
Interferon-Induced HERC5 Inhibits Ebola Virus Particle Production and Is Antagonized by Ebola Glycoprotein


 , and
, and
Abstract
{kind=link}
{kind=link}
{kind=link}
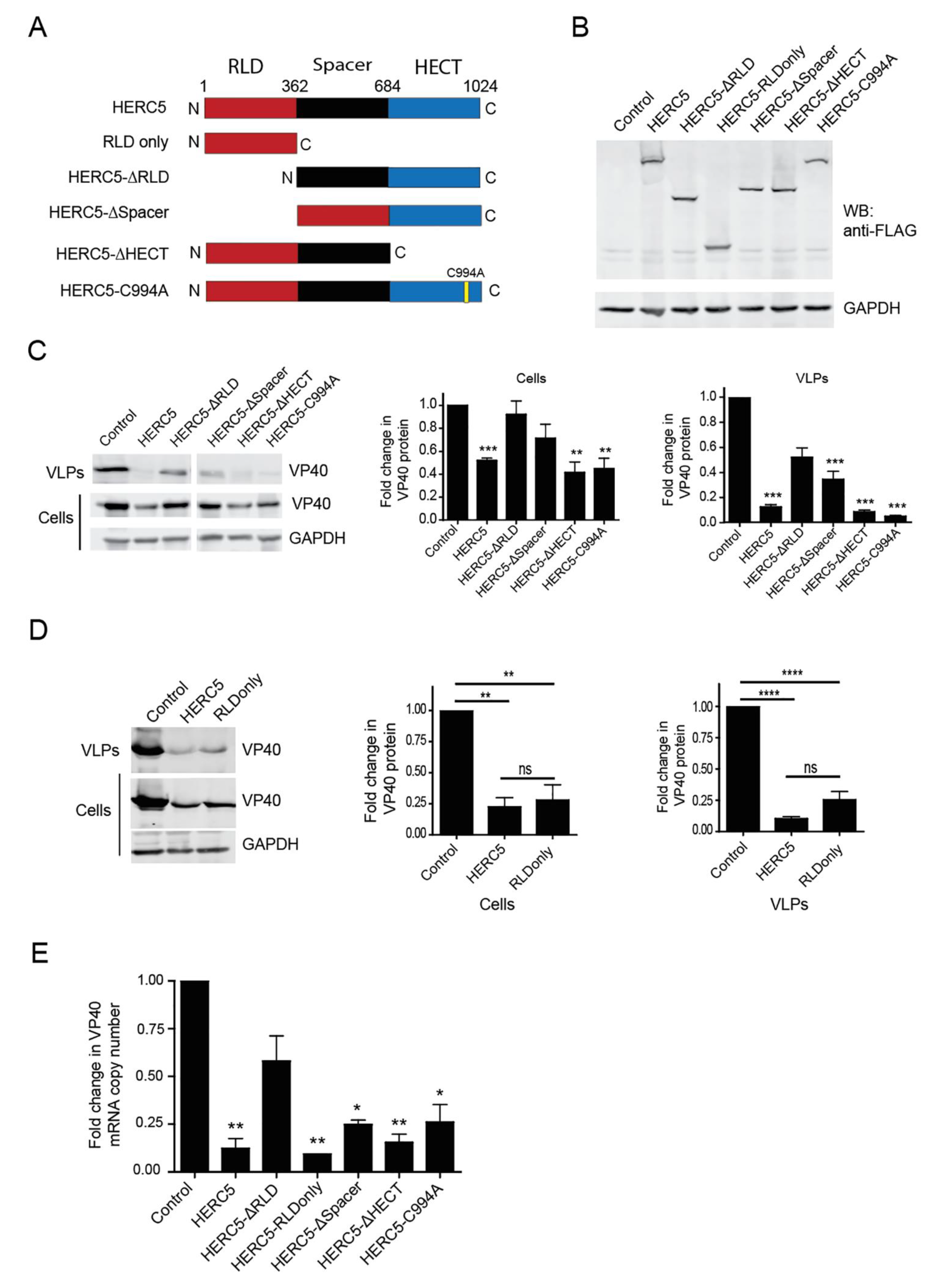{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Plasmids, Transfections, Antibodies and Quantitative Western Blotting
2.3. Confocal Immunofluorescence Microscopy
2.4. Transmission Electron Microscopy
2.5. Quantitative PCR
2.6. trVLP Assay
2.7. Cell Viability Assay
2.8. Statistical Analyses
3. Results
3.1. HERC5 Inhibits EBOV trVLP Replication
3.2. HERC5 Inhibits EBOV VP40 Particle Production
3.3. HERC5 RLD Is Necessary and Sufficient for Inhibition of VP40 Particle Production
3.4. HERC5 Depletes VP40 mRNA Independently of ZAP
3.5. EBOV GP and L Proteins Antagonize HERC5
3.6. EBOV and MARV GP Differentially Antagonize HERC5 Inhibition of EBOV trVLP Replication
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feldmann, H.; Geisbert, T.W. Ebola haemorrhagic fever. Lancet Lond. 2011, 377, 849–862. [Google Scholar] [CrossRef]
- Rubins, K.H.; Hensley, L.; Wahl-Jensen, V.; DiCaprio, K.M.D.; Young, H.; Reed, D.S.; Jahrling, P.B.; O Brown, P.; Relman, D.; Geisbert, T.W. The temporal program of peripheral blood gene expression in the response of nonhuman primates to Ebola hemorrhagic fever. Genome Biol. 2007, 8, R174. [Google Scholar] [CrossRef]
- Caballero, I.S.; Honko, A.N.; Gire, S.K.; Winnicki, S.M.; Melé, M.; Gerhardinger, C.; Lin, A.E.; Rinn, J.L.; Sabeti, P.C.; Hensley, L.E.; et al. In vivo Ebola virus infection leads to a strong innate response in circulating immune cells. BMC Genom. 2016, 17, 707. [Google Scholar] [CrossRef] [PubMed]
- Kash, J.C.; Walters, K.-A.; Kindrachuk, J.; Baxter, D.; Scherler, K.; Janosko, K.B.; Adams, R.D.; Herbert, A.S.; James, R.M.; Stonier, S.W.; et al. Longitudinal peripheral blood transcriptional analysis of a patient with severe Ebola virus disease. Sci. Transl. Med. 2017, 9, eaai9321. [Google Scholar] [CrossRef] [PubMed]
- Speranza, E.; Bixler, S.L.; Altamura, L.A.; Arnold, C.E.; Pratt, W.D.; Taylor-Howell, C.; Burrows, C.; Aguilar, W.; Rossi, F.; Shamblin, J.D.; et al. A conserved transcriptional response to intranasal Ebola virus exposure in nonhuman primates prior to onset of fever. Sci. Transl. Med. 2018, 10, eaaq1016. [Google Scholar] [CrossRef]
- Versteeg, K.; Menicucci, A.R.; Woolsey, C.; Mire, C.E.; Geisbert, J.B.; Cross, R.W.; Agans, K.N.; Jeske, D.; Messaoudi, I.; Geisbert, T.W. Infection with the Makona variant results in a delayed and distinct host immune response compared to previous Ebola virus variants. Sci. Rep. 2017, 7, 9730. [Google Scholar] [CrossRef]
- Cilloniz, C.; Ebihara, H.; Ni, C.; Neumann, G.; Korth, M.J.; Kelly, S.M.; Kawaoka, Y.; Feldmann, H.; Katze, M.G. Functional genomics reveals the induction of inflammatory response and metalloproteinase gene expression during lethal ebola virus infection. J. Virol. 2011, 85, 9060–9068. [Google Scholar] [CrossRef]
- Speranza, E.; Altamura, L.A.; Kulcsar, K.; Bixler, S.L.; Rossi, C.A.; Schoepp, R.J.; Nagle, E.; Aguilar, W.; Douglas, C.E.; Delp, K.L.; et al. Comparison of transcriptomic platforms for analysis of whole blood from ebola-infected cynomolgus macaques. Sci. Rep. 2017, 7, 14756. [Google Scholar] [CrossRef]
- Garamszegi, S.; Yen, J.Y.; Honko, A.N.; Geisbert, J.B.; Rubins, K.H.; Geisbert, T.W.; Xia, Y.; Hensley, L.E.; Connor, J.H. Transcriptional correlates of disease outcome in anticoagulant-treated non-human primates infected with ebolavirus. PLoS Negl. Trop. Dis. 2014, 8, e3061. [Google Scholar] [CrossRef]
- Liu, X.; Speranza, E.; Muñoz-Fontela, C.; Haldenby, S.; Rickett, N.Y.; Garcia-Dorival, I.; Fang, Y.; Hall, Y.; Zekeng, E.-G.; Lüdtke, A.; et al. Transcriptomic signatures differentiate survival from fatal outcomes in humans infected with Ebola virus. Genome Biol. 2017, 18, 4. [Google Scholar] [CrossRef]
- Eisfeld, A.J.; Halfmann, P.J.; Wendler, J.P.; Kyle, J.E.; Burnum-Johnson, K.E.; Peralta, Z.; Maemura, T.; Walters, K.B.; Watanabe, T.; Fukuyama, S.; et al. Multi-platform ’omics analysis of human Ebola virus disease pathogenesis. Cell Host Microbe 2017, 22, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Yen, J.Y.; Garamszegi, S.; Geisbert, J.B.; Rubins, K.H.; Geisbert, T.W.; Honko, A.; Xia, Y.; Connor, J.H.; Hensley, L.E. Therapeutics of Ebola hemorrhagic fever: Whole-genome transcriptional analysis of successful disease mitigation. J. Infect. Dis. 2011, 204, S1043–S1052. [Google Scholar] [CrossRef]
- Kash, J.C.; Mühlberger, E.; Carter, V.; Grosch, M.; Perwitasari, O.; Proll, S.C.; Thomas, M.J.; Weber, F.; Klenk, H.-D.; Katze, M.G. Global suppression of the host antiviral response by Ebola- and Marburgviruses: Increased antagonism of the type I interferon response is associated with enhanced virulence. J. Virol. 2006, 80, 3009–3020. [Google Scholar] [CrossRef] [PubMed]
- Hartman, A.L.; Ling, L.; Nichol, S.T.; Hibberd, M.L. Whole-genome expression profiling reveals that inhibition of host innate immune response pathways by Ebola virus can be reversed by a single amino acid change in the VP35 protein. J. Virol. 2008, 82, 5348–5358. [Google Scholar] [CrossRef]
- Hartman, A.L.; Bird, B.H.; Towner, J.S.; Antoniadou, Z.-A.; Zaki, S.R.; Nichol, S.T. Inhibition of IRF-3 activation by VP35 is critical for the high level of virulence of ebola virus. J. Virol. 2008, 82, 2699–2704. [Google Scholar] [CrossRef] [PubMed]
- Prins, K.C.; Delpeut, S.; Leung, D.W.; Reynard, O.; Volchkova, V.A.; Reid, S.P.; Ramanan, P.; Cárdenas, W.B.; Amarasinghe, G.K.; Volchkov, V.E.; et al. Mutations abrogating VP35 interaction with double-stranded RNA render Ebola virus avirulent in guinea pigs. J. Virol. 2010, 84, 3004–3015. [Google Scholar] [CrossRef] [PubMed]
- Basler, C.F.; Amarasinghe, G.K. Evasion of interferon responses by Ebola and Marburg viruses. J. Interf. Cytokine Res. 2009, 29, 511–520. [Google Scholar] [CrossRef]
- Messaoudi, I.; Amarasinghe, G.K.; Basler, C.F. Filovirus pathogenesis and immune evasion: Insights from Ebola virus and Marburg virus. Nat. Rev. Microbiol. 2015, 13, 663–676. [Google Scholar] [CrossRef]
- Luthra, P.; Ramanan, P.; Mire, C.E.; Weisend, C.; Tsuda, Y.; Yen, B.; Liu, G.; Leung, D.W.; Geisbert, T.W.; Ebihara, H.; et al. Mutual antagonism between the Ebola Virus VP35 protein and the RIG-I activator PACT determines infection outcome. Cell Host Microbe 2013, 14, 74–84. [Google Scholar] [CrossRef]
- Bieniasz, P.D. Intrinsic immunity: A front-line defense against viral attack. Nat. Immunol. 2004, 5, 1109–1115. [Google Scholar] [CrossRef]
- Neil, S.J.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef]
- Jouvenet, N.; Neil, S.J.D.; Zhadina, M.; Zang, T.; Kratovac, Z.; Lee, Y.; McNatt, M.; Hatziioannou, T.; Bieniasz, P.D. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J. Virol. 2009, 83, 1837–1844. [Google Scholar] [CrossRef] [PubMed]
- Kaletsky, R.L.; Francica, J.R.; Agrawal-Gamse, C.; Bates, P. Tetherin-mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proc. Natl. Acad. Sci. USA 2009, 106, 2886–2891. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, T.; Noda, T.; Urata, S.; Kawaoka, Y.; Yasuda, J. Inhibition of Lassa and Marburg virus production by tetherin. J. Virol. 2009, 83, 2382–2385. [Google Scholar] [CrossRef] [PubMed]
- Radoshitzky, S.R.; Dong, L.; Chi, X.; Clester, J.C.; Retterer, C.; Spurgers, K.; Kuhn, J.H.; Sandwick, S.; Ruthel, G.; Kota, K.; et al. Infectious Lassa virus, but not filoviruses, is restricted by BST-2/tetherin. J. Virol. 2010, 84, 10569–10580. [Google Scholar] [CrossRef] [PubMed]
- Huang, I.-C.; Bailey, C.C.; Weyer, J.L.; Radoshitzky, S.R.; Becker, M.M.; Chiang, J.J.; Brass, A.L.; Ahmed, A.A.; Chi, X.; Dong, L.; et al. Distinct Patterns of IFITM-Mediated Restriction of Filoviruses, SARS Coronavirus, and Influenza A Virus. PLoS Pathog. 2011, 7, e1001258. [Google Scholar] [CrossRef]
- Wrensch, F.; Karsten, C.B.; Gnirß, K.; Hoffmann, M.; Lu, K.; Takada, A.; Winkler, M.; Simmons, G.; Pöhlmann, S. Interferon-Induced transmembrane protein–mediated inhibition of host cell entry of Ebolaviruses. J. Infect. Dis. 2015, 212, S210–S218. [Google Scholar] [CrossRef] [PubMed]
- Menicucci, A.R.; Versteeg, K.; Woolsey, C.; Mire, C.E.; Geisbert, J.B.; Cross, R.W.; Agans, K.N.; Jankeel, A.; Geisbert, T.W.; Messaoudi, I. Transcriptome analysis of circulating immune cell subsets highlight the role of Monocytes in zaire Ebola virus Makona pathogenesis. Front. Immunol. 2017, 8, 1372. [Google Scholar] [CrossRef]
- Woods, M.W.; Tong, J.G.; Tom, S.K.; Szabo, P.A.; Cavanagh, P.C.; Dikeakos, J.D.; Haeryfar, S.M.M.; Barr, S.D. Interferon-induced HERC5 is evolving under positive selection and inhibits HIV-1 particle production by a novel mechanism targeting Rev/RRE-dependent RNA nuclear export. Retrovirology 2014, 11, 27. [Google Scholar] [CrossRef]
- Woods, M.W.; Kelly, J.N.; Hattlmann, C.J.; Tong, J.G.K.; Xu, L.S.; Coleman, M.D.; Quest, G.R.; Smiley, J.R.; Barr, S.D. Human HERC5 restricts an early stage of HIV-1 assembly by a mechanism correlating with the ISGylation of Gag. Retrovirology 2011, 8, 95. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Zhong, G.; Zhu, L.; Liu, X.; Shan, Y.; Feng, H.; Bu, Z.; Chen, H.; Wang, C. Herc5 attenuates influenza A virus by catalyzing ISGylation of viral NS1 protein. J. Immunol. 2010, 184, 5777–5790. [Google Scholar] [CrossRef] [PubMed]
- Versteeg, G.A.; Hale, B.G.; van Boheemen, S.; Wolff, T.; Lenschow, D.J.; Garcia-Sastre, A. Species-specific antagonism of host ISGylation by the influenza B virus NS1 protein. J. Virol. 2010, 84, 5423–5430. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Hsiang, T.Y.; Kuo, R.L.; Krug, R.M. ISG15 conjugation system targets the viral NS1 protein in influenza A virus-infected cells. Proc. Natl. Acad. Sci. USA 2010, 107, 2253–2258. [Google Scholar] [CrossRef]
- Durfee, L.A.; Lyon, N.; Seo, K.; Huibregtse, J.M. The ISG15 conjugation system broadly targets newly synthesized proteins: Implications for the antiviral function of ISG15. Mol. Cell 2010, 38, 722–732. [Google Scholar] [CrossRef]
- Paparisto, E.; Woods, M.W.; Coleman, M.D.; Moghadasi, S.A.; Kochar, D.S.; Tom, S.K.; Kohio, H.P.; Gibson, R.M.; Rohringer, T.J.; Hunt, N.R.; et al. Evolution-guided structural and functional analyses of the HERC family reveal an ancient marine origin and determinants of antiviral activity. J. Virol. 2018, 92, e0052818. [Google Scholar] [CrossRef]
- Wong, J.J.Y.; Pung, Y.F.; Sze, N.S.; Chin, K.C. HERC5 is an IFN-induced HECT-type E3 protein ligase that mediates type I IFN-induced ISGylation of protein targets. Proc. Natl. Acad. Sci. USA 2006, 103, 10735–10740. [Google Scholar] [CrossRef]
- Dastur, A.; Beaudenon, S.; Kelley, M.; Krug, R.M.; Huibregtse, J.M. Herc5, an interferon-induced HECT E3 enzyme, is required for conjugation of ISG15 in human cells. J. Biol. Chem. 2006, 281, 4334–4338. [Google Scholar] [CrossRef]
- Watanabe, S.; Watanabe, T.; Noda, T.; Takada, A.; Feldmann, H.; Jasenosky, L.D.; Kawaoka, Y. Production of novel ebola virus-like particles from cDNAs: An alternative to ebola virus generation by reverse genetics. J. Virol. 2004, 78, 999–1005. [Google Scholar] [CrossRef]
- Watt, A.; Moukambi, F.; Banadyga, L.; Groseth, A.; Callison, J.; Herwig, A.; Ebihara, H.; Feldmann, H.; Hoenen, T. A novel life cycle modeling system for Ebola virus shows a genome length-dependent role of VP24 in virus infectivity. J. Virol. 2014, 88, 10511–10524. [Google Scholar] [CrossRef]
- Wendt, L.; Kämper, L.; Schmidt, M.L.; Mettenleiter, T.C.; Hoenen, T. Analysis of a putative late domain using an Ebola virus transcription and replication-competent virus-like particle system. J. Infect. Dis. 2018, 218, S355–S359. [Google Scholar] [CrossRef] [PubMed]
- Côté, M.; Misasi, J.; Ren, T.; Bruchez, A.; Lee, K.; Filone, C.M.; Hensley, L.; Li, Q.; Ory, D.; Chandran, K.; et al. Small molecule inhibitors reveal Niemann–Pick C1 is essential for Ebola virus infection. Nature 2011, 477, 344–348. [Google Scholar] [CrossRef]
- Chandran, K.; Sullivan, N.J.; Felbor, U.; Whelan, S.P.; Cunningham, J.M. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science 2005, 308, 1643–1645. [Google Scholar] [CrossRef]
- Schmidt, M.L.; Hoenen, T. Characterization of the catalytic center of the Ebola virus L polymerase. PLoS Negl. Trop. Dis. 2017, 11, e0005996. [Google Scholar] [CrossRef] [PubMed]
- Hoenen, T.; Watt, A.; Mora, A.; Feldmann, H. Modeling the lifecycle of Ebola virus under biosafety level 2 conditions with virus-like particles containing tetracistronic minigenomes. J. Vis. Exp. 2014, 52381. [Google Scholar] [CrossRef]
- Frick, C.; Ollmann-Saphire, E.; Stahelin, R. Live-cell imaging of Ebola virus matrix protein VP40. FASEB J. 2015, 29, 886.4. [Google Scholar] [CrossRef]
- Noda, T.; Sagara, H.; Suzuki, E.; Takada, A.; Kida, H.; Kawaoka, Y. Ebola virus VP40 drives the formation of virus-like filamentous particles along with GP. J. Virol. 2002, 76, 4855–4865. [Google Scholar] [CrossRef]
- Johnson, K.A.; Taghon, G.J.F.; Scott, J.L.; Stahelin, R.V. The Ebola Virus matrix protein, VP40, requires phosphatidylinositol extensive oligomerization at the plasma membrane and viral egress. Sci. Rep. 2016, 6, 19125. [Google Scholar] [CrossRef]
- Bick, M.J.; Carroll, J.W.; Gao, G.; Goff, S.P.; Rice, C.M.; MacDonald, M.R. Expression of the zinc-finger antiviral protein inhibits alphavirus replication. J. Virol. 2003, 77, 11555–11562. [Google Scholar] [CrossRef]
- Kerns, J.A.; Emerman, M.; Malik, H.S. Positive selection and increased antiviral activity associated with the PARP-containing isoform of human zinc-finger antiviral protein. PLoS Genet. 2008, 4, e21. [Google Scholar] [CrossRef]
- Mao, R.; Nie, H.; Cai, D.; Zhang, J.; Liu, H.; Yan, R.; Cuconati, A.; Block, T.M.; Guo, J.-T.; Guo, H. Inhibition of Hepatitis B virus replication by the host zinc finger antiviral protein. PLoS Pathog. 2013, 9, e1003494. [Google Scholar] [CrossRef]
- Muller, S.; Moller, P.; Bick, M.J.; Wurr, S.; Becker, S.; Gunther, S.; Kummerer, B.M. Inhibition of filovirus replication by the zinc finger antiviral protein. J. Virol. 2007, 81, 2391–2400. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tu, F.; Zhu, Y.; Gao, G. Zinc-finger antiviral protein inhibits XMRV infection. PLoS ONE 2012, 7, e39159. [Google Scholar] [CrossRef][Green Version]
- Zhang, Y.; Burke, C.W.; Ryman, K.D.; Klimstra, W.B. Identification and characterization of interferon-induced proteins that inhibit alphavirus replication. J. Virol. 2007, 81, 11246–11255. [Google Scholar] [CrossRef]
- Zhu, Y.; Chen, G.; Lv, F.; Wang, X.; Ji, X.; Xu, Y.; Sun, J.; Wu, L.; Zheng, Y.-T.; Gao, G. Zinc-finger antiviral protein inhibits HIV-1 infection by selectively targeting multiply spliced viral mRNAs for degradation. Proc. Natl. Acad. Sci. USA 2011, 108, 15834–15839. [Google Scholar] [CrossRef]
- Li, M.M.H.; Aguilar, E.G.; Michailidis, E.; Pabon, J.; Park, P.; Wu, X.; de Jong, Y.P.; Schneider, W.M.; Molina, H.; Rice, C.M.; et al. Characterization of novel splice variants of zinc finger antiviral protein (ZAP). J. Virol. 2019, 93, e00715–e00719. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, S.; Shiratori, S.; Yamato, H.; Kameyama, T.; Kitatsuji, C.; Kashigi, F.; Goto, S.; Kameoka, S.; Fujikura, D.; Yamada, T.; et al. ZAPS is a potent stimulator of signaling mediated by the RNA helicase RIG-I during antiviral responses. Nat. Immunol. 2011, 12, 37–44. [Google Scholar] [CrossRef]
- Sánchez-Tena, S.; Cubillos-Rojas, M.; Schneider, T.; Rosa, J.L. Functional and pathological relevance of HERC family proteins: A decade later. Cell. Mol. Life Sci. 2016, 73, 1955–1968. [Google Scholar] [CrossRef]
- Hochrainer, K.; Mayer, H.; Baranyi, U.; Binder, B.; Lipp, J.; Kroismayr, R. The human HERC family of ubiquitin ligases: Novel members, genomic organization, expression profiling, and evolutionary aspects. Genomics 2005, 85, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Jacquet, S.; Pontier, D.; Etienne, L. Rapid evolution of HERC6 and duplication of a chimeric HERC5/6 gene in rodents and bats suggest an overlooked role of HERCs in mammalian immunity. Front. Immunol. 2020, 11, 605270. [Google Scholar] [CrossRef]
- Duggal, N.K.; Emerman, M. Evolutionary conflicts between viruses and restriction factors shape immunity. Nat. Rev. 2012, 12, 687–695. [Google Scholar] [CrossRef]
- Daugherty, M.D.; Young, J.M.; Kerns, J.A.; Malik, H.S. Rapid evolution of PARP genes suggests a broad role for ADP-Ribosylation in host-virus conflicts. PLoS Genet. 2014, 10, e1004403. [Google Scholar] [CrossRef] [PubMed]
- Xuan, Y.; Gong, D.; Qi, J.; Han, C.; Deng, H.; Gao, G. ZAP inhibits murine gammaherpesvirus 68 ORF64 expression and is antagonized by RTA. J. Virol. 2013, 87, 2735–2743. [Google Scholar] [CrossRef]
- Guo, X.; Carroll, J.-W.N.; MacDonald, M.R.; Goff, S.P.; Gao, G. The zinc finger antiviral protein directly binds to specific viral mRNAs through the CCCH zinc finger motifs. J. Virol. 2004, 78, 12781–12787. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wang, X.; Goff, S.P.; Gao, G. Translational repression precedes and is required for ZAP-mediated mRNA decay. EMBO J. 2012, 31, 4236–4246. [Google Scholar] [CrossRef]
- Karki, S.; Li, M.M.H.; Schoggins, J.W.; Tian, S.; Rice, C.M.; MacDonald, M.R. Multiple interferon stimulated genes synergize with the zinc finger antiviral protein to mediate anti-alphavirus activity. PLoS ONE 2012, 7, e37398. [Google Scholar] [CrossRef]
- Chen, G.; Guo, X.; Lv, F.; Xu, Y.; Gao, G. p72 DEAD box RNA helicase is required for optimal function of the zinc-finger antiviral protein. Proc. Natl. Acad. Sci. USA 2008, 105, 4352–4357. [Google Scholar] [CrossRef]
- Guo, X.; Ma, J.; Sun, J.; Gao, G. The zinc-finger antiviral protein recruits the RNA processing exosome to degrade the target mRNA. Proc. Natl. Acad. Sci. USA 2007, 104, 151–156. [Google Scholar] [CrossRef]
- Okumura, A.; Pitha, P.M.; Harty, R.N. ISG15 inhibits Ebola VP40 VLP budding in an L-domain-dependent manner by blocking Nedd4 ligase activity. Proc. Natl. Acad. Sci. USA 2008, 105, 3974–3979. [Google Scholar] [CrossRef]
- Bray, M. The role of the Type I interferon response in the resistance of mice to filovirus infection. J. Gen. Virol. 2001, 82, 1365–1373. [Google Scholar] [CrossRef]
- Ebihara, H.; Takada, A.; Kobasa, D.; Jones, S.; Neumann, G.; Theriault, S.; Bray, M.; Feldmann, H.; Kawaoka, Y. Molecular determinants of Ebola virus virulence in mice. PLoS Pathog. 2006, 2, e73. [Google Scholar] [CrossRef] [PubMed]
- Guzzo, C.; Jung, M.; Graveline, A.; Banfield, B.W.; Gee, K. IL-27 increases BST-2 expression in human monocytes and T cells independently of type I IFN. Sci. Rep. 2012, 2, 974. [Google Scholar] [CrossRef]
- Bailey, C.C.; Huang, I.-C.; Kam, C.; Farzan, M. Ifitm3 limits the severity of acute influenza in mice. PLoS Pathog. 2012, 8, e1002909. [Google Scholar] [CrossRef]
- Kroismayr, R.; Baranyi, U.; Stehlik, C.; Dorfleutner, A.; Binder, B.R.; Lipp, J. HERC5, a HECT E3 ubiquitin ligase tightly regulated in LPS activated endothelial cells. J. Cell Sci. 2004, 117, 4749–4756. [Google Scholar] [CrossRef]
- Kühl, A.; Banning, C.; Marzi, A.; Votteler, J.; Steffen, I.; Bertram, S.; Glowacka, I.; Konrad, A.; Stürzl, M.; Guo, J.-T.; et al. The Ebola virus glycoprotein and HIV-1 Vpu employ different strategies to counteract the antiviral factor Tetherin. J. Infect. Dis. 2011, 204, S850–S860. [Google Scholar] [CrossRef] [PubMed]
- Lopez, L.A.; Yang, S.J.; Hauser, H.; Exline, C.M.; Haworth, K.G.; Oldenburg, J.; Cannon, P.M. Ebola virus glycoprotein counteracts BST-2/tetherin restriction in a sequence-independent manner that does not require tetherin surface removal. J. Virol. 2010, 84, 7243–7255. [Google Scholar] [CrossRef] [PubMed]
- Lopez, L.A.; Yang, S.J.; Exline, C.M.; Rengarajan, S.; Haworth, K.G.; Cannon, P.M. Anti-tetherin activities of HIV-1 Vpu and Ebola virus glycoprotein do not involve removal of Tetherin from lipid rafts. J. Virol. 2012, 86, 5467–5480. [Google Scholar] [CrossRef]
- González-Hernández, M.; Hoffmann, M.; Brinkmann, C.; Nehls, J.; Winkler, M.; Schindler, M.; Pöhlmann, S. A GXXXA motif in the transmembrane domain of the ebola virus glycoprotein is required for Tetherin antagonism. J. Virol. 2018, 92, e00403–e00418. [Google Scholar] [CrossRef]
- Burgt, N.H.V.; Kaletsky, R.L.; Bates, P. Requirements within the Ebola Viral Glycoprotein for tetherin antagonism. Viruses 2015, 7, 5587–5602. [Google Scholar] [CrossRef]
- Gustin, J.K.; Bai, Y.; Moses, A.V.; Douglas, J.L. Ebola virus glycoprotein promotes enhanced viral egress by preventing Ebola VP40 from associating with the host restriction factor BST2/Tetherin. J. Infect. Dis. 2015, 212, S181–S190. [Google Scholar] [CrossRef]
- Brinkmann, C.; Nehlmeier, I.; Walendy-Gnirß, K.; Nehls, J.; González Hernández, M.; Hoffmann, M.; Qiu, X.; Takada, A.; Schindler, M.; Pöhlmann, S. The Tetherin antagonism of the Ebola virus glycoprotein requires an intact receptor-binding domain and can be blocked by GP1-specific antibodies. J. Virol. 2016, 90, 11075–11086. [Google Scholar] [CrossRef]
- Gnirß, K.; Fiedler, M.; Krämer-Kühl, A.; Bolduan, S.; Mittler, E.; Becker, S.; Schindler, M.; Pöhlmann, S. Analysis of determinants in filovirus glycoproteins required for tetherin antagonism. Viruses 2014, 6, 1654–1671. [Google Scholar] [CrossRef]
- Wang, M.K.; Lim, S.-Y.; Lee, S.M.; Cunningham, J.M. Biochemical basis for increased activity of Ebola Glycoprotein in the 2013-16 epidemic. Cell Host Microbe 2017, 21, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Marzi, A.; Chadinah, S.; Haddock, E.; Sow, S.; Massaquoi, M.; Feldmann, H. Recently identified mutations in the Ebola Virus-makona genome do not alter pathogenicity in animal models. Cell Rep. 2018, 23, 1806–1816. [Google Scholar] [CrossRef]
- Sullivan, N.J.; Peterson, M.; Yang, Z.; Kong, W.; Duckers, H.; Nabel, E.; Nabel, G.J. Ebola virus glycoprotein toxicity is mediated by a dynamin-dependent protein-trafficking pathway. J. Virol. 2005, 79, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Iampietro, M.; Younan, P.; Nishida, A.; Dutta, M.; Lubaki, N.M.; Santos, R.I.; Koup, R.A.; Katze, M.G.; Bukreyev, A. Ebola virus glycoprotein directly triggers T lymphocyte death despite of the lack of infection. PLoS Pathog. 2017, 13, e1006397. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.M.; Phan, A.; Bo, Y.; LeBlond, N.D.; Smith, T.K.T.; Laroche, G.; Giguère, P.M.; Fullerton, M.D.; Pelchat, M.; Kobasa, D.; et al. Ebola virus triggers receptor tyrosine kinase-dependent signaling to promote the delivery of viral particles to entry-conducive intracellular compartments. PLoS Pathog. 2021, 17, e1009275. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paparisto, E.; Hunt, N.R.; Labach, D.S.; Coleman, M.D.; Di Gravio, E.J.; Dodge, M.J.; Friesen, N.J.; Côté, M.; Müller, A.; Hoenen, T.; et al. Interferon-Induced HERC5 Inhibits Ebola Virus Particle Production and Is Antagonized by Ebola Glycoprotein. Cells 2021, 10, 2399. https://doi.org/10.3390/cells10092399
Paparisto E, Hunt NR, Labach DS, Coleman MD, Di Gravio EJ, Dodge MJ, Friesen NJ, Côté M, Müller A, Hoenen T, et al. Interferon-Induced HERC5 Inhibits Ebola Virus Particle Production and Is Antagonized by Ebola Glycoprotein. Cells. 2021; 10(9):2399. https://doi.org/10.3390/cells10092399
Chicago/Turabian StylePaparisto, Ermela, Nina R. Hunt, Daniel S. Labach, Macon D. Coleman, Eric J. Di Gravio, Mackenzie J. Dodge, Nicole J. Friesen, Marceline Côté, Andreas Müller, Thomas Hoenen, and et al. 2021. "Interferon-Induced HERC5 Inhibits Ebola Virus Particle Production and Is Antagonized by Ebola Glycoprotein" Cells 10, no. 9: 2399. https://doi.org/10.3390/cells10092399
APA StylePaparisto, E., Hunt, N. R., Labach, D. S., Coleman, M. D., Di Gravio, E. J., Dodge, M. J., Friesen, N. J., Côté, M., Müller, A., Hoenen, T., & Barr, S. D. (2021). Interferon-Induced HERC5 Inhibits Ebola Virus Particle Production and Is Antagonized by Ebola Glycoprotein. Cells, 10(9), 2399. https://doi.org/10.3390/cells10092399