
Dynamic Regulation of Cysteine Oxidation and Phosphorylation in Myocardial Ischemia–Reperfusion Injury



Abstract
1. Introduction
2. Myocardial Ischemia–Reperfusion Injury
2.1. Pathophysiology of Ischemia
2.2. Pathophysiology of Reperfusion
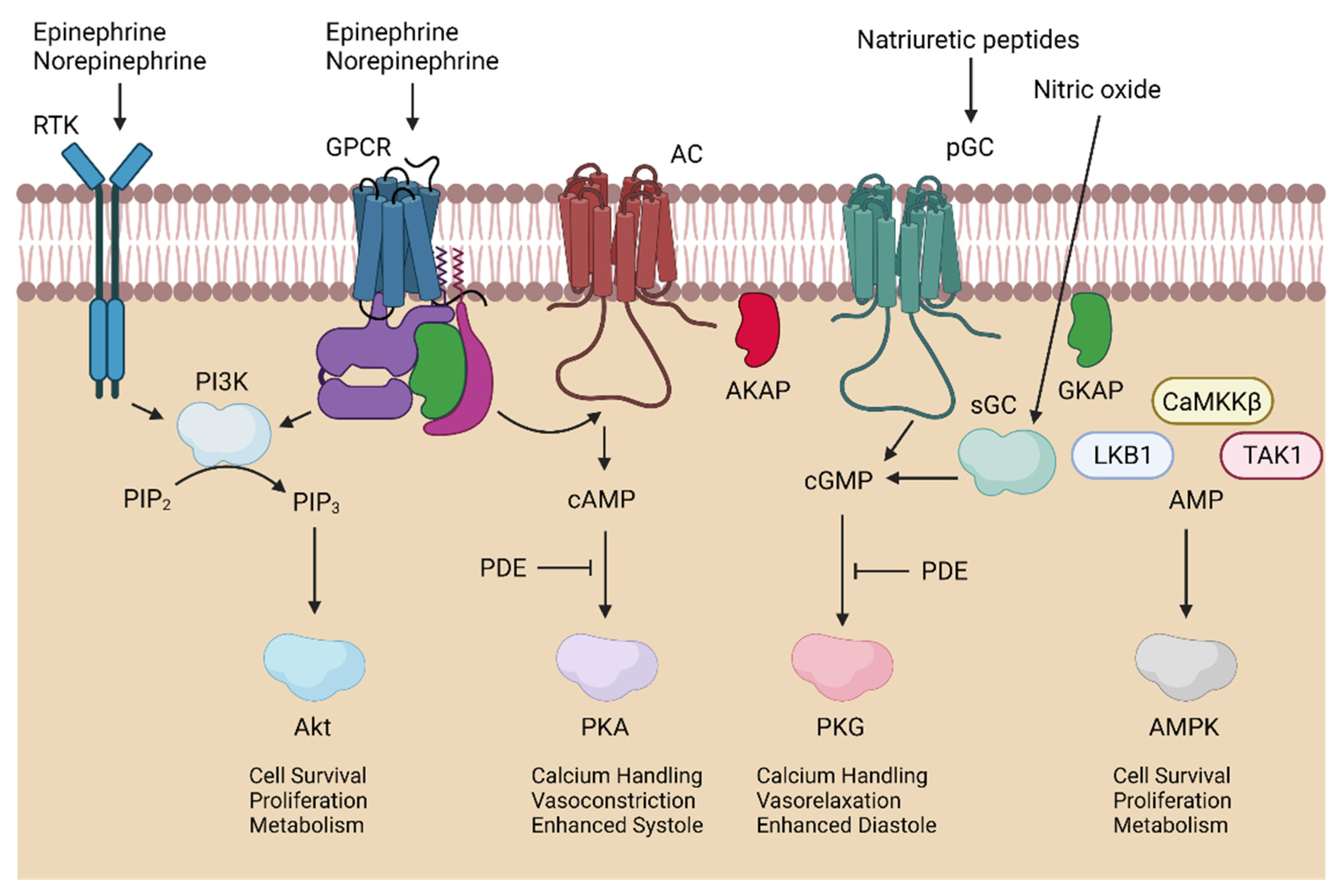
3. Kinases in Cardioprotection
3.1. Cardioprotection
3.2. Regulation of Oxidant Proteins by Kinases
3.3. PI3K/Akt in Myocardial Ischemia–Reperfusion Injury
3.4. AMPK in Myocardial Ischemia–Reperfusion Injury
3.5. PKA in Myocardial Ischemia–Reperfusion Injury
3.6. PKG in Myocardial Ischemia–Reperfusion Injury
4. Oxidation of Kinases and Pathological Consequences
4.1. Brief Overview of Redox Biology
4.2. Kinase Oxidation in Myocardial Ischemia–Reperfusion Injury
4.3. PI3K/Akt Oxidation
4.4. AMPK Oxidation
4.5. PKA/PKG Oxidation
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AC | Adenylyl cyclase |
| AKAP | A-kinase-anchoring protein |
| Akt | Protein kinase B |
| AMP | Adenosine monophosphate |
| AMPK | Adenosine monophosphate kinase |
| ATP | Adenosine triphosphate |
| CaMKKb | Calmodulin-dependent protein kinase kinase |
| cAMP | Cyclic adenosine monophosphate |
| cGMP | Cyclic guanosine monophosphate |
| eNOS | Endothelial nitric oxide synthase |
| ETC | Electron transport chain |
| GC | Guanylyl cyclase |
| GKAP | G-protein-anchoring protein |
| GSK | Glycogen synthase kinase |
| H2O2 | Hydrogen peroxide |
| I/R | Ischemia-reperfusion |
| LKB1 | Liver kinase B1 |
| mTORC | Mammalian target of rapamycin complex |
| NAPDH | Nicotinamide adenine dinucleotide phosphate |
| NOS | Nitric oxide synthase |
| NOX | NAPDH oxidase |
| PDE | Phosphodiesterase |
| pGC | Particulate guanylyl cyclase |
| PI3K | Phosphoinositide 3-kinase |
| PIP2 | Phosphatidylinositol-(4,5) bisphosphate |
| PIP3 | Phosphatidylinositol-(3,4,5) triphosphate |
| PKA | Protein kinase A |
| PKG | Protein kinase G |
| RISK | Reperfusion injury salvage kinase |
| ROS | Reactive oxygen species |
| SERCA | Sarcoplasmic reticulum calcium transport ATPase |
| sGC | Soluble guanylyl cyclase |
| SR | Sacroplasmic reticulum |
| TAK1 | Transforming growth factor-b-activated kinase-1 |
| Trx | Thioredoxin |
References
- Jennings, R.B. Historical perspective on the pathology of myocardial ischemia/reperfusion injury. Circ. Res. 2013, 113, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.L.; Morrow, D.A. Acute Myocardial Infarction. N. Engl. J. Med. 2017, 376, 2053–2064. [Google Scholar] [CrossRef]
- Gunata, M.; Parlakpinar, H. A review of myocardial ischaemia/reperfusion injury: Pathophysiology, experimental models, biomarkers, genetics and pharmacological treatment. Cell Biochem. Funct. 2021, 39, 190–217. [Google Scholar] [CrossRef] [PubMed]
- Truong, T.H.; Carroll, K.S. Redox regulation of protein kinases. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 332–356. [Google Scholar] [CrossRef] [PubMed]
- Erickson, J.R.; Joiner, M.L.; Guan, X.; Kutschke, W.; Yang, J.; Oddis, C.V.; Bartlett, R.K.; Lowe, J.S.; O’Donnell, S.E.; Aykin-Burns, N.; et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 2008, 133, 462–474. [Google Scholar] [CrossRef]
- Moskovitz, J.; Smith, A. Methionine sulfoxide and the methionine sulfoxide reductase system as modulators of signal transduction pathways: A review. Amino Acids 2021, 53, 1011–1020. [Google Scholar] [CrossRef]
- Kumar, H.; Choi, D.K. Hypoxia Inducible Factor Pathway and Physiological Adaptation: A Cell Survival Pathway? Mediat. Inflamm. 2015, 2015, 584758. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Stanley, W.C. Glucose metabolism in the ischemic heart. Circulation 1997, 95, 313–315. [Google Scholar] [CrossRef] [PubMed]
- Clausen, M.V.; Hilbers, F.; Poulsen, H. The Structure and Function of the Na,K-ATPase Isoforms in Health and Disease. Front. Physiol. 2017, 8, 371. [Google Scholar] [CrossRef]
- Bobulescu, I.A.; Di Sole, F.; Moe, O.W. Na+/H+ exchangers: Physiology and link to hypertension and organ ischemia. Curr. Opin. Nephrol. Hypertens. 2005, 14, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Fuller, W.; Parmar, V.; Eaton, P.; Bell, J.R.; Shattock, M.J. Cardiac ischemia causes inhibition of the Na/K ATPase by a labile cytosolic compound whose production is linked to oxidant stress. Cardiovasc. Res. 2003, 57, 1044–1051. [Google Scholar] [CrossRef]
- Pierce, G.N.; Czubryt, M.P. The contribution of ionic imbalance to ischemia/reperfusion-induced injury. J. Mol. Cell Cardiol. 1995, 27, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Kubasiak, L.A.; Hernandez, O.M.; Bishopric, N.H.; Webster, K.A. Hypoxia and acidosis activate cardiac myocyte death through the Bcl-2 family protein BNIP3. Proc. Natl. Acad. Sci. USA 2002, 99, 12825–12830. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.M.; Frazier, D.P.; Thompson, J.W.; Haliko, S.; Li, H.; Wasserlauf, B.J.; Spiga, M.G.; Bishopric, N.H.; Webster, K.A. A unique pathway of cardiac myocyte death caused by hypoxia-acidosis. J. Exp. Biol. 2004, 207, 3189–3200. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Pathophysiology of Myocardial Infarction. Compr. Physiol. 2015, 5, 1841–1875. [Google Scholar] [CrossRef]
- DiFrancesco, D. The role of the funny current in pacemaker activity. Circ. Res. 2010, 106, 434–446. [Google Scholar] [CrossRef]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef]
- Bodi, I.; Mikala, G.; Koch, S.E.; Akhter, S.A.; Schwartz, A. The L-type calcium channel in the heart: The beat goes on. J. Clin. Investig. 2005, 115, 3306–3317. [Google Scholar] [CrossRef]
- Lehnart, S.E.; Wehrens, X.H.; Kushnir, A.; Marks, A.R. Cardiac ryanodine receptor function and regulation in heart disease. Ann. N. Y. Acad. Sci. 2004, 1015, 144–159. [Google Scholar] [CrossRef]
- Marques, M.A.; de Oliveira, G.A. Cardiac Troponin and Tropomyosin: Structural and Cellular Perspectives to Unveil the Hypertrophic Cardiomyopathy Phenotype. Front. Physiol. 2016, 7, 429. [Google Scholar] [CrossRef]
- Zucchi, R.; Ronca, F.; Ronca-Testoni, S. Modulation of sarcoplasmic reticulum function: A new strategy in cardioprotection? Pharmacol. Ther. 2001, 89, 47–65. [Google Scholar] [CrossRef]
- Inserte, J.; Hernando, V.; Garcia-Dorado, D. Contribution of calpains to myocardial ischaemia/reperfusion injury. Cardiovasc. Res. 2012, 96, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Neuhof, C.; Neuhof, H. Calpain system and its involvement in myocardial ischemia and reperfusion injury. World J. Cardiol. 2014, 6, 638–652. [Google Scholar] [CrossRef] [PubMed]
- McNamara, R.L.; Wang, Y.; Herrin, J.; Curtis, J.P.; Bradley, E.H.; Magid, D.J.; Peterson, E.D.; Blaney, M.; Frederick, P.D.; Krumholz, H.M. Effect of door-to-balloon time on mortality in patients with ST-segment elevation myocardial infarction. J. Am. Coll. Cardiol. 2006, 47, 2180–2186. [Google Scholar] [CrossRef]
- Berger, P.B.; Ellis, S.G.; Holmes, D.R., Jr.; Granger, C.B.; Criger, D.A.; Betriu, A.; Topol, E.J.; Califf, R.M. Relationship between delay in performing direct coronary angioplasty and early clinical outcome in patients with acute myocardial infarction: Results from the global use of strategies to open occluded arteries in Acute Coronary Syndromes (GUSTO-IIb) trial. Circulation 1999, 100, 14–20. [Google Scholar] [CrossRef]
- Kiernan, T.J.; Gersh, B.J. Thrombolysis in acute myocardial infarction: Current status. Med. Clin. N. Am. 2007, 91, 617–637. [Google Scholar] [CrossRef] [PubMed]
- White, H.D.; Van de Werf, F.J. Thrombolysis for acute myocardial infarction. Circulation 1998, 97, 1632–1646. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.N.; Elgendy, I.Y.; Mentias, A.; Saad, M.; Ibrahim, W.; Mojadidi, M.K.; Nairooz, R.; Eshtehardi, P.; David Anderson, R.; Samady, H. Percutaneous coronary intervention or coronary artery bypass grafting for unprotected left main coronary artery disease. Catheter. Cardiovasc. Interv. 2017, 90, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Konijnenberg, L.S.F.; Damman, P.; Duncker, D.J.; Kloner, R.A.; Nijveldt, R.; van Geuns, R.M.; Berry, C.; Riksen, N.P.; Escaned, J.; van Royen, N. Pathophysiology and diagnosis of coronary microvascular dysfunction in ST-elevation myocardial infarction. Cardiovasc. Res. 2020, 116, 787–805. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.H.; Smith, P.K. Coronary-Artery Bypass Grafting. N. Engl. J. Med. 2016, 374, 1954–1964. [Google Scholar] [CrossRef]
- Ambrosio, G.; Zweier, J.L.; Flaherty, J.T. The relationship between oxygen radical generation and impairment of myocardial energy metabolism following post-ischemic reperfusion. J. Mol. Cell Cardiol. 1991, 23, 1359–1374. [Google Scholar] [CrossRef]
- Bolli, R.; Jeroudi, M.O.; Patel, B.S.; DuBose, C.M.; Lai, E.K.; Roberts, R.; McCay, P.B. Direct evidence that oxygen-derived free radicals contribute to postischemic myocardial dysfunction in the intact dog. Proc. Natl. Acad. Sci. USA 1989, 86, 4695–4699. [Google Scholar] [CrossRef]
- Taverne, Y.J.; Bogers, A.J.; Duncker, D.J.; Merkus, D. Reactive oxygen species and the cardiovascular system. Oxid. Med. Cell Longev. 2013, 2013, 862423. [Google Scholar] [CrossRef]
- Abdallah, Y.; Kasseckert, S.A.; Iraqi, W.; Said, M.; Shahzad, T.; Erdogan, A.; Neuhof, C.; Gündüz, D.; Schlüter, K.D.; Tillmanns, H.; et al. Interplay between Ca2+ cycling and mitochondrial permeability transition pores promotes reperfusion-induced injury of cardiac myocytes. J. Cell. Mol. Med. 2011, 15, 2478–2485. [Google Scholar] [CrossRef]
- Bernardi, P.; Di Lisa, F. The mitochondrial permeability transition pore: Molecular nature and role as a target in cardioprotection. J. Mol. Cell. Cardiol. 2015, 78, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Steenbergen, C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev. 2008, 88, 581–609. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; He, L.; Lemasters, J.J. Mitochondrial permeability transition: A common pathway to necrosis and apoptosis. Biochem. Biophys. Res. Commun. 2003, 304, 463–470. [Google Scholar] [CrossRef]
- Bauer, T.M.; Murphy, E. Role of Mitochondrial Calcium and the Permeability Transition Pore in Regulating Cell Death. Circ. Res. 2020, 126, 280–293. [Google Scholar] [CrossRef]
- Bonora, M.; Patergnani, S.; Ramaccini, D.; Morciano, G.; Pedriali, G.; Kahsay, A.E.; Bouhamida, E.; Giorgi, C.; Wieckowski, M.R.; Pinton, P. Physiopathology of the Permeability Transition Pore: Molecular Mechanisms in Human Pathology. Biomolecules 2020, 10, 998. [Google Scholar] [CrossRef] [PubMed]
- Rasola, A.; Sciacovelli, M.; Pantic, B.; Bernardi, P. Signal transduction to the permeability transition pore. FEBS Lett. 2010, 584, 1989–1996. [Google Scholar] [CrossRef]
- Hurst, S.; Gonnot, F.; Dia, M.; Crola Da Silva, C.; Gomez, L.; Sheu, S.S. Phosphorylation of cyclophilin D at serine 191 regulates mitochondrial permeability transition pore opening and cell death after ischemia-reperfusion. Cell Death Dis. 2020, 11, 661. [Google Scholar] [CrossRef]
- Reimer, K.A.; Murry, C.E.; Yamasawa, I.; Hill, M.L.; Jennings, R.B. Four brief periods of myocardial ischemia cause no cumulative ATP loss or necrosis. Am. J. Physiol. 1986, 251, H1306–H1315. [Google Scholar] [CrossRef]
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef]
- Mentias, A.; Mahmoud, A.N.; Elgendy, I.Y.; Elgendy, A.Y.; Barakat, A.F.; Abuzaid, A.S.; Saad, M.; Kapadia, S.R. Ischemic postconditioning during primary percutaneous coronary intervention. Catheter. Cardiovasc. Interv. 2017, 90, 1059–1067. [Google Scholar] [CrossRef]
- Tapuria, N.; Kumar, Y.; Habib, M.M.; Abu Amara, M.; Seifalian, A.M.; Davidson, B.R. Remote ischemic preconditioning: A novel protective method from ischemia reperfusion injury--a review. J. Surg. Res. 2008, 150, 304–330. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Cohen, M.V.; Downey, J.M. Mechanism of cardioprotection by early ischemic preconditioning. Cardiovasc. Drugs Ther. 2010, 24, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Altamirano, F.; Wang, Z.V.; Hill, J.A. Cardioprotection in ischaemia-reperfusion injury: Novel mechanisms and clinical translation. J. Physiol. 2015, 593, 3773–3788. [Google Scholar] [CrossRef]
- Crestanello, J.A.; Lingle, D.M.; Kamelgard, J.; Millili, J.; Whitman, G.J. Ischemic preconditioning decreases oxidative stress during reperfusion: A chemiluminescence study. J. Surg. Res. 1996, 65, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.B.; Dongworth, R.K.; Cabrera-Fuentes, H.A.; Hausenloy, D.J. Role of the MPTP in conditioning the heart—ranslatability and mechanism. Br. J. Pharmacol. 2015, 172, 2074–2084. [Google Scholar] [CrossRef] [PubMed]
- Müller, B.A.; Dhalla, N.S. Mechanisms of the beneficial actions of ischemic preconditioning on subcellular remodeling in ischemic-reperfused heart. Curr. Cardiol. Rev. 2010, 6, 255–264. [Google Scholar] [CrossRef]
- Andreadou, I.; Cabrera-Fuentes, H.A.; Devaux, Y.; Frangogiannis, N.G.; Frantz, S.; Guzik, T.; Liehn, E.A.; Gomes, C.P.C.; Schulz, R.; Hausenloy, D.J. Immune cells as targets for cardioprotection: New players and novel therapeutic opportunities. Cardiovasc. Res. 2019, 115, 1117–1130. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Yellon, D.M. Reperfusion injury salvage kinase signalling: Taking a RISK for cardioprotection. Heart Fail. Rev. 2007, 12, 217–234. [Google Scholar] [CrossRef]
- Heusch, G. Molecular basis of cardioprotection: Signal transduction in ischemic pre-, post-, and remote conditioning. Circ. Res. 2015, 116, 674–699. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Fernhoff, N.B.; Derbyshire, E.R.; Marletta, M.A. A nitric oxide/cysteine interaction mediates the activation of soluble guanylate cyclase. Proc. Natl. Acad. Sci. USA 2009, 106, 21602–21607. [Google Scholar] [CrossRef]
- Stomberski, C.T.; Hess, D.T.; Stamler, J.S. Protein S-Nitrosylation: Determinants of Specificity and Enzymatic Regulation of S-Nitrosothiol-Based Signaling. Antioxid. Redox Signal. 2019, 30, 1331–1351. [Google Scholar] [CrossRef]
- Fulton, D.; Gratton, J.P.; McCabe, T.J.; Fontana, J.; Fujio, Y.; Walsh, K.; Franke, T.F.; Papapetropoulos, A.; Sessa, W.C. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 1999, 399, 597–601. [Google Scholar] [CrossRef]
- Dimmeler, S.; Fleming, I.; Fisslthaler, B.; Hermann, C.; Busse, R.; Zeiher, A.M. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 1999, 399, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Mount, P.F.; Kemp, B.E.; Power, D.A. Regulation of endothelial and myocardial NO synthesis by multi-site eNOS phosphorylation. J. Mol. Cell. Cardiol. 2007, 42, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.P.; Mitchelhill, K.I.; Michell, B.J.; Stapleton, D.; Rodriguez-Crespo, I.; Witters, L.A.; Power, D.A.; Ortiz de Montellano, P.R.; Kemp, B.E. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett. 1999, 443, 285–289. [Google Scholar] [CrossRef]
- Michell, B.J.; Harris, M.B.; Chen, Z.P.; Ju, H.; Venema, V.J.; Blackstone, M.A.; Huang, W.; Venema, R.C.; Kemp, B.E. Identification of regulatory sites of phosphorylation of the bovine endothelial nitric-oxide synthase at serine 617 and serine 635. J. Biol. Chem. 2002, 277, 42344–42351. [Google Scholar] [CrossRef]
- Thors, B.; Halldórsson, H.; Thorgeirsson, G. eNOS activation mediated by AMPK after stimulation of endothelial cells with histamine or thrombin is dependent on LKB1. Biochim. Biophys. Acta 2011, 1813, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Cave, A.C.; Brewer, A.C.; Narayanapanicker, A.; Ray, R.; Grieve, D.J.; Walker, S.; Shah, A.M. NADPH oxidases in cardiovascular health and disease. Antioxid. Redox Signal. 2006, 8, 691–728. [Google Scholar] [CrossRef]
- Bokoch, G.M.; Diebold, B.; Kim, J.S.; Gianni, D. Emerging evidence for the importance of phosphorylation in the regulation of NADPH oxidases. Antioxid. Redox Signal. 2009, 11, 2429–2441. [Google Scholar] [CrossRef]
- Matsushima, S.; Kuroda, J.; Ago, T.; Zhai, P.; Ikeda, Y.; Oka, S.; Fong, G.H.; Tian, R.; Sadoshima, J. Broad suppression of NADPH oxidase activity exacerbates ischemia/reperfusion injury through inadvertent downregulation of hypoxia-inducible factor-1α and upregulation of peroxisome proliferator-activated receptor-α. Circ. Res. 2013, 112, 1135–1149. [Google Scholar] [CrossRef]
- Maejima, Y.; Kuroda, J.; Matsushima, S.; Ago, T.; Sadoshima, J. Regulation of myocardial growth and death by NADPH oxidase. J. Mol. Cell. Cardiol. 2011, 50, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Powell, D.W.; Rane, M.J.; Singh, S.; Butt, W.; Klein, J.B.; McLeish, K.R. Akt phosphorylates p47phox and mediates respiratory burst activity in human neutrophils. J. Immunol. 2003, 170, 5302–5308. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.X.; Zeng, H.; Tuo, Q.H.; Yu, H.; Meyrick, B.; Aschner, J.L. NADPH oxidase modulates myocardial Akt, ERK1/2 activation, and angiogenesis after hypoxia-reoxygenation. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1664–H1674. [Google Scholar] [CrossRef]
- Zhao, Q.D.; Viswanadhapalli, S.; Williams, P.; Shi, Q.; Tan, C.; Yi, X.; Bhandari, B.; Abboud, H.E. NADPH oxidase 4 induces cardiac fibrosis and hypertrophy through activating Akt/mTOR and NFκB signaling pathways. Circulation 2015, 131, 643–655. [Google Scholar] [CrossRef]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol. Ther. 2015, 148, 114–131. [Google Scholar] [CrossRef]
- Gonzalez, E.; McGraw, T.E. The Akt kinases: Isoform specificity in metabolism and cancer. Cell Cycle 2009, 8, 2502–2508. [Google Scholar] [CrossRef] [PubMed]
- Mullonkal, C.J.; Toledo-Pereyra, L.H. Akt in ischemia and reperfusion. J. Invest. Surg. 2007, 20, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Shiojima, I.; Walsh, K. Role of Akt signaling in vascular homeostasis and angiogenesis. Circ. Res. 2002, 90, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Kandel, E.S.; Hay, N. The regulation and activities of the multifunctional serine/threonine kinase Akt/PKB. Exp. Cell Res. 1999, 253, 210–229. [Google Scholar] [CrossRef] [PubMed]
- Fujio, Y.; Nguyen, T.; Wencker, D.; Kitsis, R.N.; Walsh, K. Akt promotes survival of cardiomyocytes in vitro and protects against ischemia-reperfusion injury in mouse heart. Circulation 2000, 101, 660–667. [Google Scholar] [CrossRef]
- Matsui, T.; Tao, J.; del Monte, F.; Lee, K.H.; Li, L.; Picard, M.; Force, T.L.; Franke, T.F.; Hajjar, R.J.; Rosenzweig, A. Akt activation preserves cardiac function and prevents injury after transient cardiac ischemia in vivo. Circulation 2001, 104, 330–335. [Google Scholar] [CrossRef]
- Tong, H.; Imahashi, K.; Steenbergen, C.; Murphy, E. Phosphorylation of glycogen synthase kinase-3beta during preconditioning through a phosphatidylinositol-3-kinase--dependent pathway is cardioprotective. Circ. Res. 2002, 90, 377–379. [Google Scholar] [CrossRef]
- Tong, H.; Chen, W.; Steenbergen, C.; Murphy, E. Ischemic preconditioning activates phosphatidylinositol-3-kinase upstream of protein kinase C. Circ. Res. 2000, 87, 309–315. [Google Scholar] [CrossRef]
- Uchiyama, T.; Engelman, R.M.; Maulik, N.; Das, D.K. Role of Akt signaling in mitochondrial survival pathway triggered by hypoxic preconditioning. Circulation 2004, 109, 3042–3049. [Google Scholar] [CrossRef] [PubMed]
- Devaney, J.M.; Gordish-Dressman, H.; Harmon, B.T.; Bradbury, M.K.; Devaney, S.A.; Harris, T.B.; Thompson, P.D.; Clarkson, P.M.; Price, T.B.; Angelopoulos, T.J.; et al. AKT1 polymorphisms are associated with risk for metabolic syndrome. Hum. Genet. 2011, 129, 129–139. [Google Scholar] [CrossRef]
- McKenzie, J.A.; Witkowski, S.; Ludlow, A.T.; Roth, S.M.; Hagberg, J.M. AKT1 G205T genotype influences obesity-related metabolic phenotypes and their responses to aerobic exercise training in older Caucasians. Exp. Physiol. 2011, 96, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Harmon, B.T.; Devaney, S.A.; Gordish-Dressman, H.; Reeves, E.K.; Zhao, P.; Devaney, J.M.; Hoffman, E.P. Functional characterization of a haplotype in the AKT1 gene associated with glucose homeostasis and metabolic syndrome. Hum. Genet. 2010, 128, 635–645. [Google Scholar] [CrossRef][Green Version]
- Kim, J.; Yang, G.; Kim, Y.; Kim, J.; Ha, J. AMPK activators: Mechanisms of action and physiological activities. Exp. Mol. Med. 2016, 48, e224. [Google Scholar] [CrossRef]
- Hardie, D.G. Sensing of energy and nutrients by AMP-activated protein kinase. Am. J. Clin. Nutr. 2011, 93, 891s–896. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.; Dickerson, K.; Heath, R.; Hong, S.P.; Momcilovic, M.; Johnstone, S.R.; Carlson, M.; Carling, D. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005, 2, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D. AMP-activated protein kinase control of energy metabolism in the ischemic heart. Int. J. Obes. 2008, 32 Suppl 4, S29–S35. [Google Scholar] [CrossRef][Green Version]
- Noppe, G.; Dufeys, C.; Buchlin, P.; Marquet, N.; Castanares-Zapatero, D.; Balteau, M.; Hermida, N.; Bouzin, C.; Esfahani, H.; Viollet, B.; et al. Reduced scar maturation and contractility lead to exaggerated left ventricular dilation after myocardial infarction in mice lacking AMPKα1. J. Mol. Cell. Cardiol. 2014, 74, 32–43. [Google Scholar] [CrossRef]
- Kim, A.S.; Miller, E.J.; Wright, T.M.; Li, J.; Qi, D.; Atsina, K.; Zaha, V.; Sakamoto, K.; Young, L.H. A small molecule AMPK activator protects the heart against ischemia-reperfusion injury. J. Mol. Cell Cardiol. 2011, 51, 24–32. [Google Scholar] [CrossRef]
- Gundewar, S.; Calvert, J.W.; Jha, S.; Toedt-Pingel, I.; Ji, S.Y.; Nunez, D.; Ramachandran, A.; Anaya-Cisneros, M.; Tian, R.; Lefer, D.J. Activation of AMP-activated protein kinase by metformin improves left ventricular function and survival in heart failure. Circ. Res. 2009, 104, 403–411. [Google Scholar] [CrossRef]
- Calvert, J.W.; Gundewar, S.; Jha, S.; Greer, J.J.; Bestermann, W.H.; Tian, R.; Lefer, D.J. Acute metformin therapy confers cardioprotection against myocardial infarction via AMPK-eNOS-mediated signaling. Diabetes 2008, 57, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Sato, K.; Pimentel, D.R.; Sam, F.; Shaw, R.J.; Dyck, J.R.; Walsh, K. Cardiac-specific deletion of LKB1 leads to hypertrophy and dysfunction. J. Biol. Chem. 2009, 284, 35839–35849. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Zarrinpashneh, E.; Budas, G.R.; Pouleur, A.C.; Dutta, A.; Prescott, A.R.; Vanoverschelde, J.L.; Ashworth, A.; Jovanović, A.; Alessi, D.R.; et al. Deficiency of LKB1 in heart prevents ischemia-mediated activation of AMPKalpha2 but not AMPKalpha1. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E780–E788. [Google Scholar] [CrossRef] [PubMed]
- Jessen, N.; Koh, H.J.; Folmes, C.D.; Wagg, C.; Fujii, N.; Løfgren, B.; Wolf, C.M.; Berul, C.I.; Hirshman, M.F.; Lopaschuk, G.D.; et al. Ablation of LKB1 in the heart leads to energy deprivation and impaired cardiac function. Biochim. Biophys. Acta 2010, 1802, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.R., 3rd; Li, J.; Coven, D.L.; Pypaert, M.; Zechner, C.; Palmeri, M.; Giordano, F.J.; Mu, J.; Birnbaum, M.J.; Young, L.H. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J. Clin. Investig. 2004, 114, 495–503. [Google Scholar] [CrossRef]
- Dufeys, C.; Daskalopoulos, E.P.; Castanares-Zapatero, D.; Conway, S.J.; Ginion, A.; Bouzin, C.; Ambroise, J.; Bearzatto, B.; Gala, J.L.; Heymans, S.; et al. AMPKα1 deletion in myofibroblasts exacerbates post-myocardial infarction fibrosis by a connexin 43 mechanism. Basic. Res. Cardiol. 2021, 116, 10. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, J.; Fontes, S.K.; Bautista, E.N.; Cheng, Z. Physiological And Pathological Roles Of Protein Kinase A In The Heart. Cardiovasc. Res. 2021. [Google Scholar] [CrossRef]
- Colombe, A.S.; Pidoux, G. Cardiac cAMP-PKA Signaling Compartmentalization in Myocardial Infarction. Cells 2021, 10, 922. [Google Scholar] [CrossRef]
- Fischmeister, R.; Castro, L.R.; Abi-Gerges, A.; Rochais, F.; Jurevicius, J.; Leroy, J.; Vandecasteele, G. Compartmentation of cyclic nucleotide signaling in the heart: The role of cyclic nucleotide phosphodiesterases. Circ. Res. 2006, 99, 816–828. [Google Scholar] [CrossRef]
- Xiao, R.P.; Lakatta, E.G. Beta 1-adrenoceptor stimulation and beta 2-adrenoceptor stimulation differ in their effects on contraction, cytosolic Ca2+, and Ca2+ current in single rat ventricular cells. Circ. Res. 1993, 73, 286–300. [Google Scholar] [CrossRef] [PubMed]
- Vila Petroff, M.G.; Egan, J.M.; Wang, X.; Sollott, S.J. Glucagon-like peptide-1 increases cAMP but fails to augment contraction in adult rat cardiac myocytes. Circ. Res. 2001, 89, 445–452. [Google Scholar] [CrossRef]
- Bhushan, S.; Kondo, K.; Predmore, B.L.; Zlatopolsky, M.; King, A.L.; Pearce, C.; Huang, H.; Tao, Y.X.; Condit, M.E.; Lefer, D.J. Selective β2-adrenoreceptor stimulation attenuates myocardial cell death and preserves cardiac function after ischemia-reperfusion injury. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1865–1874. [Google Scholar] [CrossRef][Green Version]
- Prabu, S.K.; Anandatheerthavarada, H.K.; Raza, H.; Srinivasan, S.; Spear, J.F.; Avadhani, N.G. Protein kinase A-mediated phosphorylation modulates cytochrome c oxidase function and augments hypoxia and myocardial ischemia-related injury. J. Biol. Chem. 2006, 281, 2061–2070. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, J.; Xia, P.; Stratakis, C.A.; Cheng, Z. Loss of PKA regulatory subunit 1α aggravates cardiomyocyte necrosis and myocardial ischemia/reperfusion injury. J. Biol. Chem. 2021, 297, 100850. [Google Scholar] [CrossRef]
- Haushalter, K.J.; Schilling, J.M.; Song, Y.; Sastri, M.; Perkins, G.A.; Strack, S.; Taylor, S.S.; Patel, H.H. Cardiac ischemia-reperfusion injury induces ROS-dependent loss of PKA regulatory subunit RIα. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H1231–h1242. [Google Scholar] [CrossRef] [PubMed]
- Cuello, F.; Nikolaev, V.O. Cardiac cGMP Signaling in Health and Disease: Location, Location, Location. J. Cardiovasc. Pharmacol. 2020, 75, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Sandner, P.; Krieg, T. cGMP at the centre of attention: Emerging strategies for activating the cardioprotective PKG pathway. Basic Res. Cardiol. 2018, 113, 24. [Google Scholar] [CrossRef] [PubMed]
- Vo, N.K.; Gettemy, J.M.; Coghlan, V.M. Identification of cGMP-dependent protein kinase anchoring proteins (GKAPs). Biochem. Biophys. Res. Commun. 1998, 246, 831–835. [Google Scholar] [CrossRef]
- Kukreja, R.C.; Salloum, F.N.; Das, A. Cyclic guanosine monophosphate signaling and phosphodiesterase-5 inhibitors in cardioprotection. J. Am. Coll. Cardiol. 2012, 59, 1921–1927. [Google Scholar] [CrossRef]
- Rybalkin, S.D.; Yan, C.; Bornfeldt, K.E.; Beavo, J.A. Cyclic GMP phosphodiesterases and regulation of smooth muscle function. Circ. Res. 2003, 93, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, S.; Radovits, T.; Barnucz, E.; Hirschberg, K.; Neugebauer, P.; Loganathan, S.; Veres, G.; Páli, S.; Seidel, B.; Zöllner, S.; et al. Pharmacological activation of soluble guanylate cyclase protects the heart against ischemic injury. Circulation 2009, 120, 677–686. [Google Scholar] [CrossRef]
- Maas, O.; Donat, U.; Frenzel, M.; Rütz, T.; Kroemer, H.K.; Felix, S.B.; Krieg, T. Vardenafil protects isolated rat hearts at reperfusion dependent on GC and PKG. Br. J. Pharmacol. 2008, 154, 25–31. [Google Scholar] [CrossRef]
- Kukreja, R.; Salloum, F.; Xi, L. Anti-ischemic effects of sildenafil, vardenafil and tadalafil in heart. Int. J. Impot. Res. 2007, 19, 226–227. [Google Scholar] [CrossRef]
- Ranek, M.J.; Oeing, C.; Sanchez-Hodge, R.; Kokkonen-Simon, K.M.; Dillard, D.; Aslam, M.I.; Rainer, P.P.; Mishra, S.; Dunkerly-Eyring, B.; Holewinski, R.J.; et al. CHIP phosphorylation by protein kinase G enhances protein quality control and attenuates cardiac ischemic injury. Nat. Commun. 2020, 11, 5237. [Google Scholar] [CrossRef]
- Wang, D.Z.; Jones, A.W.; Wang, W.Z.; Wang, M.; Korthuis, R.J. Soluble guanylate cyclase activation during ischemic injury in mice protects against postischemic inflammation at the mitochondrial level. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G747–G756. [Google Scholar] [CrossRef]
- Cohen, M.V.; Yang, X.M.; Liu, Y.; Solenkova, N.V.; Downey, J.M. Cardioprotective PKG-independent NO signaling at reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H2028–H2036. [Google Scholar] [CrossRef][Green Version]
- Rüdebusch, J.; Benkner, A.; Nath, N.; Fleuch, L.; Kaderali, L.; Grube, K.; Klingel, K.; Eckstein, G.; Meitinger, T.; Fielitz, J.; et al. Stimulation of soluble guanylyl cyclase (sGC) by riociguat attenuates heart failure and pathological cardiac remodelling. Br. J. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Loscalzo, J. Metabolic Responses to Reductive Stress. Antioxid. Redox Signal. 2020, 32, 1330–1347. [Google Scholar] [CrossRef] [PubMed]
- Fukuto, J.M.; Carrington, S.J.; Tantillo, D.J.; Harrison, J.G.; Ignarro, L.J.; Freeman, B.A.; Chen, A.; Wink, D.A. Small molecule signaling agents: The integrated chemistry and biochemistry of nitrogen oxides, oxides of carbon, dioxygen, hydrogen sulfide, and their derived species. Chem. Res. Toxicol. 2012, 25, 769–793. [Google Scholar] [CrossRef]
- Kannan, K.; Jain, S.K. Oxidative stress and apoptosis. Pathophysiology 2000, 7, 153–163. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [PubMed]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS signaling in organismal homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Case, A.J. On the Origin of Superoxide Dismutase: An Evolutionary Perspective of Superoxide-Mediated Redox Signaling. Antioxidants 2017, 6, 82. [Google Scholar] [CrossRef]
- Galasso, M.; Gambino, S.; Romanelli, M.G.; Donadelli, M.; Scupoli, M.T. Browsing the oldest antioxidant enzyme: Catalase and its multiple regulation in cancer. Free Radic. Biol. Med. 2021, 172, 264–272. [Google Scholar] [CrossRef]
- Sundaresan, M.; Yu, Z.X.; Ferrans, V.J.; Irani, K.; Finkel, T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 1995, 270, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.S.; Kang, S.W.; Seo, M.S.; Baines, I.C.; Tekle, E.; Chock, P.B.; Rhee, S.G. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. J. Biol. Chem. 1997, 272, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Byrne, D.P.; Shrestha, S.; Galler, M.; Cao, M.; Daly, L.A.; Campbell, A.E.; Eyers, C.E.; Veal, E.A.; Kannan, N.; Eyers, P.A. Aurora A regulation by reversible cysteine oxidation reveals evolutionarily conserved redox control of Ser/Thr protein kinase activity. Sci. Signal. 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, S.; Katiyar, S.; Sanz-Rodriguez, C.E.; Kemppinen, N.R.; Kim, H.W.; Kadirvelraj, R.; Panagos, C.; Keyhaninejad, N.; Colonna, M.; Chopra, P.; et al. A redox-active switch in fructosamine-3-kinases expands the regulatory repertoire of the protein kinase superfamily. Sci. Signal. 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Burchfield, J.G.; Yang, P.; Humphrey, S.J.; Yang, G.; Francis, D.; Yasmin, S.; Shin, S.-Y.; Norris, D.M.; Kearney, A.L.; et al. Global redox proteome and phosphoproteome analysis reveals redox switch in Akt. Nat. Commun. 2019, 10, 5486. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Lee, S.R.; Yang, K.S.; Ahn, Y.; Kim, Y.J.; Stadtman, E.R.; Rhee, S.G. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc. Natl. Acad. Sci. USA 2004, 101, 16419–16424. [Google Scholar] [CrossRef]
- Lee, K.S.; Kim, S.R.; Park, S.J.; Park, H.S.; Min, K.H.; Lee, M.H.; Jin, S.M.; Jin, G.Y.; Yoo, W.H.; Lee, Y.C. Hydrogen peroxide induces vascular permeability via regulation of vascular endothelial growth factor. Am. J. Respir. Cell Mol. Biol. 2006, 35, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Wani, R.; Qian, J.; Yin, L.; Bechtold, E.; King, S.B.; Poole, L.B.; Paek, E.; Tsang, A.W.; Furdui, C.M. Isoform-specific regulation of Akt by PDGF-induced reactive oxygen species. Proc. Natl. Acad. Sci. USA 2011, 108, 10550–10555. [Google Scholar] [CrossRef]
- Adluri, R.S.; Thirunavukkarasu, M.; Zhan, L.; Akita, Y.; Samuel, S.M.; Otani, H.; Ho, Y.S.; Maulik, G.; Maulik, N. Thioredoxin 1 enhances neovascularization and reduces ventricular remodeling during chronic myocardial infarction: A study using thioredoxin 1 transgenic mice. J. Mol. Cell. Cardiol. 2011, 50, 239–247. [Google Scholar] [CrossRef]
- Arslan, F.; Lai, R.C.; Smeets, M.B.; Akeroyd, L.; Choo, A.; Aguor, E.N.; Timmers, L.; van Rijen, H.V.; Doevendans, P.A.; Pasterkamp, G.; et al. Mesenchymal stem cell-derived exosomes increase ATP levels, decrease oxidative stress and activate PI3K/Akt pathway to enhance myocardial viability and prevent adverse remodeling after myocardial ischemia/reperfusion injury. Stem Cell Res. 2013, 10, 301–312. [Google Scholar] [CrossRef]
- Shao, D.; Oka, S.; Liu, T.; Zhai, P.; Ago, T.; Sciarretta, S.; Li, H.; Sadoshima, J. A redox-dependent mechanism for regulation of AMPK activation by Thioredoxin1 during energy starvation. Cell Metab. 2014, 19, 232–245. [Google Scholar] [CrossRef]
- Hinchy, E.C.; Gruszczyk, A.V.; Willows, R.; Navaratnam, N.; Hall, A.R.; Bates, G.; Bright, T.P.; Krieg, T.; Carling, D.; Murphy, M.P. Mitochondria-derived ROS activate AMP-activated protein kinase (AMPK) indirectly. J. Biol. Chem. 2018, 293, 17208–17217. [Google Scholar] [CrossRef]
- Zmijewski, J.W.; Banerjee, S.; Bae, H.; Friggeri, A.; Lazarowski, E.R.; Abraham, E. Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J. Biol. Chem. 2010, 285, 33154–33164. [Google Scholar] [CrossRef]
- Arad, M.; Benson, D.W.; Perez-Atayde, A.R.; McKenna, W.J.; Sparks, E.A.; Kanter, R.J.; McGarry, K.; Seidman, J.G.; Seidman, C.E. Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy. J. Clin. Investig. 2002, 109, 357–362. [Google Scholar] [CrossRef]
- Blair, E.; Redwood, C.; Ashrafian, H.; Oliveira, M.; Broxholme, J.; Kerr, B.; Salmon, A.; Ostman-Smith, I.; Watkins, H. Mutations in the gamma(2) subunit of AMP-activated protein kinase cause familial hypertrophic cardiomyopathy: Evidence for the central role of energy compromise in disease pathogenesis. Hum. Mol. Genet. 2001, 10, 1215–1220. [Google Scholar] [CrossRef]
- Ahmad, F.; Arad, M.; Musi, N.; He, H.; Wolf, C.; Branco, D.; Perez-Atayde, A.R.; Stapleton, D.; Bali, D.; Xing, Y.; et al. Increased alpha2 subunit-associated AMPK activity and PRKAG2 cardiomyopathy. Circulation 2005, 112, 3140–3148. [Google Scholar] [CrossRef]
- Banerjee, S.K.; Ramani, R.; Saba, S.; Rager, J.; Tian, R.; Mathier, M.A.; Ahmad, F. A PRKAG2 mutation causes biphasic changes in myocardial AMPK activity and does not protect against ischemia. Biochem. Biophys. Res. Commun. 2007, 360, 381–387. [Google Scholar] [CrossRef]
- Zou, L.; Shen, M.; Arad, M.; He, H.; Løfgren, B.; Ingwall, J.S.; Seidman, C.E.; Seidman, J.G.; Tian, R. N488I mutation of the gamma2-subunit results in bidirectional changes in AMP-activated protein kinase activity. Circ. Res. 2005, 97, 323–328. [Google Scholar] [CrossRef]
- Brennan, J.P.; Wait, R.; Begum, S.; Bell, J.R.; Dunn, M.J.; Eaton, P. Detection and mapping of widespread intermolecular protein disulfide formation during cardiac oxidative stress using proteomics with diagonal electrophoresis. J. Biol. Chem. 2004, 279, 41352–41360. [Google Scholar] [CrossRef]
- Brennan, J.P.; Bardswell, S.C.; Burgoyne, J.R.; Fuller, W.; Schröder, E.; Wait, R.; Begum, S.; Kentish, J.C.; Eaton, P. Oxidant-induced activation of type I protein kinase A is mediated by RI subunit interprotein disulfide bond formation. J. Biol. Chem. 2006, 281, 21827–21836. [Google Scholar] [CrossRef] [PubMed]
- León, D.A.; Herberg, F.W.; Banky, P.; Taylor, S.S. A stable alpha-helical domain at the N terminus of the RIalpha subunits of cAMP-dependent protein kinase is a novel dimerization/docking motif. J. Biol. Chem. 1997, 272, 28431–28437. [Google Scholar] [CrossRef]
- Simon, J.N.; Vrellaku, B.; Monterisi, S.; Chu, S.M.; Rawlings, N.; Lomas, O.; Marchal, G.A.; Waithe, D.; Syeda, F.; Gajendragadkar, P.R.; et al. Oxidation of Protein Kinase A Regulatory Subunit PKARIα Protects Against Myocardial Ischemia-Reperfusion Injury by Inhibiting Lysosomal-Triggered Calcium Release. Circulation 2021, 143, 449–465. [Google Scholar] [CrossRef] [PubMed]
- Trum, M.; Islam, M.M.T.; Lebek, S.; Baier, M.; Hegner, P.; Eaton, P.; Maier, L.S.; Wagner, S. Inhibition of cardiac potassium currents by oxidation-activated protein kinase A contributes to early afterdepolarizations in the heart. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H1347–H1357. [Google Scholar] [CrossRef] [PubMed]
- Rababa’h, A.; Craft, J.W., Jr.; Wijaya, C.S.; Atrooz, F.; Fan, Q.; Singh, S.; Guillory, A.N.; Katsonis, P.; Lichtarge, O.; McConnell, B.K. Protein kinase A and phosphodiesterase-4D3 binding to coding polymorphisms of cardiac muscle anchoring protein (mAKAP). J. Mol. Biol. 2013, 425, 3277–3288. [Google Scholar] [CrossRef]
- Suryavanshi, S.V.; Jadhav, S.M.; Anderson, K.L.; Katsonis, P.; Lichtarge, O.; McConnell, B.K. Human muscle-specific A-kinase anchoring protein polymorphisms modulate the susceptibility to cardiovascular diseases by altering cAMP/PKA signaling. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H109–H121. [Google Scholar] [CrossRef]
- Smith, F.D.; Omar, M.H.; Nygren, P.J.; Soughayer, J.; Hoshi, N.; Lau, H.T.; Snyder, C.G.; Branon, T.C.; Ghosh, D.; Langeberg, L.K.; et al. Single nucleotide polymorphisms alter kinase anchoring and the subcellular targeting of A-kinase anchoring proteins. Proc. Natl. Acad. Sci. USA 2018, 115, E11465–e11474. [Google Scholar] [CrossRef]
- Suryavanshi, S.V.; Jadhav, S.M.; McConnell, B.K. Polymorphisms/Mutations in A-Kinase Anchoring Proteins (AKAPs): Role in the Cardiovascular System. J. Cardiovasc. Dev. Dis. 2018, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Kathiresan, S.; Voight, B.F.; Purcell, S.; Musunuru, K.; Ardissino, D.; Mannucci, P.M.; Anand, S.; Engert, J.C.; Samani, N.J.; Schunkert, H.; et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat. Genet. 2009, 41, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Monken, C.E.; Gill, G.N. Structural analysis of cGMP-dependent protein kinase using limited proteolysis. J. Biol. Chem. 1980, 255, 7067–7070. [Google Scholar] [CrossRef]
- Burgoyne, J.R.; Madhani, M.; Cuello, F.; Charles, R.L.; Brennan, J.P.; Schröder, E.; Browning, D.D.; Eaton, P. Cysteine redox sensor in PKGIa enables oxidant-induced activation. Science 2007, 317, 1393–1397. [Google Scholar] [CrossRef] [PubMed]
- Scotcher, J.; Prysyazhna, O.; Boguslavskyi, A.; Kistamas, K.; Hadgraft, N.; Martin, E.D.; Worthington, J.; Rudyk, O.; Rodriguez Cutillas, P.; Cuello, F.; et al. Disulfide-activated protein kinase G Iα regulates cardiac diastolic relaxation and fine-tunes the Frank-Starling response. Nat. Commun. 2016, 7, 13187. [Google Scholar] [CrossRef] [PubMed]
- Sheehe, J.L.; Bonev, A.D.; Schmoker, A.M.; Ballif, B.A.; Nelson, M.T.; Moon, T.M.; Dostmann, W.R. Oxidation of cysteine 117 stimulates constitutive activation of the type Iα cGMP-dependent protein kinase. J. Biol. Chem. 2018, 293, 16791–16802. [Google Scholar] [CrossRef]
- Oeing, C.U.; Nakamura, T.; Pan, S.; Mishra, S.; Dunkerly-Eyring, B.L.; Kokkonen-Simon, K.M.; Lin, B.L.; Chen, A.; Zhu, G.; Bedja, D.; et al. PKG1α Cysteine-42 Redox State Controls mTORC1 Activation in Pathological Cardiac Hypertrophy. Circ. Res. 2020, 127, 522–533. [Google Scholar] [CrossRef]
- Nakamura, T.; Ranek, M.J.; Lee, D.I.; Shalkey Hahn, V.; Kim, C.; Eaton, P.; Kass, D.A. Prevention of PKG1α oxidation augments cardioprotection in the stressed heart. J. Clin. Investig. 2015, 125, 2468–2472. [Google Scholar] [CrossRef] [PubMed]
- Inserte, J.; Hernando, V.; Vilardosa, Ú.; Abad, E.; Poncelas-Nozal, M.; Garcia-Dorado, D. Activation of cGMP/protein kinase G pathway in postconditioned myocardium depends on reduced oxidative stress and preserved endothelial nitric oxide synthase coupling. J. Am. Heart Assoc. 2013, 2, e005975. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Enzyme | Cysteine (Species) | On/Off | Heart Effect | References |
|---|---|---|---|---|
| Akt | 60, 77 (Mouse) | On | Reduces I/R injury. | [110,111,112,113,114,115] |
| AMPK | 130, 174, 299, 304 (Mouse) | On (AMPKa1) Off (AMPKa2) | Decreases I/R injury. | [116,117,118] |
| PKA | 16, 37 (Rat) 17 (Mouse) | On None (Mouse) | Detrimental; reduce arrhythmias. | [71,119,120,121,122,123] |
| PKG | 42, 117 (Rat/Mouse) | On | Harmful to heart failure; protective from I/R injury. | [124,125,126,127,128,129,130] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casin, K.M.; Calvert, J.W. Dynamic Regulation of Cysteine Oxidation and Phosphorylation in Myocardial Ischemia–Reperfusion Injury. Cells 2021, 10, 2388. https://doi.org/10.3390/cells10092388
Casin KM, Calvert JW. Dynamic Regulation of Cysteine Oxidation and Phosphorylation in Myocardial Ischemia–Reperfusion Injury. Cells. 2021; 10(9):2388. https://doi.org/10.3390/cells10092388
Chicago/Turabian StyleCasin, Kevin M., and John W. Calvert. 2021. "Dynamic Regulation of Cysteine Oxidation and Phosphorylation in Myocardial Ischemia–Reperfusion Injury" Cells 10, no. 9: 2388. https://doi.org/10.3390/cells10092388
APA StyleCasin, K. M., & Calvert, J. W. (2021). Dynamic Regulation of Cysteine Oxidation and Phosphorylation in Myocardial Ischemia–Reperfusion Injury. Cells, 10(9), 2388. https://doi.org/10.3390/cells10092388