolf, E.J.; Sadeh, N.; Spielberg, J.M.; Sperbeck, E.; Hayes, S.M.; McGlinchey, R.E.; et al. Comt val158met polymorphism moderates the association between ptsd symptom severity and hippocampal volume. J. Psychiatry Neurosci. 2017, 42, 95–102. [Google Scholar] [CrossRef] [Green Version]
- Mansell, T.; Novakovic, B.; Meyer, B.; Rzehak, P.; Vuillermin, P.; Ponsonby, A.L.; Collier, F.; Burgner, D.; Saffery, R.; Ryan, J.; et al. The effects of maternal anxiety during pregnancy on igf2/h19 methylation in cord blood. Transl. Psychiatry 2016, 6, e765. [Google Scholar] [CrossRef] [Green Version]
- Stonawski, V.; Frey, S.; Golub, Y.; Rohleder, N.; Kriebel, J.; Goecke, T.W.; Fasching, P.A.; Beckmann, M.W.; Kornhuber, J.; Kratz, O.; et al. Associations of prenatal depressive symptoms with DNA methylation of hpa axis-related genes and diurnal cortisol profiles in primary school-aged children. Dev. Psychopathol. 2019, 31, 419–431. [Google Scholar] [CrossRef]
- Folger, A.T.; Ding, L.; Ji, H.; Yolton, K.; Ammerman, R.T.; Van Ginkel, J.B.; Bowers, K. Neonatal nr3c1 methylation and social-emotional development at 6 and 18 months of age. Front. Behav. Neurosci. 2019, 13, 14. [Google Scholar] [CrossRef]
- Sosnowski, D.W.; Booth, C.; York, T.P.; Amstadter, A.B.; Kliewer, W. Maternal prenatal stress and infant DNA methylation: A systematic review. Dev. Psychobiol. 2018, 6, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.; Liu, J.P.; Robertson, E.J.; Efstratiadis, A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell 1993, 75, 73–82. [Google Scholar] [CrossRef]
- Reik, W.; Walter, J. Genomic imprinting: Parental influence on the genome. Nat. Rev. Genet. 2001, 2, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, Q.; Rialdi, A.; Mystal, E.; Ly, J.; Finik, J.; Davey, T.; Lambertini, L.; Nomura, Y. Influences of maternal stress during pregnancy on the epi/genome: Comparison of placenta and umbilical cord blood. J. Depress. Anxiety 2014, 3, 152. [Google Scholar] [PubMed] [Green Version]
- Rijlaarsdam, J.; Cecil, C.A.; Walton, E.; Mesirow, M.S.; Relton, C.L.; Gaunt, T.R.; McArdle, W.; Barker, E.D. Prenatal unhealthy diet, insulin-like growth factor 2 gene (igf2) methylation, and attention deficit hyperactivity disorder symptoms in youth with early-onset conduct problems. J. Child Psychol. Psychiatry 2017, 58, 19–27. [Google Scholar] [CrossRef] [Green Version]
- Cecil, C.A.; Walton, E.; Barker, E.D. Prenatal diet and childhood adhd: Exploring the potential role of igf2 methylation. Epigenomics 2016, 8, 1573–1576. [Google Scholar] [CrossRef] [Green Version]
- Fuemmeler, B.F.; Lee, C.T.; Soubry, A.; Iversen, E.S.; Huang, Z.; Murtha, A.P.; Schildkraut, J.M.; Jirtle, R.L.; Murphy, S.K.; Hoyo, C. DNA methylation of regulatory regions of imprinted genes at birth and its relation to infant temperament. Genet. Epigenet. 2016, 8, 59–67. [Google Scholar] [CrossRef] [Green Version]
- Gabory, A.; Attig, L.; Junien, C. Sexual dimorphism in environmental epigenetic programming. Mol. Cell Endocrinol. 2009, 304, 8–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Cerezales, S.; Ramos-Ibeas, P.; Rizos, D.; Lonergan, P.; Bermejo-Alvarez, P.; Gutierrez-Adan, A. Early sex-dependent differences in response to environmental stress. Reproduction 2018, 155, R39–R51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burns, S.B.; Szyszkowicz, J.K.; Luheshi, G.N.; Lutz, P.E.; Turecki, G. Plasticity of the epigenome during early-life stress. Semin. Cell Dev. Biol. 2018, 77, 115–132. [Google Scholar] [CrossRef]
- Watamura, S.E.; Roth, T.L. Looking back and moving forward: Evaluating and advancing translation from animal models to human studies of early life stress and DNA methylation. Dev. Psychobiol. 2019, 61, 323–340. [Google Scholar] [CrossRef] [PubMed]
- Veru, F.; Laplante, D.P.; Luheshi, G.; King, S. Prenatal maternal stress exposure and immune function in the offspring. Stress 2014, 17, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.S.; Estecio, M.R.; Doshi, K.; Kondo, Y.; Tajara, E.H.; Issa, J.P. A simple method for estimating global DNA methylation using bisulfite pcr of repetitive DNA elements. Nucleic Acids Res. 2004, 32, e38. [Google Scholar] [CrossRef]
- Arrindell, W.A.; Ettema, J.H.M. Symptom Checklist (scl-90): Handleiding bij een Multidimensionale Psychopathologie-Indicator. [Symptom Checklist (scl-90): Manual for a Multidimensional Measure of Psychopathology]; Harcour Test Publishers: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Achenbach, T.M.; Rescorla, L.A. Manual for the ASEBA School-Age Forms & Profiles: An Integrated System of Mult-Informant Assessment; University of Vermont, Research Center for Children, Youth & Families: Burlington, VT, USA, 2001. [Google Scholar]
- Gries, J.; Schumacher, D.; Arand, J.; Lutsik, P.; Markelova, M.R.; Fichtner, I.; Walter, J.; Sers, C.; Tierling, S. Bi-prof: Bisulfite profiling of target regions using 454 gs flx titanium technology. Epigenetics 2013, 8, 765–771. [Google Scholar] [CrossRef] [Green Version]
- Hayes, A.F. Introduction to Mediation, Moderation, and Conditional Process Analysis; The Guilford Press: New York, NY, USA, 2013. [Google Scholar]
- Hayes, A.F.; Rockwood, N.J. Conditional process analysis: Concepts, computation, and advances in the modeling of the contingencies of mechanisms. Am. Behav. Sci. 2020, 64, 19–54. [Google Scholar] [CrossRef] [Green Version]
- Murgatroyd, C.; Quinn, J.P.; Sharp, H.M.; Pickles, A.; Hill, J. Effects of prenatal and postnatal depression, and maternal stroking, at the glucocorticoid receptor gene. Transl. Psychiatry 2015, 5, e560. [Google Scholar] [CrossRef] [Green Version]
- Mueller, B.R.; Bale, T.L. Sex-specific programming of offspring emotionality after stress early in pregnancy. J. Neurosci. 2008, 28, 9055–9065. [Google Scholar] [CrossRef] [Green Version]
- Mychasiuk, R.; Gibb, R.; Kolb, B. Prenatal stress produces sexually dimorphic and regionally specific changes in gene expression in hippocampus and frontal cortex of developing rat offspring. Dev. Neurosci. 2011, 33, 531–538. [Google Scholar] [CrossRef]
- Luoni, A.; Berry, A.; Raggi, C.; Bellisario, V.; Cirulli, F.; Riva, M.A. Sex-specific effects of prenatal stress on bdnf expression in response to an acute challenge in rats: A role for gadd45beta. Mol. Neurobiol. 2016, 53, 7037–7047. [Google Scholar] [CrossRef] [PubMed]
- Appleton, A.A.; Armstrong, D.A.; Lesseur, C.; Lee, J.; Padbury, J.F.; Lester, B.M.; Marsit, C.J. Patterning in placental 11-b hydroxysteroid dehydrogenase methylation according to prenatal socioeconomic adversity. PLoS ONE 2013, 8, e74691. [Google Scholar]
- Stroud, L.R.; Papandonatos, G.D.; Parade, S.H.; Salisbury, A.L.; Phipps, M.G.; Lester, B.M.; Padbury, J.F.; Marsit, C.J. Prenatal major depressive disorder, placenta glucocorticoid and serotonergic signaling, and infant cortisol response. Psychosom. Med. 2016, 78, 979–990. [Google Scholar] [CrossRef] [PubMed]
- Belancio, V.P.; Roy-Engel, A.M.; Pochampally, R.R.; Deininger, P. Somatic expression of LINE-1 elements in human tissues. Nucleic Acids Res 2010, 38, 3909–3922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearce, M.S.; McConnell, J.C.; Potter, C.; Barrett, L.M.; Parker, L.; Mathers, J.C.; Relton, C.L. Global LINE-1 DNA methylation is associated with blood glycaemic and lipid profiles. Int. J. Epidemiol. 2012, 41, 210–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kile, M.L.; Baccarelli, A.; Tarantini, L.; Hoffman, E.; Wright, R.O.; Christiani, D.C. Correlation of global and gene-specific DNA methylation in maternal-infant pairs. PLoS ONE 2010, 5, e13730. [Google Scholar] [CrossRef] [Green Version]
- Cao-Lei, L.; Dancause, K.N.; Elgbeili, G.; Massart, R.; Szyf, M.; Liu, A.; Laplante, D.P.; King, S. DNA methylation mediates the impact of exposure to prenatal maternal stress on bmi and central adiposity in children at age 13(1/2) years: Project ice storm. Epigenetics 2015, 1, 749–761. [Google Scholar] [CrossRef]
- Cao-Lei, L.; Dancause, K.N.; Elgbeili, G.; Laplante, D.P.; Szyf, M.; King, S. Pregnant women’s cognitive appraisal of a natural disaster affects their children’s bmi and central adiposity via DNA methylation: Project ice storm. Early Hum. Dev. 2016, 103, 189–192. [Google Scholar] [CrossRef]
- Unternaehrer, E.; Bolten, M.; Nast, I.; Staehli, S.; Meyer, A.H.; Dempster, E.; Hellhammer, D.H.; Lieb, R.; Meinlschmidt, G. Maternal adversities during pregnancy and cord blood oxytocin receptor (oxtr) DNA methylation. Soc. Cogn. Affect. Neurosci. 2016, 11, 1460–1470. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Range | Mean | Std. Deviation | |
| Mothers | |||
| Gestational age at birth (weeks) | 36.570–42.290 | 39.613 | 1.154 |
| Anxiety—Trimester 1 (SCL-90) | 10.000–35.000 | 13.794 | 4.741 |
| Anxiety—Trimester 2 (SCL-90) | 10.000–31.000 | 13.072 | 3.721 |
| Anxiety—Trimester 3 (SCL-90) | 10.000–32.000 | 13.072 | 3.581 |
| Anxiety when child was at age of 4 (SCL-90) | 10.000–32.000 | 12.771 | 4.058 |
| % | |||
| Children | |||
| Sex | |||
| Male | 44.900 | ||
| Female | 55.100 | ||
| Range | Mean | Std. Deviation | |
| Children’s birth weight (g) | 2605.000–4590.000 | 3427.360 | 408.427 |
| Children’s behavior outcomes: | |||
| -Internalizing problems (CBCL) | 0.000–26.000 | 7.256 | 6.012 |
| -Externalizing problems (CBCL) | 1.000–34.000 | 10.683 | 6.887 |
| -DSM affective problems (CBCL) | 0.000–10.000 | 1.841 | 1.815 |
| -DSM anxiety problems (CBCL) | 0.000–8.000 | 2.268 | 1.969 |
| -DSM pervasive developmental problems (CBCL) | 0.000–14.000 | 3.378 | 2.765 |
| -DSM attention deficit/hyperactivity problems (CBCL) | 0.000–9.000 | 3.494 | 2.281 |
| -DSM oppositional defiant problems (CBCL) | 0.000–12.000 | 3.866 | 2.562 |
| CBCL Subscales | |||||||
|---|---|---|---|---|---|---|---|
| Internalizing Problems | Externalizing Problems | DSM Affective Problems | DSM Anxiety Problems | DSM Pervasive Developmental Problems | DSM Attention Deficit/Hyperactivity Problems | DSM Oppositional Defiant Problems | |
| Anxiety—Trimester 1 (SCL-90) | 0.052 | 0.053 | 0.108 | −0.009 | 0.026 | 0.037 | 0.121 |
| Anxiety—Trimester 2 (SCL-90) | 0.254 a | 0.195 | 0.173 | 0.197 | 0.203 | 0.176 | 0.245 b |
| Anxiety—Trimester 3 (SCL-90) | 0.154 | 0.205 | 0.037 | 0.071 | 0.161 | 0.252 c | 0.203 |
| LINE1_motif1 | −0.061 | 0.051 | −0.026 | −0.038 | −0.059 | −0.014 | 0.087 |
| LINE1_motif2 | −0.126 | 0.078 | −0.003 | −0.097 | −0.139 | 0.116 | 0.021 |
| LINE1_motif3 | −0.013 | 0.103 | 0.039 | −0.037 | 0.008 | 0.067 | 0.123 |
| NR3C1-CpG1 | −0.008 | −0.165 | −0.026 | −0.082 | −0.028 | −0.144 | −0.105 |
| NR3C1-CpG2 | −0.042 | −0.175 | 0.045 | −0.100 | −0.111 | −0.177 | −0.092 |
| NR3C1-CpG3 | −0.017 | −0.151 | 0.019 | −0.057 | −0.086 | −0.149 | −0.085 |
| NR3C1-CpG4 | −0.016 | −0.179 | −0.009 | −0.045 | −0.044 | −0.193 | −0.136 |
| NR3C1-CpG5 | −0.057 | 0.029 | −0.070 | −0.135 | −0.053 | −0.065 | 0.006 |
| NR3C1-CpG6 | −0.079 | −0.038 | −0.090 | −0.024 | −0.070 | −0.040 | −0.088 |
| NR3C1-CpG7 | −0.143 | −0.015 | −0.147 | −0.174 | −0.064 | −0.019 | −0.052 |
| NR3C1-CpG8 | −0.106 | −0.051 | 0.012 | 0.033 | −0.100 | −0.032 | −0.023 |
| NR3C1-CpG9 | −0.076 | 0.014 | −0.005 | −0.157 | −0.067 | −0.084 | 0.081 |
| NR3C1-CpG10 | 0.059 | 0.196 | 0.005 | −0.168 | 0.066 | 0.010 | 0.201 |
| IGF2/H19 ICR -CpG1 | −0.054 | −0.055 | −0.002 | −0.094 | −0.067 | −0.137 | −0.020 |
| IGF2/H19 ICR -CpG2 | −0.091 | −0.037 | −0.039 | −0.129 | −0.104 | −0.119 | −0.001 |
| IGF2/H19 ICR -CpG3 | −0.070 | −0.041 | −0.023 | −0.124 | −0.087 | −0.105 | 0.005 |
| IGF2/H19 ICR -CpG4 | −0.057 | −0.041 | −0.041 | −0.107 | −0.072 | −0.093 | −0.007 |
| IGF2/H19 ICR -CpG5 | −0.090 | −0.062 | −0.046 | −0.123 | −0.092 | −0.104 | −0.021 |
| IGF2/H19 ICR -CpG6 | −0.072 | −0.008 | −0.014 | −0.116 | −0.106 | −0.076 | 0.028 |
| IGF2/H19 ICR -CpG7 | −0.089 | −0.044 | −0.042 | −0.147 | −0.116 | −0.123 | 0.009 |
| IGF2/H19 ICR -CpG8 | −0.073 | −0.021 | −0.006 | −0.129 | −0.103 | −0.107 | 0.036 |
| IGF2/H19 ICR -CpG9 | −0.072 | −0.037 | −0.081 | −0.155 | −0.101 | −0.079 | −0.009 |
| IGF2/H19 ICR -CpG10 | −0.092 | −0.059 | −0.111 | −0.160 | −0.113 | −0.128 | −0.018 |
| Predictor Variable | β | R | R2 | ΔR2 | F | ΔF |
|---|---|---|---|---|---|---|
| 0.157 | 0.025 | 0.946 | ||||
| Gestational age at birth | 0.001 | |||||
| Postnatal maternal anxiety | 0.001 | |||||
| 0.175 | 0.031 | 0.006 | 0.783 | 0.470 | ||
| Gestational age at birth | 0.001 | |||||
| Postnatal maternal anxiety | 0.001 | |||||
| Maternal anxiety (third trimester) | −0.002 | |||||
| 0.176 | 0.031 | 0.000 | 0.582 | 0.013 | ||
| Gestational age at birth | 0.001 | |||||
| Postnatal maternal anxiety | 0.001 | |||||
| Maternal anxiety (third trimester) | −0.002 | |||||
| Sex | 0.002 | |||||
| 0.291 | 0.085 | 0.054 | 1.336 | 4.246 a | ||
| Gestational age at birth | 0.000 | |||||
| Postnatal maternal anxiety | 0.001 | |||||
| Maternal anxiety (third trimester) | −0.007 | |||||
| Sex | −0.119 | |||||
| Maternal anxiety (third trimester) × sex | 0.009 |
| Predictor Variable | β | R | R2 | ΔR2 | F | ΔF |
|---|---|---|---|---|---|---|
| 0.133 | 0.018 | 0.680 | ||||
| Gestational age at birth | 0.001 | |||||
| Postnatal maternal anxiety | 0.001 | |||||
| 0.148 | 0.022 | 0.004 | 0.551 | 0.307 | ||
| Gestational age at birth | 0.001 | |||||
| Postnatal maternal anxiety | 0.001 | |||||
| Maternal anxiety (third trimester) | −0.001 | |||||
| 0.152 | 0.023 | 0.001 | 0.430 | 0.087 | ||
| Gestational age at birth | 0.001 | |||||
| Postnatal maternal anxiety | 0.001 | |||||
| Maternal anxiety (third trimester) | −0.001 | |||||
| Sex | 0.004 | |||||
| 0.291 | 0.085 | 0.061 | 1.329 | 4.836 a | ||
| Gestational age at birth | 0.000 | |||||
| Postnatal maternal anxiety | 0.001 | |||||
| Maternal anxiety (third trimester) | −0.007 | |||||
| Sex | −0.125 | |||||
| Maternal anxiety (third trimester) × sex | 0.010 |
| Predictor Variable | β | R | R2 | ΔR2 | F | ΔF |
|---|---|---|---|---|---|---|
| 0.169 | 0.029 | 1.101 | ||||
| Gestational age at birth | 0.001 | |||||
| Postnatal maternal anxiety | 0.000 | |||||
| 0.169 | 0.029 | 0.000 | 0.725 | 0.003 | ||
| Gestational age at birth | 0.001 | |||||
| Postnatal maternal anxiety | 0.000 | |||||
| Maternal anxiety (third trimester) | 0.000 | |||||
| 0.169 | 0.029 | 0.000 | 0.536 | 0.000 | ||
| Gestational age at birth | 0.001 | |||||
| Postnatal maternal anxiety | 0.000 | |||||
| Maternal anxiety (third trimester) | 0.000 | |||||
| Sex | 0.000 | |||||
| 0.297 | 0.088 | 0.059 | 1.390 | 4.697 a | ||
| Gestational age at birth | 0.001 | |||||
| Postnatal maternal anxiety | 0.000 | |||||
| Maternal anxiety (third trimester) | −0.005 | |||||
| Sex | −0.134 | |||||
| Maternal anxiety (third trimester) × sex | 0.011 |
| Predictor Variable | β | R | R2 | ΔR2 | F | ΔF |
|---|---|---|---|---|---|---|
| 0.137 | 0.019 | 0.724 | ||||
| Gestational age at birth | 0.000 | |||||
| Postnatal maternal anxiety | 0.000 | |||||
| 0.149 | 0.022 | 0.003 | 0.567 | 0.268 | ||
| Gestational age at birth | 0.000 | |||||
| Postnatal maternal anxiety | 0.001 | |||||
| Maternal anxiety (second trimester) | 0.000 | |||||
| 0.211 | 0.045 | 0.022 | 0.846 | 1.738 | ||
| Gestational age at birth | 0.000 | |||||
| Postnatal maternal anxiety | 0.001 | |||||
| Maternal anxiety (second trimester) | 0.000 | |||||
| Sex | −0.006 | |||||
| 0.379 | 0.143 | 0.099 | 2.445 | 8.424 a | ||
| Gestational age at birth | 0.000 | |||||
| Postnatal maternal anxiety | 0.000 | |||||
| Maternal anxiety (second trimester) | 0.003 | |||||
| Sex | 0.049 | |||||
| Maternal anxiety (second trimester) × sex | −0.004 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao-Lei, L.; van den Heuvel, M.I.; Huse, K.; Platzer, M.; Elgbeili, G.; Braeken, M.A.K.A.; Otte, R.A.; Witte, O.W.; Schwab, M.; Van den Bergh, B.R.H. Epigenetic Modifications Associated with Maternal Anxiety during Pregnancy and Children’s Behavioral Measures. Cells 2021, 10, 2421. https://doi.org/10.3390/cells10092421
Cao-Lei L, van den Heuvel MI, Huse K, Platzer M, Elgbeili G, Braeken MAKA, Otte RA, Witte OW, Schwab M, Van den Bergh BRH. Epigenetic Modifications Associated with Maternal Anxiety during Pregnancy and Children’s Behavioral Measures. Cells. 2021; 10(9):2421. https://doi.org/10.3390/cells10092421
Chicago/Turabian StyleCao-Lei, Lei, Marion I. van den Heuvel, Klaus Huse, Matthias Platzer, Guillaume Elgbeili, Marijke A. K. A. Braeken, Renée A. Otte, Otto W. Witte, Matthias Schwab, and Bea R. H. Van den Bergh. 2021. "Epigenetic Modifications Associated with Maternal Anxiety during Pregnancy and Children’s Behavioral Measures" Cells 10, no. 9: 2421. https://doi.org/10.3390/cells10092421
APA StyleCao-Lei, L., van den Heuvel, M. I., Huse, K., Platzer, M., Elgbeili, G., Braeken, M. A. K. A., Otte, R. A., Witte, O. W., Schwab, M., & Van den Bergh, B. R. H. (2021). Epigenetic Modifications Associated with Maternal Anxiety during Pregnancy and Children’s Behavioral Measures. Cells, 10(9), 2421. https://doi.org/10.3390/cells10092421