Porin 1 Modulates Autophagy in Yeast

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Yeast Strains and Growth Conditions
2.2. Growth Assays
2.3. Yeast Autophagy Measurements
2.4. Analysis of Cell Death
2.5. Assessment of Vacuole Morphology and Function
2.6. Lipid Extraction and Quantification by Shotgun Mass Spectrometry
2.7. Lipid Extraction and Quantification by Thin-Layer Chromatography
2.8. Statistical Analysis
3. Results
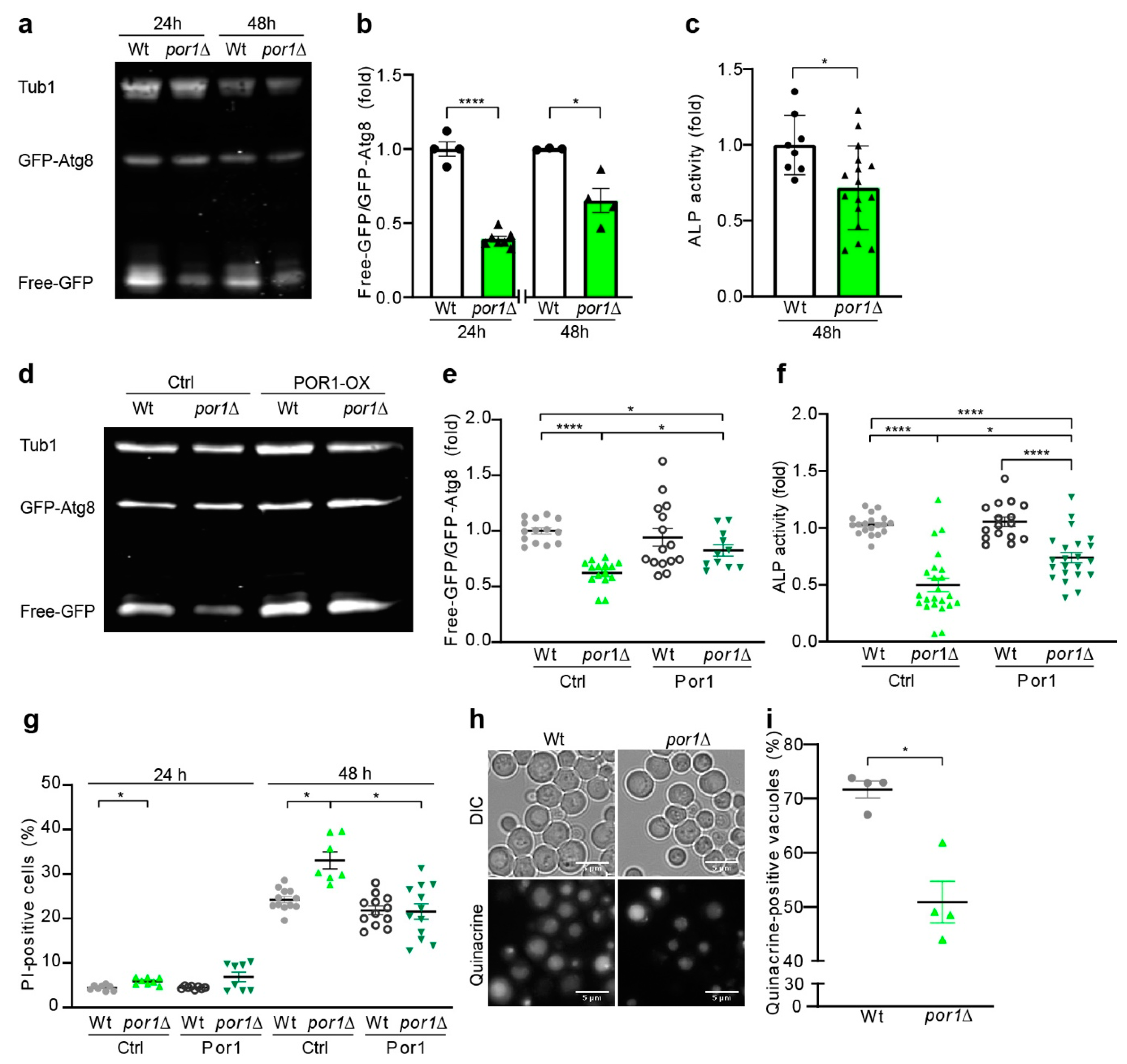
3.1. Loss of POR1 Reduced Cellular Autophagy Levels
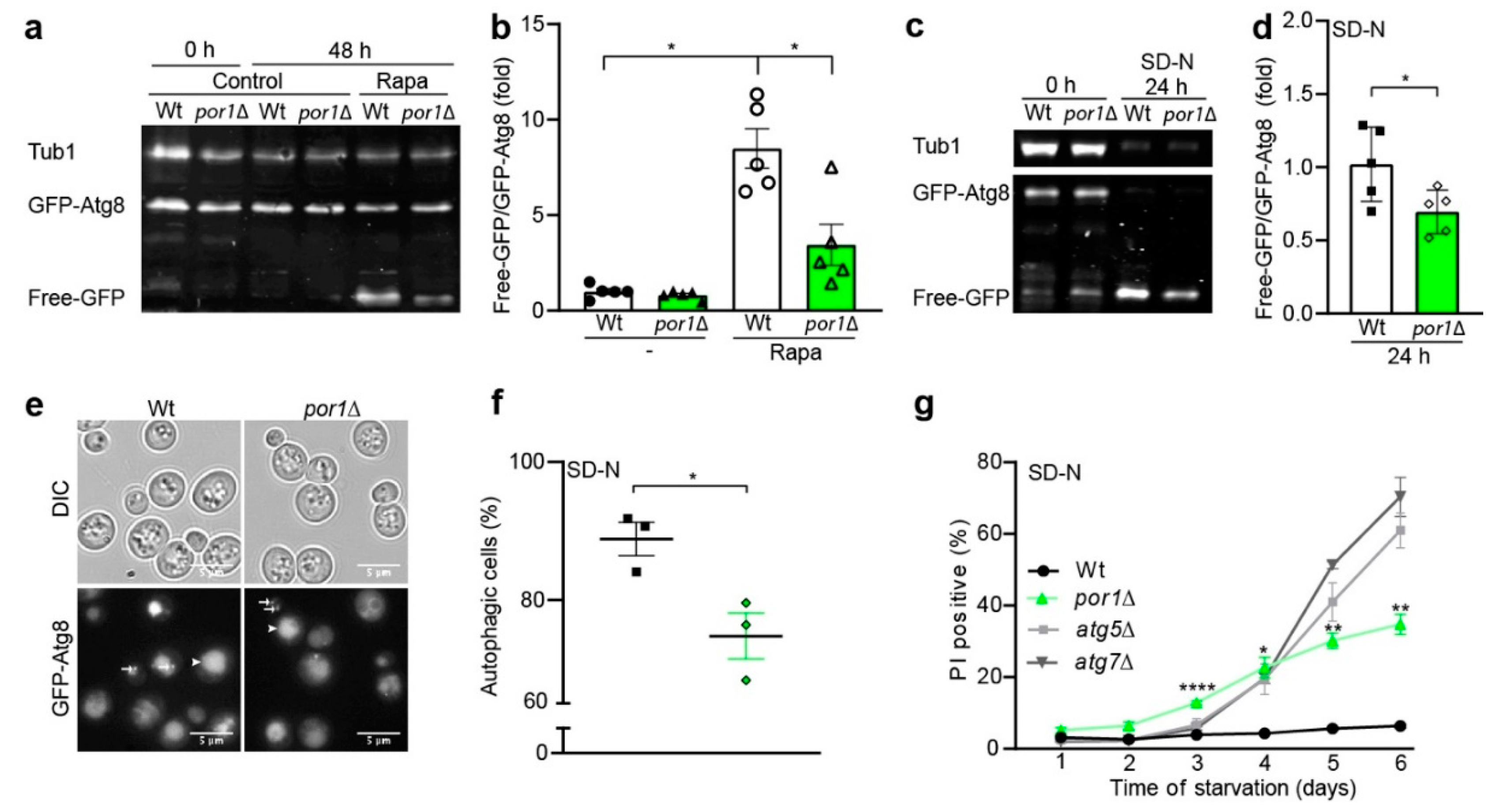
3.2. POR1 Deficiency Reduced the Autophagic Capacity upon Induction with Rapamycin or Nitrogen Starvation
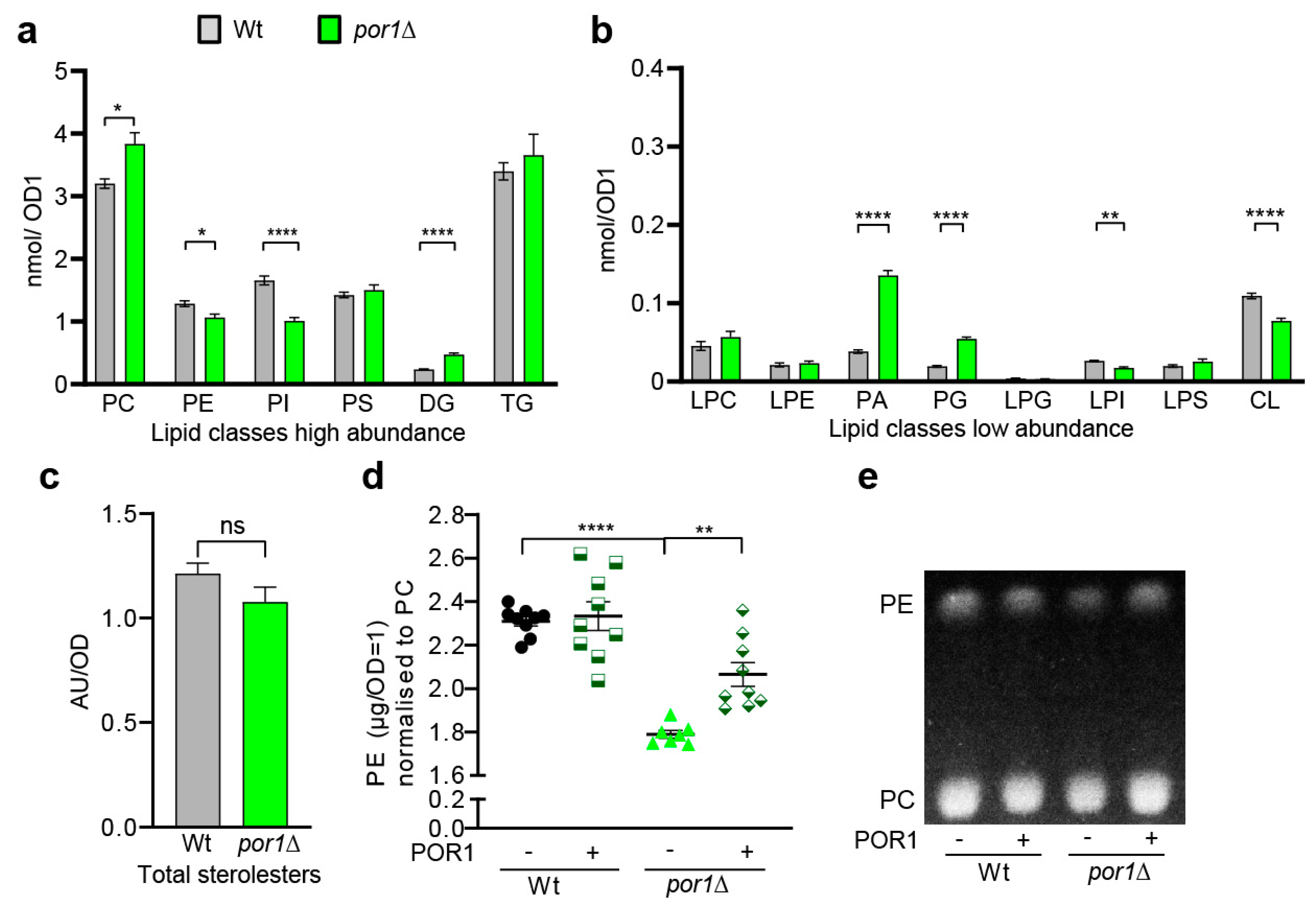
3.3. POR1 Deficiency Was Associated with Specific Changes in the Lipid Profile, Particularly Affecting Phospholipids
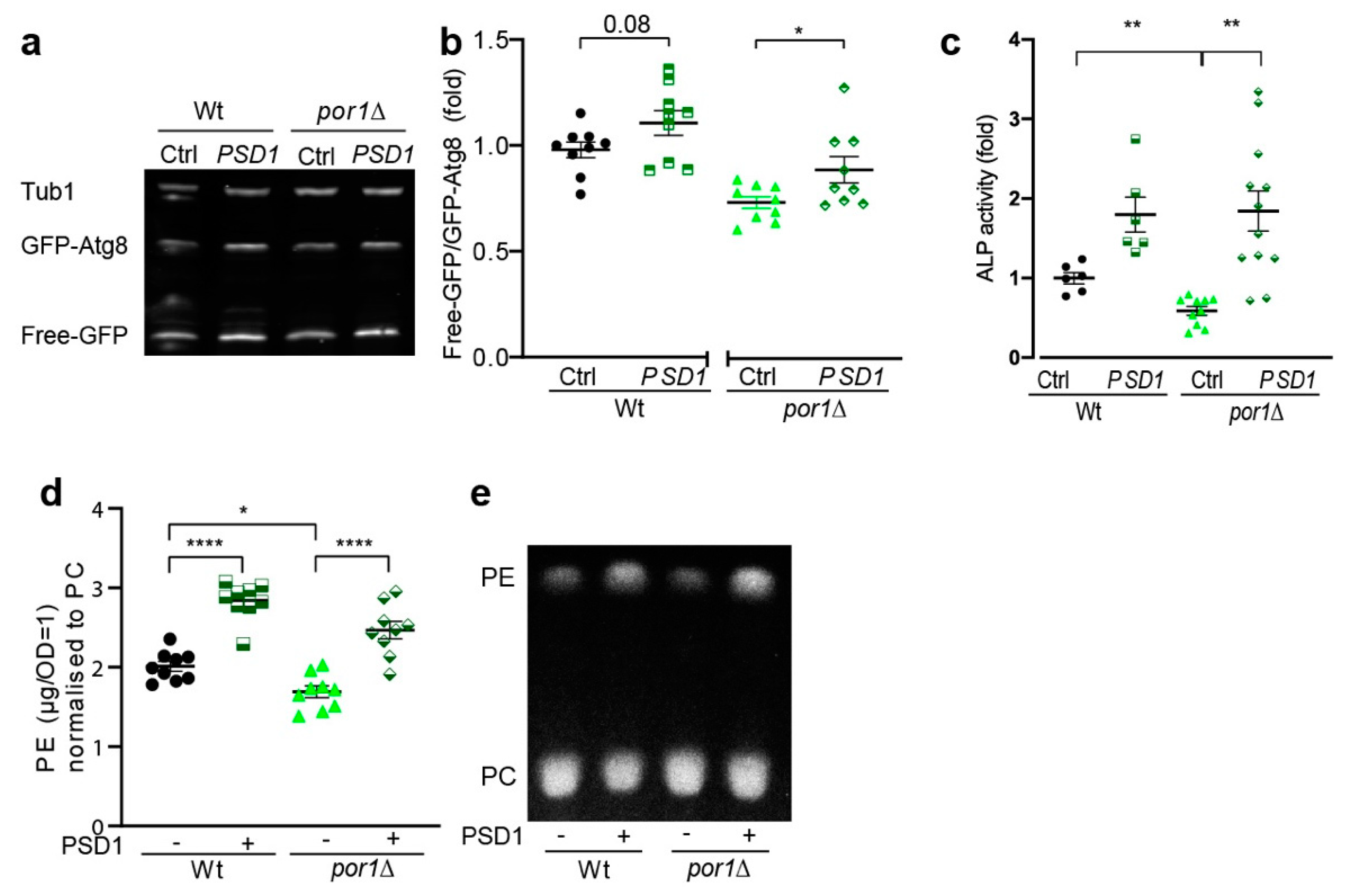
3.4. PSD1 Overexpression Could Partially Compensate for the Loss of POR1
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The Machinery of Macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [Green Version]
- Hansen, M.; Rubinsztein, D.C.; Walker, D.W. Autophagy as a Promoter of Longevity: Insights from Model Organisms. Nat. Rev. Mol. Cell Biol. 2018, 19, 579–593. [Google Scholar] [CrossRef]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome Formation from Membrane Compartments Enriched in Phosphatidylinositol 3-Phosphate and Dynamically Connected to the Endoplasmic Reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [Green Version]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria Supply Membranes for Autophagosome Biogenesis during Starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef] [Green Version]
- Geng, J.; Nair, U.; Yasumura-Yorimitsu, K.; Klionsky, D.J. Post-Golgi Sec Proteins Are Required for Autophagy in Saccharomyces Cerevisiae. Mol. Biol. Cell 2010, 21, 2257–2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Vaart, A.; Griffith, J.; Reggiori, F. Exit from the Golgi Is Required for the Expansion of the Autophagosomal Phagophore in Yeast Saccharomyces Cerevisiae. Mol. Biol. Cell 2010, 21, 2270–2284. [Google Scholar] [CrossRef] [Green Version]
- Nascimbeni, A.C.; Giordano, F.; Dupont, N.; Grasso, D.; Vaccaro, M.I.; Codogno, P.; Morel, E. ER–Plasma Membrane Contact Sites Contribute to Autophagosome Biogenesis by Regulation of Local PI3P Synthesis. EMBO J. 2017, 36, 2018–2033. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Moreau, K.; Jahreiss, L.; Puri, C.; Rubinsztein, D.C. Plasma Membrane Contributes to the Formation of Pre-Autophagosomal Structures. Nat. Cell Biol. 2010, 12, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Longatti, A.; Lamb, C.A.; Razi, M.; Yoshimura, S.; Barr, F.A.; Tooze, S.A. TBC1D14 Regulates Autophagosome Formation via Rab11- and ULK1-Positive Recycling Endosomes. J. Cell Biol. 2012, 197, 659–675. [Google Scholar] [CrossRef] [Green Version]
- Puri, C.; Vicinanza, M.; Ashkenazi, A.; Gratian, M.J.; Zhang, Q.; Bento, C.F.; Renna, M.; Menzies, F.M.; Rubinsztein, D.C. The RAB11A-Positive Compartment Is a Primary Platform for Autophagosome Assembly Mediated by WIPI2 Recognition of PI3P-RAB11A. Dev. Cell 2018, 45, 114–131.e8. [Google Scholar] [CrossRef] [Green Version]
- Dupont, N.; Chauhan, S.; Arko-Mensah, J.; Castillo, E.F.; Masedunskas, A.; Weigert, R.; Robenek, H.; Proikas-Cezanne, T.; Deretic, V. Neutral Lipid Stores and Lipase PNPLA5 Contribute to Autophagosome Biogenesis. Curr. Biol. 2014, 24, 609–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shpilka, T.; Welter, E.; Borovsky, N.; Amar, N.; Mari, M.; Reggiori, F.; Elazar, Z. Lipid Droplets and Their Component Triglycerides and Steryl Esters Regulate Autophagosome Biogenesis. EMBO J. 2015, 34, 2117–2131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schütter, M.; Giavalisco, P.; Brodesser, S.; Graef, M. Local Fatty Acid Channeling into Phospholipid Synthesis Drives Phagophore Expansion during Autophagy. Cell 2020, 180, 135–149.e14. [Google Scholar] [CrossRef] [PubMed]
- Gross, A.S.; Zimmermann, A.; Pendl, T.; Schroeder, S.; Schoenlechner, H.; Knittelfelder, O.; Lamplmayr, L.; Santiso, A.; Aufschnaiter, A.; Waltenstorfer, D.; et al. Acetyl-CoA Carboxylase 1-Dependent Lipogenesis Promotes Autophagy Downstream of AMPK. J. Biol. Chem. 2019, 294, 12020–12039. [Google Scholar] [CrossRef] [Green Version]
- Niso-Santano, M.; Malik, S.A.; Pietrocola, F.; Bravo-San Pedro, J.M.; Mariño, G.; Cianfanelli, V.; Ben-Younès, A.; Troncoso, R.; Markaki, M.; Sica, V.; et al. Unsaturated Fatty Acids Induce Non-Canonical Autophagy. EMBO J. 2015, 34, 1025–1041. [Google Scholar] [CrossRef]
- Sauvat, A.; Chen, G.; Müller, K.; Tong, M.; Aprahamian, F.; Durand, S.; Cerrato, G.; Bezu, L.; Leduc, M.; Franz, J.; et al. Trans-Fats Inhibit Autophagy Induced by Saturated Fatty Acids. EBioMedicine 2018, 30, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Rockenfeller, P.; Koska, M.; Pietrocola, F.; Minois, N.; Knittelfelder, O.; Sica, V.; Franz, J.; Carmona-Gutierrez, D.; Kroemer, G.; Madeo, F. Phosphatidylethanolamine Positively Regulates Autophagy and Longevity. Cell Death Differ. 2015, 22, 499–508. [Google Scholar] [CrossRef] [Green Version]
- Tatsuta, T.; Langer, T. Intramitochondrial Phospholipid Trafficking. Biochim. Biophys. Acta BBA-Mol. Cell Biol. Lipids 2017, 1862, 81–89. [Google Scholar] [CrossRef]
- Kuchler, K.; Daum, G.; Paltauf, F. Subcellular and Submitochondrial Localization of Phospholipid-Synthesizing Enzymes in Saccharomyces Cerevisiae. J. Bacteriol. 1986, 165, 901–910. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S.E.; Böttinger, L.; Vögtle, F.-N.; Wiedemann, N.; Meisinger, C.; Becker, T.; Daum, G. Processing and Topology of the Yeast Mitochondrial Phosphatidylserine Decarboxylase 1. J. Biol. Chem. 2012, 287, 36744–36755. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.R.; Kannan, M.; Toulmay, A.; Jan, C.H.; Weissman, J.S.; Prinz, W.A.; Nunnari, J. Lipid Homeostasis Is Maintained by Dual Targeting of the Mitochondrial PE Biosynthesis Enzyme to the ER. Dev. Cell 2018, 44, 261–270.e6. [Google Scholar] [CrossRef] [Green Version]
- Bürgermeister, M.; Birner-Grünberger, R.; Nebauer, R.; Daum, G. Contribution of Different Pathways to the Supply of Phosphatidylethanolamine and Phosphatidylcholine to Mitochondrial Membranes of the Yeast Saccharomyces Cerevisiae. Biochim. Biophys. Acta BBA-Mol. Cell Biol. Lipids 2004, 1686, 161–168. [Google Scholar] [CrossRef]
- Miliara, X.; Tatsuta, T.; Berry, J.-L.; Rouse, S.L.; Solak, K.; Chorev, D.S.; Wu, D.; Robinson, C.V.; Matthews, S.; Langer, T. Structural Determinants of Lipid Specificity within Ups/PRELI Lipid Transfer Proteins. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Harner, M.; Körner, C.; Walther, D.; Mokranjac, D.; Kaesmacher, J.; Welsch, U.; Griffith, J.; Mann, M.; Reggiori, F.; Neupert, W. The Mitochondrial Contact Site Complex, a Determinant of Mitochondrial Architecture. EMBO J. 2011, 30, 4356–4370. [Google Scholar] [CrossRef] [Green Version]
- Connerth, M.; Tatsuta, T.; Haag, M.; Klecker, T.; Westermann, B.; Langer, T. Intramitochondrial Transport of Phosphatidic Acid in Yeast by a Lipid Transfer Protein. Science 2012, 338, 815–818. [Google Scholar] [CrossRef] [PubMed]
- AhYoung, A.P.; Jiang, J.; Zhang, J.; Dang, X.K.; Loo, J.A.; Zhou, Z.H.; Egea, P.F. Conserved SMP Domains of the ERMES Complex Bind Phospholipids and Mediate Tether Assembly. Proc. Natl. Acad. Sci. USA 2015, 112, E3179–E3188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornmann, B.; Osman, C.; Walter, P. The Conserved GTPase Gem1 Regulates Endoplasmic Reticulum–Mitochondria Connections. Proc. Natl. Acad. Sci. USA 2011, 108, 14151–14156. [Google Scholar] [CrossRef] [Green Version]
- Lahiri, S.; Chao, J.T.; Tavassoli, S.; Wong, A.K.O.; Choudhary, V.; Young, B.P.; Loewen, C.J.R.; Prinz, W.A. A Conserved Endoplasmic Reticulum Membrane Protein Complex (EMC) Facilitates Phospholipid Transfer from the ER to Mitochondria. PLoS Biol. 2014, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombini, M. The VDAC Channel: Molecular Basis for Selectivity. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2016, 1863, 2498–2502. [Google Scholar] [CrossRef]
- Bayrhuber, M.; Meins, T.; Habeck, M.; Becker, S.; Giller, K.; Villinger, S.; Vonrhein, C.; Griesinger, C.; Zweckstetter, M.; Zeth, K. Structure of the Human Voltage-Dependent Anion Channel. Proc. Natl. Acad. Sci. USA 2008, 105, 15370–15375. [Google Scholar] [CrossRef] [Green Version]
- Prinz, W.A.; Toulmay, A.; Balla, T. The Functional Universe of Membrane Contact Sites. Nat. Rev. Mol. Cell Biol. 2020, 21, 7–24. [Google Scholar] [CrossRef]
- Cherry, J.M.; Hong, E.L.; Amundsen, C.; Balakrishnan, R.; Binkley, G.; Chan, E.T.; Christie, K.R.; Costanzo, M.C.; Dwight, S.S.; Engel, S.R.; et al. Saccharomyces Genome Database: The Genomics Resource of Budding Yeast. Nucleic Acids Res. 2012, 40, D700–D705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costanzo, M.; Baryshnikova, A.; Bellay, J.; Kim, Y.; Spear, E.D.; Sevier, C.S.; Ding, H.; Koh, J.L.Y.; Toufighi, K.; Mostafavi, S.; et al. The Genetic Landscape of a Cell. Science 2010, 327, 425–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoppins, S.; Collins, S.R.; Cassidy-Stone, A.; Hummel, E.; DeVay, R.M.; Lackner, L.L.; Westermann, B.; Schuldiner, M.; Weissman, J.S.; Nunnari, J. A Mitochondrial-Focused Genetic Interaction Map Reveals a Scaffold-like Complex Required for Inner Membrane Organization in Mitochondria. J. Cell Biol. 2011, 195, 323–340. [Google Scholar] [CrossRef] [Green Version]
- Murley, A.; Sarsam, R.D.; Toulmay, A.; Yamada, J.; Prinz, W.A.; Nunnari, J. Ltc1 Is an ER-Localized Sterol Transporter and a Component of ER–Mitochondria and ER–Vacuole Contacts. J. Cell Biol. 2015, 209, 539–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgenstern, M.; Stiller, S.B.; Lübbert, P.; Peikert, C.D.; Dannenmaier, S.; Drepper, F.; Weill, U.; Höß, P.; Feuerstein, R.; Gebert, M.; et al. Definition of a High-Confidence Mitochondrial Proteome at Quantitative Scale. Cell Rep. 2017, 19, 2836–2852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janke, C.; Magiera, M.M.; Rathfelder, N.; Taxis, C.; Reber, S.; Maekawa, H.; Moreno-Borchart, A.; Doenges, G.; Schwob, E.; Schiebel, E.; et al. A Versatile Toolbox for PCR-Based Tagging of Yeast Genes: New Fluorescent Proteins, More Markers and Promoter Substitution Cassettes. Yeast 2004, 21, 947–962. [Google Scholar] [CrossRef]
- Eisenberg, T.; Schroeder, S.; Andryushkova, A.; Pendl, T.; Küttner, V.; Bhukel, A.; Mariño, G.; Pietrocola, F.; Harger, A.; Zimmermann, A.; et al. Nucleocytosolic Depletion of the Energy Metabolite Acetyl-Coenzyme A Stimulates Autophagy and Prolongs Lifespan. Cell Metab. 2014, 19, 431–444. [Google Scholar] [CrossRef] [Green Version]
- Daniel Gietz, R.; Woods, R.A. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. In Methods in Enzymology; Guthrie, C., Fink, G.R., Eds.; Guide to Yeast Genetics and Molecular and Cell Biology—Part B; Academic Press: Cambridge, MA, USA, 2002; Volume 350, pp. 87–96. [Google Scholar]
- Bernard, A.; Jin, M.; Xu, Z.; Klionsky, D.J. A Large-Scale Analysis of Autophagy-Related Gene Expression Identifies New Regulators of Autophagy. Autophagy 2015, 11, 2114–2122. [Google Scholar] [CrossRef]
- Kirisako, T.; Baba, M.; Ishihara, N.; Miyazawa, K.; Ohsumi, M.; Yoshimori, T.; Noda, T.; Ohsumi, Y. Formation Process of Autophagosome Is Traced with Apg8/Aut7p in Yeast. J. Cell Biol. 1999, 147, 435–446. [Google Scholar] [CrossRef] [Green Version]
- Kainz, K.; Tadic, J.; Zimmermann, A.; Pendl, T.; Carmona-Gutierrez, D.; Ruckenstuhl, C.; Eisenberg, T.; Madeo, F. Chapter Nineteen—Methods to Assess Autophagy and Chronological Aging in Yeast. In Methods in Enzymology; Galluzzi, L., Bravo-San Pedro, J.M., Kroemer, G., Eds.; Molecular Characterization of Autophagic Responses, Part B; Academic Press: Cambridge, MA, USA, 2017; Volume 588, pp. 367–394. [Google Scholar]
- Riezman, H.; Hase, T.; van Loon, A.P.; Grivell, L.A.; Suda, K.; Schatz, G. Import of Proteins into Mitochondria: A 70 Kilodalton Outer Membrane Protein with a Large Carboxy-Terminal Deletion Is Still Transported to the Outer Membrane. EMBO J. 1983, 2, 2161–2168. [Google Scholar] [CrossRef]
- Noda, T.; Klionsky, D.J. Chapter 3 The Quantitative Pho8Δ60 Assay of Nonspecific Autophagy. In Methods in Enzymology; Autophagy: Lower Eukaryotes and Non-Mammalian Systems, Part A; Academic Press: Cambridge, MA, USA, 2008; Volume 451, pp. 33–42. [Google Scholar]
- Hughes, A.L.; Hughes, C.E.; Henderson, K.A.; Yazvenko, N.; Gottschling, D.E. Selective Sorting and Destruction of Mitochondrial Membrane Proteins in Aged Yeast. eLife 2016, 5, e13943. [Google Scholar] [CrossRef]
- Ruckenstuhl, C.; Netzberger, C.; Entfellner, I.; Carmona-Gutierrez, D.; Kickenweiz, T.; Stekovic, S.; Gleixner, C.; Schmid, C.; Klug, L.; Sorgo, A.G.; et al. Lifespan Extension by Methionine Restriction Requires Autophagy-Dependent Vacuolar Acidification. PLoS Genet. 2014, 10, e1004347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sales, S.; Knittelfelder, O.; Shevchenko, A. Lipidomics of Human Blood Plasma by High-Resolution Shotgun Mass Spectrometry. Methods Mol. Biol. 2017, 1619, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Sales, S.; Graessler, J.; Ciucci, S.; Al-Atrib, R.; Vihervaara, T.; Schuhmann, K.; Kauhanen, D.; Sysi-Aho, M.; Bornstein, S.R.; Bickle, M.; et al. Gender, Contraceptives and Individual Metabolic Predisposition Shape a Healthy Plasma Lipidome. Sci. Rep. 2016, 6, 27710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuhmann, K.; Almeida, R.; Baumert, M.; Herzog, R.; Bornstein, S.R.; Shevchenko, A. Shotgun Lipidomics on a LTQ Orbitrap Mass Spectrometer by Successive Switching between Acquisition Polarity Modes. J. Mass Spectrom. JMS 2012, 47, 96–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuhmann, K.; Thomas, H.; Ackerman, J.M.; Nagornov, K.O.; Tsybin, Y.O.; Shevchenko, A. Intensity-Independent Noise Filtering in FT MS and FT MS/MS Spectra for Shotgun Lipidomics. Anal. Chem. 2017, 89, 7046–7052. [Google Scholar] [CrossRef] [PubMed]
- Schuhmann, K.; Srzentić, K.; Nagornov, K.O.; Thomas, H.; Gutmann, T.; Coskun, Ü.; Tsybin, Y.O.; Shevchenko, A. Monitoring Membrane Lipidome Turnover by Metabolic 15N Labeling and Shotgun Ultra-High-Resolution Orbitrap Fourier Transform Mass Spectrometry. Anal. Chem. 2017, 89, 12857–12865. [Google Scholar] [CrossRef] [PubMed]
- Herzog, R.; Schuhmann, K.; Schwudke, D.; Sampaio, J.L.; Bornstein, S.R.; Schroeder, M.; Shevchenko, A. LipidXplorer: A Software for Consensual Cross-Platform Lipidomics. PLoS ONE 2012, 7, e29851. [Google Scholar] [CrossRef] [Green Version]
- Folch, J.; Lees, M.; Stanley, G.H.S. A Simple Method for the Isolation and Purification of Total Lipides from Animal Tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Schneiter, R.; Daum, G. Analysis of Yeast Lipids. Methods Mol. Biol. 2006, 313, 75–84. [Google Scholar] [CrossRef]
- Knittelfelder, O.L.; Kohlwein, S.D. Thin-Layer Chromatography to Separate Phospholipids and Neutral Lipids from Yeast. Cold Spring Harb. Protoc. 2017, 2017. [Google Scholar] [CrossRef]
- Mangold, H.K. Thin-Layer Chromatography of Lipids. J. Am. Oil Chem. Soc. 1961, 38, 708–727. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji—An Open Source Platform for Biological Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Magrì, A.; Di Rosa, M.C.; Orlandi, I.; Guarino, F.; Reina, S.; Guarnaccia, M.; Morello, G.; Spampinato, A.; Cavallaro, S.; Messina, A.; et al. Deletion of Voltage-Dependent Anion Channel 1 Knocks Mitochondria down Triggering Metabolic Rewiring in Yeast. Cell. Mol. Life Sci. 2019. [Google Scholar] [CrossRef]
- Dihanich, M.; Suda, K.; Schatz, G. A Yeast Mutant Lacking Mitochondrial Porin Is Respiratory-Deficient, but Can Recover Respiration with Simultaneous Accumulation of an 86-Kd Extramitochondrial Protein. EMBO J. 1987, 6, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Miyata, N.; Fujii, S.; Kuge, O. Porin Proteins Have Critical Functions in Mitochondrial Phospholipid Metabolism in Yeast. J. Biol. Chem. 2018, 293, 17593–17605. [Google Scholar] [CrossRef] [Green Version]
- Blachly-Dyson, E.; Song, J.; Wolfgang, W.J.; Colombini, M.; Forte, M. Multicopy Suppressors of Phenotypes Resulting from the Absence of Yeast VDAC Encode a VDAC-like Protein. Mol. Cell. Biol. 1997, 17, 5727–5738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graef, M.; Nunnari, J. Mitochondria Regulate Autophagy by Conserved Signalling Pathways. EMBO J. 2011, 30, 2101–2114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shintani, T.; Reggiori, F. Chapter Four Fluorescence Microscopy-Based Assays for Monitoring Yeast Atg Protein Trafficking. In Methods in Enzymology; Autophagy: Lower Eukaryotes and Non-Mammalian Systems, Part A; Academic Press: Cambridge, MA, USA, 2008; Volume 451, pp. 43–56. [Google Scholar]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Arozena, A.A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy (3rd Edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakatogawa, H.; Suzuki, K.; Kamada, Y.; Ohsumi, Y. Dynamics and Diversity in Autophagy Mechanisms: Lessons from Yeast. Nat. Rev. Mol. Cell Biol. 2009, 10, 458–467. [Google Scholar] [CrossRef] [Green Version]
- Loewen, C.J.R.; Gaspar, M.L.; Jesch, S.A.; Delon, C.; Ktistakis, N.T.; Henry, S.A.; Levine, T.P. Phospholipid Metabolism Regulated by a Transcription Factor Sensing Phosphatidic Acid. Science 2004, 304, 1644–1647. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.C.; Xu, X.; Blachly-Dyson, E.; Forte, M.; Colombini, M. The Role of Yeast VDAC Genes on the Permeability of the Mitochondrial Outer Membrane. J. Membr. Biol. 1998, 161, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Sakaue, H.; Shiota, T.; Ishizaka, N.; Kawano, S.; Tamura, Y.; Tan, K.S.; Imai, K.; Motono, C.; Hirokawa, T.; Taki, K.; et al. Porin Associates with Tom22 to Regulate the Mitochondrial Protein Gate Assembly. Mol. Cell 2019, 73, 1044–1055.e8. [Google Scholar] [CrossRef] [Green Version]
- Ellenrieder, L.; Dieterle, M.P.; Doan, K.N.; Mårtensson, C.U.; Floerchinger, A.; Campo, M.L.; Pfanner, N.; Becker, T. Dual Role of Mitochondrial Porin in Metabolite Transport across the Outer Membrane and Protein Transfer to the Inner Membrane. Mol. Cell 2019, 73, 1056–1065.e7. [Google Scholar] [CrossRef] [Green Version]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-Mediated Coupling of Endoplasmic Reticulum and Mitochondrial Ca2+ Channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maldonado, E.N.; Sheldon, K.L.; DeHart, D.N.; Patnaik, J.; Manevich, Y.; Townsend, D.M.; Bezrukov, S.M.; Rostovtseva, T.K.; Lemasters, J.J. Voltage-Dependent Anion Channels Modulate Mitochondrial Metabolism in Cancer Cells. J. Biol. Chem. 2013, 288, 11920–11929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosc, C.; Broin, N.; Fanjul, M.; Saland, E.; Farge, T.; Courdy, C.; Batut, A.; Masoud, R.; Larrue, C.; Skuli, S.; et al. Autophagy Regulates Fatty Acid Availability for Oxidative Phosphorylation through Mitochondria-Endoplasmic Reticulum Contact Sites. Nat. Commun. 2020, 11, 4056. [Google Scholar] [CrossRef]
- Strogolova, V.; Orlova, M.; Shevade, A.; Kuchin, S. Mitochondrial Porin Por1 and Its Homolog Por2 Contribute to the Positive Control of Snf1 Protein Kinase in Saccharomyces Cerevisiae. Eukaryot. Cell 2012, 11, 1568–1572. [Google Scholar] [CrossRef] [Green Version]
- Pujol-Carrion, N.; Petkova, M.I.; Serrano, L.; de la Torre-Ruiz, M.A. The MAP Kinase Slt2 Is Involved in Vacuolar Function and Actin Remodeling in Saccharomyces Cerevisiae Mutants Affected by Endogenous Oxidative Stress. Appl. Environ. Microbiol. 2013, 79, 6459–6471. [Google Scholar] [CrossRef] [Green Version]
- Kornmann, B.; Currie, E.; Collins, S.R.; Schuldiner, M.; Nunnari, J.; Weissman, J.S.; Walter, P. An ER-Mitochondria Tethering Complex Revealed by a Synthetic Biology Screen. Science 2009, 325, 477–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nebauer, R.; Rosenberger, S.; Daum, G. Phosphatidylethanolamine, a Limiting Factor of Autophagy in Yeast Strains Bearing a Defect in the Carboxypeptidase Y Pathway of Vacuolar Targeting. J. Biol. Chem. 2007, 282, 16736–16743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigié, P.; Cougouilles, E.; Bhatia-Kiššová, I.; Salin, B.; Blancard, C.; Camougrand, N. Mitochondrial Phosphatidylserine Decarboxylase 1 (Psd1) Is Involved in Nitrogen Starvation-Induced Mitophagy in Yeast. J. Cell Sci. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Broeskamp, F.; Edrich, E.S.M.; Knittelfelder, O.; Neuhaus, L.; Meyer, T.; Heyden, J.; Habernig, L.; Kreppel, F.; Gourlay, C.W.; Rockenfeller, P. Porin 1 Modulates Autophagy in Yeast. Cells 2021, 10, 2416. https://doi.org/10.3390/cells10092416
Broeskamp F, Edrich ESM, Knittelfelder O, Neuhaus L, Meyer T, Heyden J, Habernig L, Kreppel F, Gourlay CW, Rockenfeller P. Porin 1 Modulates Autophagy in Yeast. Cells. 2021; 10(9):2416. https://doi.org/10.3390/cells10092416
Chicago/Turabian StyleBroeskamp, Filomena, Elizabeth S. M. Edrich, Oskar Knittelfelder, Lisa Neuhaus, Thorsten Meyer, Jonas Heyden, Lukas Habernig, Florian Kreppel, Campbell W. Gourlay, and Patrick Rockenfeller. 2021. "Porin 1 Modulates Autophagy in Yeast" Cells 10, no. 9: 2416. https://doi.org/10.3390/cells10092416
APA StyleBroeskamp, F., Edrich, E. S. M., Knittelfelder, O., Neuhaus, L., Meyer, T., Heyden, J., Habernig, L., Kreppel, F., Gourlay, C. W., & Rockenfeller, P. (2021). Porin 1 Modulates Autophagy in Yeast. Cells, 10(9), 2416. https://doi.org/10.3390/cells10092416