
Intertwined and Finely Balanced: Endoplasmic Reticulum Morphology, Dynamics, Function, and Diseases


Abstract
:1. Introduction
2. Morphology
2.1. Morphology-Regulating Proteins
2.2. Structural and Functional ER Subdomains
2.2.1. Protein Factory and Quality Control
2.2.2. ERES: Export Checks
2.2.3. MCSs: Lipid Manufacture
2.2.4. MCSs: Lipid Exchange
2.2.5. MCSs: Calcium Control
2.2.6. MCSs: Control of Membrane Fission and Fusion
3. ER Dynamics
3.1. Cytoskeletal Control of ER Dynamics
3.1.1. Microtubule Motors Drive ER Dynamics
3.1.2. MCS-Mediated ER Dynamics
3.1.3. Motor-Independent ER-Microtubule Interactions
3.1.4. ER Interactions with the Actin Cytoskeleton
3.2. Network Fluctuations
3.3. Dynamics of Membrane and Lumenal Components
3.4. Computational Analysis of ER Dynamics
4. Morphology, Dynamics & Disease
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Valm, A.M.; Cohen, S.; Legant, W.R.; Melunis, J.; Hershberg, U.; Wait, E.; Cohen, A.R.; Davidson, M.W.; Betzig, E.; Lippincott-Schwartz, J. Applying systems-level spectral imaging and analysis to reveal the organelle interactome. Nature 2017, 546, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Voeltz, G.K.; Prinz, W.A.; Shibata, Y.; Rist, J.M.; Rapoport, T.A. A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell 2006, 124, 573–586. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Shibata, Y.; Voss, C.; Shemesh, T.; Li, Z.; Coughlin, M.; Kozlov, M.M.; Rapoport, T.A.; Prinz, W.A. Membrane proteins of the endoplasmic reticulum induce high-curvature tubules. Science 2008, 319, 1247–1250. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Shibata, Y.; Zhu, P.P.; Voss, C.; Rismanchi, N.; Prinz, W.A.; Rapoport, T.A.; Blackstone, C. A Class of Dynamin-like GTPases Involved in the Generation of the Tubular ER Network. Cell 2009, 138, 549–561. [Google Scholar] [CrossRef] [Green Version]
- Orso, G.; Pendin, D.; Liu, S.; Tosetto, J.; Moss, T.J.; Faust, J.E.; Micaroni, M.; Egorova, A.; Martinuzzi, A.; McNew, J.A.; et al. Homotypic fusion of ER membranes requires the dynamin-like GTPase Atlastin. Nature 2009, 460, 978–983. [Google Scholar] [CrossRef]
- Wang, S.; Tukachinsky, H.; Romano, F.B.; Rapoport, T.A. Cooperation of the ER-shaping proteins atlastin, lunapark, and reticulons to generate a tubular membrane network. Elife 2016, 5, 1–29. [Google Scholar] [CrossRef]
- English, A.R.; Voeltz, G.K. Rab10 GTPase regulates ER dynamics and morphology. Nat. Cell Biol. 2013, 15, 169–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerondopoulos, A.; Bastos, R.N.; Yoshimura, S.I.; Anderson, R.; Carpanini, S.; Aligianis, I.; Handley, M.T.; Barr, F.A. Rab18 and a Rab18 GEF complex are required for normal ER structure. J. Cell Biol. 2014, 205, 707–720. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Novick, P.; Ferro-Novick, S. ER network formation requires a balance of the dynamin-like GTPase Sey1p and the Lunapark family member Lnp1p. Nat. Cell Biol. 2012, 14, 707–716. [Google Scholar] [CrossRef]
- Chen, S.; Desai, T.; McNew, J.A.; Gerard, P.; Novick, P.J.; Ferro-Novick, S. Lunapark stabilizes nascent three-way junctions in the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2015, 112, 418–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Zhang, T.; Huo, H.; Ye, Y.; Liu, Y. Lunapark is a component of a ubiquitin ligase complex localized to the endoplasmic reticulum three-way junctions. J. Biol. Chem. 2016, 291, 18252–18262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klopfenstein, D.R.; Klumperman, J.; Lustig, A.; Kammerer, R.A.; Oorschot, V.; Hauri, H.-P. Subdomain-specific Localisation of CLIMP-63 (p63) in the Endoplamic Reticulum is Mediated by its Luminal a-helical Segment. J. Cell Biol. 2001, 153, 1287–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, Y.; Shemesh, T.; Prinz, W.A.; Palazzo, A.F.; Kozlov, M.M.; Rapoport, T.A. Mechanisms determining the morphology of the peripheral ER. Cell 2010, 143, 774–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.; Sun, S.; Hu, J. Molecular basis for sculpting the endoplasmic reticulum membrane. Int. J. Biochem. Cell Biol. 2012, 44, 1436–1443. [Google Scholar] [CrossRef] [PubMed]
- Klopfenstein, D.R.C.; Kappeler, F.; Hauri, H.P. A novel direct interaction of endoplasmic reticulum with microtubules. EMBO J. 1998, 17, 6168–6177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa-Goto, K.; Tanaka, K.; Ueno, T.; Tanaka, K.; Kurata, T.; Sata, T.; Irie, S. p180 is Involved in the Interaction between the Endoplasmic Reticulum and Microtubules through a Novel Microtubule-binding and Bundling Domain. Mol. Biol. Cell 2007, 18, 3741–3751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.H.; Zhu, P.P.; Parker, R.L.; Blackstone, C. Hereditary spastic paraplegia proteins REEP1, spastin, and atlastin-1 coordinate microtubule interactions with the tubular ER network. J. Clin. Investig. 2010, 120, 1097–1110. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; He, Y.; Huang, X.; Guo, Y.; Li, D.; Hu, J. Reciprocal regulation between lunapark and atlastin facilitates ER three-way junction formation. Protein Cell 2019, 10, 510–525. [Google Scholar] [CrossRef] [Green Version]
- Allan, V. Protein phosphatase 1 regulates the cytoplasmic dynein-driven formation of endoplasmic reticulum networks in vitro. J. Cell Biol. 1995, 128, 879–891. [Google Scholar] [CrossRef] [PubMed]
- Woźniak, M.J.; Bola, B.; Brownhill, K.; Yang, Y.-C.; Levakova, V.; Allan, V.J. Role of kinesin-1 and cytoplasmic dynein in endoplasmic reticulum movement in VERO cells. J. Cell Sci. 2009, 122, 1979–1989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grigoriev, I.; Gouveia, S.M.; van der Vaart, B.; Demmers, J.; Smyth, J.T.; Honnappa, S.; Splinter, D.; Steinmetz, M.O.; Putney, J.W.; Hoogenraad, C.C.; et al. STIM1 is a MT-Plus-End-Tracking Protein Involved in Remodeling of the ER. Curr. Biol. 2008, 18, 177–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, J.R.; Webster, B.M.; Mastronarde, D.N.; Verhey, K.J.; Voeltz, G.K. ER sliding dynamics and ER-mitochondrial contacts occur on acetylated microtubules. J. Cell Biol. 2010, 190, 363–375. [Google Scholar] [CrossRef] [Green Version]
- Waterman-Storer, C.M.; Salmon, E.D. Endoplasmic reticulum membrane tubules are distributed by microtubules in living cells using three distinct mechanisms. Curr. Biol. 1998, 8, 798–806. [Google Scholar] [CrossRef] [Green Version]
- Pain, C.; Kriechbaumer, V.; Kittelmann, M.; Hawes, C.; Fricker, M. Quantitative analysis of plant ER architecture and dynamics. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgiades, P.; Allan, V.J.; Wright, G.D.; Woodman, P.G.; Udommai, P.; Chung, M.A.; Waigh, T.A. The flexibility and dynamics of the tubules in the endoplasmic reticulum. Sci. Rep. 2017, 7, 16474. [Google Scholar] [CrossRef]
- Spits, M.; Heesterbeek, I.T.; Voortman, L.M.; Akkermans, J.J.; Wijdeven, R.H.; Cabukusta, B.; Neefjes, J. Mobile late endosomes modulate peripheral endoplasmic reticulum network architecture. EMBO Rep. 2021, 22, 1–14. [Google Scholar] [CrossRef]
- Perkins, H.T.; Allan, V.J.; Waigh, T.A. Network Organisation and the Dynamics of Tubules in the Endoplasmic Reticulum. Sci. Rep. 2021, 11, 1–13. [Google Scholar]
- Del Castillo, U.; Gnazzo, M.M.; Sorensen Turpin, C.G.; Nguyen, K.C.Q.; Semaya, E.; Lam, Y.; de Cruz, M.A.; Bembenek, J.N.; Hall, D.H.; Riggs, B.; et al. Conserved role for Ataxin-2 in mediating endoplasmic reticulum dynamics. Traffic 2019, 20, 436–447. [Google Scholar] [CrossRef] [Green Version]
- Bem, D.; Yoshimura, S.-I.; Nunes-Bastos, R.; Bond, F.F.; Kurian, M.A.; Rahman, F.; Handley, M.T.W.; Hadzhiev, Y.; Masood, I.; Straatman-Iwanowska, A.A.; et al. Loss-of-Function Mutations in RAB18 Cause Warburg Micro Syndrome. Am. J. Hum. Genet. 2011, 88, 499–507. [Google Scholar] [CrossRef] [Green Version]
- Park, S.H.; Blackstone, C. Further assembly required: Construction and dynamics of the endoplasmic reticulum network. EMBO Rep. 2010, 11, 515–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Hu, J. Shaping the Endoplasmic Reticulum into a Social Network. Trends Cell Biol. 2016, 26, 934–943. [Google Scholar] [CrossRef] [Green Version]
- Sree, S.; Parkkinen, I.; Their, A.; Airavaara, M.; Jokitalo, E. Morphological Heterogeneity of the Endoplasmic Reticulum within Neurons and Its Implications in Neurodegeneration. Cells 2021, 10, 970. [Google Scholar] [CrossRef]
- Schroeder, L.K.; Barentine, A.E.S.; Merta, H.; Schweighofer, S.; Zhang, Y.; Baddeley, D.; Bewersdorf, J.; Bahmanyar, S. Dynamic nanoscale morphology of the ER surveyed by STED microscopy. J. Cell Biol. 2019, 218, 83–96. [Google Scholar] [CrossRef] [Green Version]
- Nixon-Abell, J.; Obara, C.J.; Weigel, A.V.; Li, D.; Legant, W.R.; Xu, C.S.; Pasolli, H.A.; Harvey, K.; Hess, H.F.; Betzig, E.; et al. Increased spatiotemporal resolution reveals highly dynamic dense tubular matrices in the peripheral ER. Science 2016, 354, aaf3928. [Google Scholar] [CrossRef] [Green Version]
- Ferencz, C.M.; Guigas, G.; Veres, A.; Neumann, B.; Stemmann, O.; Weiss, M. Shaping the endoplasmic reticulum in vitro. Biochim. Biophys. Acta-Biomembr. 2016, 1858, 2035–2040. [Google Scholar] [CrossRef]
- Terasaki, M.; Shemesh, T.; Kasthuri, N.; Klemm, R.W.; Schalek, R.; Hayworth, K.J.; Hand, A.R.; Yankova, M.; Huber, G.; Lichtman, J.W.; et al. Stacked Endoplasmic Reticulum Sheets Are Connected by Helicoidal Membrane Motifs. Cell 2013, 154, 285–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.; Ladinsky, M.S.; Kirchhausen, T. Cisternal Organization of the Endoplasmic Reticulum during Mitosis. Mol. Biol. Cell 2009, 20, 3471–3480. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Li, D.; Zhang, S.; Yang, Y.; Liu, J.J.; Wang, X.; Liu, C.; Milkie, D.E.; Moore, R.P.; Tulu, U.S.; et al. Visualizing Intracellular Organelle and Cytoskeletal Interactions at Nanoscale Resolution on Millisecond Timescales. Cell 2018, 175, 1430–1442.e17. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Prinz, W.A.; Rapoport, T.A. Weaving the web of ER tubules. Cell 2011, 147, 1226–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westrate, L.M.; Lee, J.E.; Prinz, W.A.; Voeltz, G.K. Form Follows Function: The Importance of Endoplasmic Reticulum Shape. Annu. Rev. Biochem. 2015, 84, 791–811. [Google Scholar] [CrossRef]
- Bian, X.; Klemm, R.W.; Liu, T.Y.; Zhang, M.; Sun, S.; Sui, X.; Liu, X.; Rapoport, T.A.; Hu, J. Structures of the atlastin GTPase provide insight into homotypic fusion of endoplasmic reticulum membranes. Proc. Natl. Acad. Sci. USA 2011, 108, 3976–3981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adachi, Y.; Kato, T.; Yamada, T.; Murata, D.; Arai, K.; Stahelin, R.V.; Chan, D.C.; Iijima, M.; Sesaki, H. Drp1 Tubulates the ER in a GTPase-Independent Manner. Mol. Cell 2020, 80, 621–632.e6. [Google Scholar] [CrossRef]
- Shemesh, T.; Klemm, R.W.; Romano, F.B.; Wang, S.; Vaughan, J.; Zhuang, X.; Tukachinsky, H.; Kozlov, M.M.; Rapoport, T.A. A model for the generation and interconversion of ER morphologies. Proc. Natl. Acad. Sci. USA 2014, 111, e5243–e5251. [Google Scholar] [CrossRef] [Green Version]
- Shibata, Y.; Voeltz, G.K.; Rapoport, T.A. Rough Sheets and Smooth Tubules. Cell 2006, 126, 435–439. [Google Scholar] [CrossRef] [Green Version]
- Puhka, M.; Vihinen, H.; Joensuu, M.; Jokitalo, E. Endoplasmic reticulum remains continuous and undergoes sheet-to-tubule transformation during cell division in mammalian cells. J. Cell Biol. 2007, 179, 895–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palade, G.E. Studies on the Endoplasmic Reticulum. J. Biophys. Biochem. Cytol. 1955, 1, 567–582. [Google Scholar] [CrossRef]
- Palade, G.E. The Endoplasmic Reticulum. J. Biophys. Biochem. Cytol. 1956, 2, 85–98. [Google Scholar] [CrossRef] [Green Version]
- Sicking, M.; Lang, S.; Bochen, F.; Roos, A.; Drenth, J.P.H.; Zakaria, M.; Zimmermann, R.; Linxweiler, M. Complexity and Specificity of Sec61-Channelopathies: Human Diseases Affecting Gating of the Sec61 Complex. Cells 2021, 10, 1036. [Google Scholar] [CrossRef]
- West, M.; Zurek, N.; Hoenger, A.; Voeltz, G.K. A 3D analysis of yeast ER structure reveals how ER domains are organized by membrane curvature. J. Cell Biol. 2011, 193, 333–346. [Google Scholar] [CrossRef]
- Baumann, O.; Walz, B. Endoplasmic reticulum of animal cells and its organization into structural and functional domains. Int. Rev. Cytol. 2001, 205, 149–214. [Google Scholar] [CrossRef] [PubMed]
- Caramelo, J.J.; Parodi, A.J. A sweet code for glycoprotein folding. FEBS Lett. 2015, 589, 3379–3387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenkman, M.; Lederkremer, G.Z. Compartmentalization and Selective Tagging for Disposal of Misfolded Glycoproteins. Trends Biochem. Sci. 2019, 44, 827–836. [Google Scholar] [CrossRef]
- Read, A.; Schröder, M. The unfolded protein response: An overview. Biology 2021, 10, 384. [Google Scholar] [CrossRef] [PubMed]
- Wakana, Y.; Takai, S.; Nakajima, K.; Tani, K.; Yamamoto, A.; Watson, P.; Stephens, D.J.; Hauri, H.-P.; Tagaya, M. Bap31 is an Itinerant Protein That Moves between the Peripheral Endoplasmic Reticulum (ER) and a Juxtanuclear Compartment Related to ER-associated Degradation. Mol. Biol. Cell 2008, 19, 1825–1836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, F.; Du, W.; Zou, Q.; Wang, Y.; Zhang, X.; Xing, X.; Li, Y.; Zhang, D.; Wang, H.; Zhang, W.; et al. COPII mitigates ER stress by promoting formation of ER whorls. Cell Res. 2021, 31, 141–156. [Google Scholar] [CrossRef]
- Borgese, N.; Francolini, M.; Snapp, E. Endoplasmic reticulum architecture: Structures in flux. Curr. Opin. Cell Biol. 2006, 18, 358–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snapp, E.L.; Hegde, R.S.; Francolini, M.; Lombardo, F.; Colombo, S.; Pedrazzini, E.; Borgese, N.; Lippincott-Schwartz, J. Formation of stacked ER cisternae by low affinity protein interactions. J. Cell Biol. 2003, 163, 257–269. [Google Scholar] [CrossRef]
- Bannykh, S.I.; Rowe, T.; Balch, W.E. The organization of endoplasmic reticulum export complexes. J. Cell Biol. 1996, 135, 19–35. [Google Scholar] [CrossRef]
- Shomron, O.; Hirschberg, K.; Burakov, A.; Kamentseva, R.; Kornilova, E.; Nadezhdina, E.; Brodsky, I. Positioning of endoplasmic reticulum exit sites around the Golgi depends on BicaudalD2 and Rab6 activity. Traffic 2021, 22, 64–77. [Google Scholar] [CrossRef]
- Presley, J.F.; Cole, N.B.; Schroer, T.A.; Hirschberg, K.; Zaal, K.J.M.; Lippincott-Schwartz, J. ER-to-Golgi transport visualized in living cells. Nature 1997, 389, 81–85. [Google Scholar] [CrossRef]
- Gupta, V.; Palmer, K.J.; Spence, P.; Hudson, A.; Stephens, D.J. Kinesin-1 (uKHC/KIF5B) is required for bidirectional motility of ER exit sites and efficient ER-to-Golgi transport. Traffic 2008, 9, 1850–1866. [Google Scholar] [CrossRef] [PubMed]
- Peotter, J.; Kasberg, W.; Pustova, I.; Audhya, A. COPII-mediated trafficking at the ER/ERGIC interface. Traffic 2019, 20, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Weigel, A.V.; Chang, C.L.; Shtengel, G.; Xu, C.S.; Hoffman, D.P.; Freeman, M.; Iyer, N.; Aaron, J.; Khuon, S.; Bogovic, J.; et al. ER-to-Golgi protein delivery through an interwoven, tubular network extending from ER. Cell 2021, 184, 2412–2429.e16. [Google Scholar] [CrossRef]
- Shomron, O.; Nevo-Yassaf, I.; Aviad, T.; Yaffe, Y.; Zahavi, E.E.; Dukhovny, A.; Perlson, E.; Brodsky, I.; Yeheskel, A.; Pasmanik-Chor, M.; et al. COPII collar defines the boundary between ER and ER exit site and does not coat cargo containers. J. Cell Biol. 2021, 220, e201907224. [Google Scholar] [CrossRef] [PubMed]
- Westrate, L.M.; Hoyer, M.J.; Nash, M.J.; Voeltz, G.K. Vesicular and uncoated Rab1-dependent cargo carriers facilitate ER to Golgi transport. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef]
- Stephens, D.J.; Lin-Marq, N.; Pagano, A.; Pepperkok, R.; Paccaud, J.P. COPI-coated ER-to-Golgi transport complexes segregate from COPII in close proximity to ER exit sites. J. Cell Sci. 2000, 113, 2177–2185. [Google Scholar] [CrossRef]
- Aridor, M.; Bannykh, S.I.; Rowe, T.; Balch, W.E. Sequential coupling between copII and copI vesicle coats in endoplasmic reticulum to Golgi transport. J. Cell Biol. 1995, 131, 875–893. [Google Scholar] [CrossRef] [PubMed]
- Bar-Ziv, R.; Moses, E. Instability and “Pearling” States Produced in Tubular Membranes by Competition of Curvature and Tension. Phys. Rev. Lett. 1994, 73, 1392–1395. [Google Scholar] [CrossRef] [PubMed]
- Jelerčič, U.; Gov, N.S. Pearling instability of membrane tubes driven by curved proteins and actin polymerization. Phys. Biol. 2015, 12, 066022. [Google Scholar] [CrossRef]
- Watson, P.; Forster, R.; Palmer, K.J.; Pepperkok, R.; Stephens, D.J. Coupling of ER exit to microtubules through direct interaction of COPII with dynactin. Nat. Cell Biol. 2005, 7, 48–55. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.K.; Hunt, S.D.; Stephens, D.J. Opposing microtubule motors control motility, morphology and cargo segregation during ER-to-Golgi transport. Biol. Open 2014, 3, 307–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fawcett, D. The Cell, 2nd ed.; W. B. Saunders Company: Philadelphia, PA, USA, 1981; ISBN 9780721635842. [Google Scholar]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zajac, A.L.; Goldman, Y.E.; Holzbaur, E.L.F.; Ostap, E.M. Local cytoskeletal and organelle interactions impact molecular-motor-driven early endosomal trafficking. Curr. Biol. 2013, 23, 1173–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, J.R.; DiBenedetto, J.R.; West, M.; Rowland, A.A.; Voeltz, G.K. Endoplasmic reticulum-endosome contact increases as endosomes traffic and mature. Mol. Biol. Cell 2013, 24, 1030–1040. [Google Scholar] [CrossRef] [PubMed]
- Damenti, M.; Coceano, G.; Pennacchietti, F.; Bodén, A.; Testa, I. STED and parallelized RESOLFT optical nanoscopy of the tubular endoplasmic reticulum and its mitochondrial contacts in neuronal cells. Neurobiol. Dis. 2021, 155, 105361. [Google Scholar] [CrossRef]
- Chen, F.; Yan, B.; Ren, J.; Lyu, R.; Wu, Y.; Guo, Y.; Li, D.; Zhang, H.; Hu, J. FIT2 organizes lipid droplet biogenesis with ER tubule-forming proteins and septins. J. Cell Biol. 2021, 220, e201907183. [Google Scholar] [CrossRef]
- Fernández-Busnadiego, R.; Saheki, Y.; De Camilli, P. Three-dimensional architecture of extended synaptotagmin-mediated endoplasmic reticulum-plasma membrane contact sites. Proc. Natl. Acad. Sci. USA 2015, 112, e2004–e2013. [Google Scholar] [CrossRef] [Green Version]
- Csordás, G.; Renken, C.; Várnai, P.; Walter, L.; Weaver, D.; Buttle, K.F.; Balla, T.; Mannella, C.A.; Hajnóczky, G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 2006, 174, 915–921. [Google Scholar] [CrossRef] [Green Version]
- Zaman, M.F.; Nenadic, A.; Radojičić, A.; Rosado, A.; Beh, C.T. Sticking With It: ER-PM Membrane Contact Sites as a Coordinating Nexus for Regulating Lipids and Proteins at the Cell Cortex. Front. Cell Dev. Biol. 2020, 8, 1–23. [Google Scholar] [CrossRef]
- Wu, Y.; Whiteus, C.; Xu, C.S.; Hayworth, K.J.; Weinberg, R.J.; Hess, H.F.; De Camilli, P. Contacts between the endoplasmic reticulum and other membranes in neurons. Proc. Natl. Acad. Sci. USA 2017, 114, e4859–e4867. [Google Scholar] [CrossRef] [Green Version]
- Alpy, F.; Rousseau, A.; Schwab, Y.; Legueux, F.; Stoll, I.; Wendling, C.; Spiegelhalter, C.; Kessler, P.; Mathelin, C.; Rio, M.C.; et al. STARD3 or STARD3NL and VAP form a novel molecular tether between late endosomes and the ER. J. Cell Sci. 2013, 126, 5500–5512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lev, S. Non-vesicular lipid transport by lipid-transfer proteins and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Carvalho, P.; Voeltz, G.K. Here, there, and everywhere: The importance of ER membrane contact sites. Science 2018, 361, eaan5835. [Google Scholar] [CrossRef] [Green Version]
- Prinz, W.A.; Toulmay, A.; Balla, T. The functional universe of membrane contact sites. Nat. Rev. Mol. Cell Biol. 2020, 21, 7–24. [Google Scholar] [CrossRef]
- Silva, B.S.C.; DiGiovanni, L.; Kumar, R.; Carmichael, R.E.; Kim, P.K.; Schrader, M. Maintaining social contacts: The physiological relevance of organelle interactions. Biochim. Biophys. Acta-Mol. Cell Res. 2020, 1867, 118800. [Google Scholar] [CrossRef]
- Vance, J.E. Phospholipid Synthesis and Transport in Mammalian Cells. Traffic 2015, 16, 1–18. [Google Scholar] [CrossRef]
- Bell, R.M.; Ballas, L.M.; Coleman, R.A. Lipid topogenesis. J. Lipid Res. 1981, 22, 391–403. [Google Scholar] [CrossRef]
- Vance, J.E. Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 1990, 265, 7248–7256. [Google Scholar] [CrossRef]
- Gaigg, B.; Simbeni, R.; Hrastnik, C.; Paltauf, F.; Daum, G. Characterization of a microsomal subfraction associated with mitochondria of the yeast, Saccharomyces cerevisiae. Involvement in synthesis and import of phospholipids into mitochondria. Biochim. Biophys. Acta-Biomembr. 1995, 1234, 214–220. [Google Scholar] [CrossRef] [Green Version]
- Cui, Z.; Vance, J.E.; Chen, M.H.; Voelker, D.R.; Vance, D.E. Cloning and expression of a novel phosphatidylethanolamine N-methyltransferase. A specific biochemical and cytological marker for a unique membrane fraction in rat liver. J. Biol. Chem. 1993, 268, 16655–16663. [Google Scholar] [CrossRef]
- Pichler, H.; Gaigg, B.; Hrastnik, C.; Achleitner, G.; Kohlwein, S.D.; Zellnig, G.; Perktold, A.; Daum, G. A subfraction of the yeast endoplasmic reticulum associates with the plasma membrane and has a high capacity to synthesize lipids. Eur. J. Biochem. 2001, 268, 2351–2361. [Google Scholar] [CrossRef]
- Tavassoli, S.; Chao, J.T.; Young, B.P.; Cox, R.C.; Prinz, W.A.; De Kroon, A.I.P.M.; Loewen, C.J.R. Plasma membrane-Endoplasmic reticulum contact sites regulate phosphatidylcholine synthesis. EMBO Rep. 2013, 14, 434–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.J.; Guzman-Hernandez, M.L.; Balla, T. A highly dynamic ER-derived phosphatidylinositol-synthesizing organelle supplies phosphoinositides to cellular membranes. Dev. Cell 2011, 21, 813–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buhman, K.K.; Chen, H.C.; Farese, R.V. The Enzymes of Neutral Lipid Synthesis. J. Biol. Chem. 2001, 276, 40369–40372. [Google Scholar] [CrossRef] [Green Version]
- Jacquier, N.; Choudhary, V.; Mari, M.; Toulmay, A.; Reggiori, F.; Schneiter, R. Lipid droplets are functionally connected to the endoplasmic reticulum in Saccharomyces cerevisiae. J. Cell Sci. 2011, 124, 2424–2437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhary, V.; Ojha, N.; Golden, A.; Prinz, W.A. A conserved family of proteins facilitates nascent lipid droplet budding from the ER. J. Cell Biol. 2015, 211, 261–271. [Google Scholar] [CrossRef]
- Kassan, A.; Herms, A.; Fernández-Vidal, A.; Bosch, M.; Schieber, N.L.; Reddy, B.J.N.; Fajardo, A.; Gelabert-Baldrich, M.; Tebar, F.; Enrich, C.; et al. Acyl-CoA synthetase 3 promotes lipid droplet biogenesis in ER microdomains. J. Cell Biol. 2013, 203, 985–1001. [Google Scholar] [CrossRef] [Green Version]
- Walther, T.C.; Chung, J.; Farese, R.V. Lipid Droplet Biogenesis. Annu. Rev. Cell Dev. Biol. 2017, 33, 491–510. [Google Scholar] [CrossRef] [Green Version]
- Shockey, J.M.; Gidda, S.K.; Chapital, D.C.; Kuan, J.C.; Dhanoa, P.K.; Bland, J.M.; Rothstein, S.J.; Mullen, R.T.; Dyer, J.M. Tung tree DGAT1 and DGAT2 have nonredundant functions in triacylglycerol biosynthesis and are localized to different subdomains of the endoplasmic reticulum. Plant Cell 2006, 18, 2294–2313. [Google Scholar] [CrossRef] [Green Version]
- Wilfling, F.; Thiam, A.R.; Olarte, M.J.; Wang, J.; Beck, R.; Gould, T.J.; Allgeyer, E.S.; Pincet, F.; Bewersdorf, J.; Farese, R.V.; et al. Arf1/COPI machinery acts directly on lipid droplets and enables their connection to the ER for protein targeting. Elife 2014, 2014, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Soni, K.G.; Mardones, G.A.; Sougrat, R.; Smirnova, E.; Jackson, C.L.; Bonifacino, J.S. Coatomer-dependent protein delivery to lipid droplets. J. Cell Sci. 2009, 122, 1834–1841. [Google Scholar] [CrossRef] [Green Version]
- Beller, M.; Sztalryd, C.; Southall, N.; Bell, M.; Jäckle, H.; Auld, D.S.; Oliver, B. COPI Complex is a Regulator of Lipid Homeostasis. PLoS Biol. 2008, 6, e292. [Google Scholar] [CrossRef] [Green Version]
- Wilfling, F.; Wang, H.; Haas, J.T.; Krahmer, N.; Gould, T.J.; Uchida, A.; Cheng, J.X.; Graham, M.; Christiano, R.; Fröhlich, F.; et al. Triacylglycerol synthesis enzymes mediate lipid droplet growth by relocalizing from the ER to lipid droplets. Dev. Cell 2013, 24, 384–399. [Google Scholar] [CrossRef] [Green Version]
- De Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef]
- Lim, Y.; Cho, I.T.; Schoel, L.J.; Cho, G.; Golden, J.A. Hereditary spastic paraplegia-linked REEP1 modulates endoplasmic reticulum/mitochondria contacts. Ann. Neurol. 2015, 78, 679–696. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E.; Tasseva, G. Formation and function of phosphatidylserine and phosphatidylethanolamine in mammalian cells. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2013, 1831, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Funato, K.; Riezman, H.; Muñiz, M. Vesicular and non-vesicular lipid export from the ER to the secretory pathway. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2020, 1865, 158453. [Google Scholar] [CrossRef]
- Fryer, L.G.D.; Jones, B.; Duncan, E.J.; Hutchison, C.E.; Ozkan, T.; Williams, P.A.; Alder, O.; Nieuwdorp, M.; Townley, A.K.; Mensenkamp, A.R.; et al. The endoplasmic reticulum coat protein II transport machinery coordinates cellular lipid secretion and cholesterol biosynthesis. J. Biol. Chem. 2014, 289, 4244–4261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saheki, Y.; Bian, X.; Schauder, C.M.; Sawaki, Y.; Surma, M.A.; Klose, C.; Pincet, F.; Reinisch, K.M.; De Camilli, P. Control of plasma membrane lipid homeostasis by the extended synaptotagmins. Nat. Cell Biol. 2016, 18, 504–515. [Google Scholar] [CrossRef]
- Yu, H.; Liu, Y.; Gulbranson, D.R.; Paine, A.; Rathore, S.S.; Shen, J. Extended synaptotagmins are Ca2+-dependent lipid transfer proteins at membrane contact sites. Proc. Natl. Acad. Sci. USA 2016, 113, 4362–4367. [Google Scholar] [CrossRef] [Green Version]
- Lees, J.A.; Messa, M.; Sun, E.W.; Wheeler, H.; Torta, F.; Wenk, M.R.; De Camilli, P.; Reinisch, K.M. Lipid transport by TMEM24 at ER-plasma membrane contacts regulates pulsatile insulin secretion. Science 2017, 355, eaah6171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.L.; Liou, J. Phosphatidylinositol 4, 5-bisphosphate homeostasis regulated by Nir2 and Nir3 proteins at endoplasmic reticulum-plasma membrane junctions. J. Biol. Chem. 2015, 290, 14289–14301. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.J.; Guzman-Hernandez, M.L.; Wisniewski, E.; Balla, T. Phosphatidylinositol-Phosphatidic Acid Exchange by Nir2 at ER-PM Contact Sites Maintains Phosphoinositide Signaling Competence. Dev. Cell 2015, 33, 549–561. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.; Torta, F.; Masai, K.; Lucast, L.; Czapla, H.; Tanner, L.B.; Narayanaswamy, P.; Wenk, M.R.; Nakatsu, F.; De Camilli, P. PI4P/phosphatidylserine countertransport at ORP5- and ORP8-mediated ER-Plasma membrane contacts. Science 2015, 349, 428–432. [Google Scholar] [CrossRef] [Green Version]
- Ghai, R.; Du, X.; Wang, H.; Dong, J.; Ferguson, C.; Brown, A.J.; Parton, R.G.; Wu, J.W.; Yang, H. ORP5 and ORP8 bind phosphatidylinositol-4, 5-biphosphate (PtdIns(4,5)P 2) and regulate its level at the plasma membrane. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hanada, K.; Kumagai, K.; Yasuda, S.; Miura, Y.; Kawano, M.; Fukasawa, M.; Nishijima, M. Molecular machinery for non-vesicular trafficking of ceramide. Nature 2003, 426, 803–809. [Google Scholar] [CrossRef]
- Peretti, D.; Dahan, N.; Shimoni, E.; Hirschberg, K.; Lev, S. Coordinated Lipid Transfer between the Endoplasmic Reticulum and the Golgi Complex Requires the VAP Proteins and is Essential for Golgi-mediated Transport. Mol. Biol. Cell 2008, 19, 3871–3884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesmin, B.; Bigay, J.; Moser von Filseck, J.; Lacas-Gervais, S.; Drin, G.; Antonny, B. A Four-Step Cycle Driven by PI(4)P Hydrolysis Directs Sterol/PI(4)P Exchange by the ER-Golgi Tether OSBP. Cell 2013, 155, 830–843. [Google Scholar] [CrossRef] [Green Version]
- Eden, E.R.; Sanchez-Heras, E.; Tsapara, A.; Sobota, A.; Levine, T.P.; Futter, C.E. Annexin A1 Tethers Membrane Contact Sites that Mediate ER to Endosome Cholesterol Transport. Dev. Cell 2016, 37, 473–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.; Kumar, J.; Ferguson, C.; Schulz, T.A.; Ong, Y.S.; Hong, W.; Prinz, W.A.; Parton, R.G.; Brown, A.J.; Yang, H. A role for oxysterol-binding protein-related protein 5 in endosomal cholesterol trafficking. J. Cell Biol. 2011, 192, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Ouimet, M.; Hennessy, E.J.; Van Solingen, C.; Koelwyn, G.J.; Hussein, M.A.; Ramkhelawon, B.; Rayner, K.J.; Temel, R.E.; Perisic, L.; Hedin, U.; et al. MiRNA targeting of oxysterol-binding protein-like 6 regulates cholesterol trafficking and efflux. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 942–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelm, L.P.; Wendling, C.; Védie, B.; Kobayashi, T.; Chenard, M.; Tomasetto, C.; Drin, G.; Alpy, F. STARD 3 mediates endoplasmic reticulum-to-endosome cholesterol transport at membrane contact sites. EMBO J. 2017, 36, 1412–1433. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Leonzino, M.; Hancock-Cerutti, W.; Horenkamp, F.A.; Li, P.Q.; Lees, J.A.; Wheeler, H.; Reinisch, K.M.; De Camilli, P. VPS13A and VPS13C are lipid transport proteins differentially localized at ER contact sites. J. Cell Biol. 2018, 217, 3625–3639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirane, M.; Wada, M.; Morita, K.; Hayashi, N.; Kunimatsu, R.; Matsumoto, Y.; Matsuzaki, F.; Nakatsumi, H.; Ohta, K.; Tamura, Y.; et al. Protrudin and PDZD8 contribute to neuronal integrity by promoting lipid extraction required for endosome maturation. Nat. Commun. 2020, 11, 4576. [Google Scholar] [CrossRef]
- Elbaz-Alon, Y.; Guo, Y.; Segev, N.; Harel, M.; Quinnell, D.E.; Geiger, T.; Avinoam, O.; Li, D.; Nunnari, J. PDZD8 interacts with Protrudin and Rab7 at ER-late endosome membrane contact sites associated with mitochondria. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Saheki, Y.; Swarup, S.; Lucast, L.; Harper, J.W.; De Camilli, P. Endosome-ER Contacts Control Actin Nucleation and Retromer Function through VAP-Dependent Regulation of PI4P. Cell 2016, 166, 408–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabukusta, B.; Neefjes, J. Mechanisms of lysosomal positioning and movement. Traffic 2018, 19, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Lie, P.P.Y.; Nixon, R.A. Lysosome trafficking and signaling in health and neurodegenerative diseases. Neurobiol. Dis. 2019, 122, 94–105. [Google Scholar] [CrossRef]
- Eden, E.R.; White, I.J.; Tsapara, A.; Futter, C.E. Membrane contacts between endosomes and ER provide sites for PTP1B-epidermal growth factor receptor interaction. Nat. Cell Biol. 2010, 12, 267–272. [Google Scholar] [CrossRef]
- Ridgway, N.D.; Zhao, K. Cholesterol transfer at endosomal-organelle membrane contact sites. Curr. Opin. Lipidol. 2018, 29, 212–217. [Google Scholar] [CrossRef]
- Ballas, L.M.; Lazarow, P.B.; Bell, R.M. Glycerolipid synthetic capacity of rat liver peroxisomes. Biochim. Biophys. Acta (BBA)/Lipids Lipid Metab. 1984, 795, 297–300. [Google Scholar] [CrossRef]
- Hajra, A.K.; Bishop, J.E. Glycerolipid Biosynthesis in Peroxisomes Via the Acyl Dihydroxyacetone Phosphate Pathway. Ann. N. Y. Acad. Sci. 1982, 386, 170–182. [Google Scholar] [CrossRef] [Green Version]
- Guillén-Samander, A.; Leonzino, M.; Hanna, M.G.; Tang, N.; Shen, H.; De Camilli, P. VPS13D bridges the ER to mitochondria and peroxisomes via Miro. J. Cell Biol. 2021, 220, e202010004. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, H.A.; Wang, C.; Kanfer, G.; Shah, H.V.; Velayos-Baeza, A.; Dulovic-Mahlow, M.; Brüggemann, N.; Anding, A.; Baehrecke, E.H.; Maric, D.; et al. VPS13D promotes peroxisome biogenesis. J. Cell Biol. 2021, 220. [Google Scholar] [CrossRef]
- Costello, J.L.; Castro, I.G.; Hacker, C.; Schrader, T.A.; Metz, J.; Zeuschner, D.; Azadi, A.S.; Godinho, L.F.; Costina, V.; Findeisen, P.; et al. ACBD5 and VAPB mediate membrane associations between peroxisomes and the ER. J. Cell Biol. 2017, 216, 331–342. [Google Scholar] [CrossRef] [Green Version]
- Raychaudhuri, S.; Prinz, W.A. Nonvesicular phospholipid transfer between peroxisomes and the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2008, 105, 15785–15790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiao, Y.J.; Lupo, G.; Vance, J.E. Evidence that phosphatidylserine is imported into mitochondria via a mitochondria-associated membrane and that the majority of mitochondrial phosphatidylethanolamine is derived from decarboxylation of phosphatidylserine. J. Biol. Chem. 1995, 270, 11190–11198. [Google Scholar] [CrossRef] [Green Version]
- Voelker, D.R. Reconstitution of phosphatidylserine import into rat liver mitochondria. J. Biol. Chem. 1989, 264, 8019–8025. [Google Scholar] [CrossRef]
- Vance, J.E. Newly made phosphatidylserine and phosphatidylethanolamine are preferentially translocated between rat liver mitochondria and endoplasmic reticulum. J. Biol. Chem. 1991, 266, 89–97. [Google Scholar] [CrossRef]
- Kornmann, B.; Currie, E.; Collins, S.R.; Schuldiner, M.; Nunnari, J.; Weissman, J.S.; Walter, P. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science 2009, 325, 477–481. [Google Scholar] [CrossRef] [Green Version]
- AhYoung, A.P.; Jiang, J.; Zhang, J.; Dang, X.K.; Loo, J.A.; Zhou, Z.H.; Egea, P.F. Conserved SMP domains of the ERMES complex bind phospholipids and mediate tether assembly. Proc. Natl. Acad. Sci. USA 2015, 112, e3179–e3188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kojima, R.; Endo, T.; Tamura, Y. A phospholipid transfer function of ER-mitochondria encounter structure revealed in vitro. Sci. Rep. 2016, 6, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Jeong, H.; Park, J.; Jun, Y.; Lee, C. Crystal structures of Mmm1 and Mdm12–Mmm1 reveal mechanistic insight into phospholipid trafficking at ER-mitochondria contact sites. Proc. Natl. Acad. Sci. USA 2017, 114, e9502–e9511. [Google Scholar] [CrossRef] [Green Version]
- Murley, A.; Sarsam, R.D.; Toulmay, A.; Yamada, J.; Prinz, W.A.; Nunnari, J. Ltc1 is an ER-localized sterol transporter and a component of ER-mitochondria and ER-vacuole contacts. J. Cell Biol. 2015, 209, 539–548. [Google Scholar] [CrossRef] [Green Version]
- Elbaz-Alon, Y.; Eisenberg-Bord, M.; Shinder, V.; Stiller, S.B.; Shimoni, E.; Wiedemann, N.; Geiger, T.; Schuldiner, M. Lam6 Regulates the Extent of Contacts between Organelles. Cell Rep. 2015, 12, 7–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatta, A.T.; Wong, L.H.; Sere, Y.Y.; Calderón-Noreña, D.M.; Cockcroft, S.; Menon, A.K.; Levine, T.P. A new family of StART domain proteins at membrane contact sites has a role in ER-PM sterol transport. Elife 2015, 4, 1–46. [Google Scholar] [CrossRef] [PubMed]
- Sleight, R.G.; Pagano, R.E. Rapid appearance of newly synthesized phosphatidylethanolamine at the plasma membrane. J. Biol. Chem. 1983, 258, 9050–9058. [Google Scholar] [CrossRef]
- Kaplan, M.R.; Simoni, R.D. Intracellular transport of phosphatidylcholine to the plasma membrane. J. Cell Biol. 1985, 101, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, M.R.; Simoni, R.D. Transport of cholesterol from the endoplasmic reticulum to the plasma membrane. J. Cell Biol. 1985, 101, 446–453. [Google Scholar] [CrossRef] [Green Version]
- Warnock, D.E.; Lutz, M.S.; Blackburn, W.A.; Young, W.W.; Baenziger, J.U. Transport of newly synthesized glucosylceramide to the plasma membrane by a non-Golgi pathway. Proc. Natl. Acad. Sci. USA 1994, 91, 2708–2712. [Google Scholar] [CrossRef] [Green Version]
- Jouaville, L.S.; Pinton, P.; Bastianutto, C.; Rutter, G.A.; Rizzuto, R. Regulation of mitochondrial ATP synthesis by calcium: Evidence for a long-term metabolic priming. Proc. Natl. Acad. Sci. USA 1999, 96, 13807–13812. [Google Scholar] [CrossRef] [Green Version]
- Pitter, J.G.; Maechler, P.; Wollheim, C.B.; Spät, A. Mitochondria respond to Ca2+ already in the submicromolar range: Correlation with redox state. Cell Calcium 2002, 31, 97–104. [Google Scholar] [CrossRef] [Green Version]
- Scorrano, L.; Oakes, S.A.; Opferman, J.T.; Cheng, E.H.; Sorcinelli, M.D.; Pozzan, T.; Korsmeyer, S.J. BAX and BAK regulation of endoplasmic reticulum Ca2+: A control point for apoptosis. Science 2003, 300, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Wada, I.; Rindress, D.; Cameron, P.H.; Ou, W.J.; Doherty, J.J.; Louvard, D.; Bell, A.W.; Dignard, D.; Thomas, D.Y.; Bergeron, J.J.M. SSRα and associated calnexin are major calcium binding proteins of the endoplasmic reticulum membrane. J. Biol. Chem. 1991, 266, 19599–19610. [Google Scholar] [CrossRef]
- Baksh, S.; Michalak, M. Expression of calreticulin in Escherichia coli and identification of its Ca2+ binding domains. J. Biol. Chem. 1991, 266, 21458–21465. [Google Scholar] [CrossRef]
- Macer, D.R.J.; Koch, G.L.E. Identification of a set of calcium-binding proteins in reticuloplasm, the luminal content of the endoplasmic reticulum. J. Cell Sci. 1988, 91, 61–70. [Google Scholar] [CrossRef]
- Lodish, H.F.; Kong, N.; Wikstrom, L. Calcium is required for folding of newly made subunits of the asialoglycoprotein receptor within the endoplasmic reticulum. J. Biol. Chem. 1992, 267, 12753–12760. [Google Scholar] [CrossRef]
- Vassilakos, A.; Michalak, M.; Lehrman, M.A.; Williams, D.B. Oligosaccharide binding characteristics of the molecular chaperones calnexin and calreticulin. Biochemistry 1998, 37, 3480–3490. [Google Scholar] [CrossRef]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling—From basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef]
- Csordás, G.; Thomas, A.P.; Hajnóczky, G. Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. EMBO J. 1999, 18, 96–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gincel, D.; Zaid, H.; Shoshan-Barmatz, V. Calcium binding and translocation by the voltage-dependent anion channel: A possible regulatory mechanism in mitochondrial function. Biochem. J. 2001, 358, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Rapizzi, E.; Pinton, P.; Szabadkai, G.; Wieckowski, M.R.; Vandecasteele, G.; Baird, G.; Tuft, R.A.; Fogarty, K.E.; Rizzuto, R. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J. Cell Biol. 2002, 159, 613–624. [Google Scholar] [CrossRef] [Green Version]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [Green Version]
- Filadi, R.; Greotti, E.; Pizzo, P. Highlighting the endoplasmic reticulum-mitochondria connection: Focus on Mitofusin 2. Pharmacol. Res. 2018, 128, 42–51. [Google Scholar] [CrossRef]
- Atakpa, P.; Thillaiappan, N.B.; Mataragka, S.; Prole, D.L.; Taylor, C.W. IP3 Receptors Preferentially Associate with ER-Lysosome Contact Sites and Selectively Deliver Ca2+ to Lysosomes. Cell Rep. 2018, 25, 3180–3193.e7. [Google Scholar] [CrossRef] [Green Version]
- Roos, J.; DiGregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef] [Green Version]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.M.; Buchanan, J.; Luik, R.M.; Lewis, R.S. Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J. Cell Biol. 2006, 174, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.M.; Covington, E.D.; Lewis, R.S. Single-molecule analysis of diffusion and trapping of STIM1 and Orai1 at endoplasmic reticulum-plasma membrane junctions. Mol. Biol. Cell 2014, 25, 3672–3685. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.P.; Zeng, W.; Dorwart, M.R.; Choi, Y.J.; Worley, P.F.; Muallem, S. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat. Cell Biol. 2009, 11, 337–343. [Google Scholar] [CrossRef]
- Sampieri, A.; Zepeda, A.; Asanov, A.; Vaca, L. Visualizing the store-operated channel complex assembly in real time: Identification of SERCA2 as a new member. Cell Calcium 2009, 45, 439–446. [Google Scholar] [CrossRef]
- Manjarrés, I.M.; Rodríguez-García, A.; Alonso, M.T.; García-Sancho, J. The sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) is the third element in capacitative calcium entry. Cell Calcium 2010, 47, 412–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jousset, H.; Frieden, M.; Demaurex, N. STIM1 knockdown reveals that store-operated Ca2+ channels located close to sarco/endoplasmic Ca2+ ATPases (SERCA) pumps silently refill the endoplasmic reticulum. J. Biol. Chem. 2007, 282, 11456–11464. [Google Scholar] [CrossRef] [Green Version]
- Rowland, A.A.; Chitwood, P.J.; Phillips, M.J.; Voeltz, G.K. ER contact sites define the position and timing of endosome fission. Cell 2014, 159, 1027–1041. [Google Scholar] [CrossRef] [Green Version]
- Allison, R.; Edgar, J.R.; Pearson, G.; Rizo, T.; Newton, T.; Günther, S.; Berner, F.; Hague, J.; Connell, J.W.; Winkler, J.; et al. Defects in ER-endosome contacts impact lysosome function in hereditary spastic paraplegia. J. Cell Biol. 2017, 216, 1337–1355. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Voeltz, G.K. Reticulon-3 Promotes Endosome Maturation at ER Membrane Contact Sites. Dev. Cell 2021, 56, 52–66.e7. [Google Scholar] [CrossRef]
- Hoyer, M.J.; Chitwood, P.J.; Ebmeier, C.C.; Striepen, J.F.; Qi, R.Z.; Old, W.M.; Voeltz, G.K. A Novel Class of ER Membrane Proteins Regulates ER-Associated Endosome Fission. Cell 2018, 175, 254–265.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadler, L.; Speckner, K.; Weiss, M. Diffusion of Exit Sites on the Endoplasmic Reticulum: A Random Walk on a Shivering Backbone. Biophys. J. 2018, 115, 1552–1560. [Google Scholar] [CrossRef] [Green Version]
- Holcman, D.; Parutto, P.; Chambers, J.E.; Fantham, M.; Young, L.J.; Marciniak, S.J.; Kaminski, C.F.; Ron, D.; Avezov, E. Single particle trajectories reveal active endoplasmic reticulum luminal flow. Nat. Cell Biol. 2018, 20, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Chen, L.B. Dynamic behavior of endoplasmic reticulum in living cells. Cell 1988, 54, 37–46. [Google Scholar] [CrossRef]
- Dailey, M.E.; Bridgman, P.C. Dynamics of the endoplasmic reticulum and other membranous organelles in growth cones of cultured neurons. J. Neurosci. 1989, 9, 1897–1909. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Romano, F.B.; Field, C.M.; Mitchison, T.J.; Rapoport, T.A. Multiple mechanisms determine ER network morphology during the cell cycle in Xenopus egg extracts. J. Cell Biol. 2013, 203, 801–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, D.; Golchoubian, B.; Belevich, I.; Jokitalo, E.; Schlaitz, A.L. REEP3 and REEP4 determine the tubular morphology of the endoplasmic reticulum during mitosis. Mol. Biol. Cell 2019, 30, 1377–1389. [Google Scholar] [CrossRef] [PubMed]
- Terasaki, M. Microtubules and the endoplasmic reticulum are highly interdependent structures. J. Cell Biol. 1986, 103, 1557–1568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, J.D.; Allan, V.J. Microtubule-based endoplasmic reticulum motility in Xenopus laevis: Activation of membrane-associated kinesin during development. Mol. Biol. Cell 1999, 10, 1909–1922. [Google Scholar] [CrossRef] [Green Version]
- Joensuu, M.; Belevich, I.; Rämö, O.; Nevzorov, I.; Vihinen, H.; Puhka, M.; Witkos, T.M.; Lowe, M.; Vartiainen, M.K.; Jokitalo, E. ER sheet persistence is coupled to myosin 1c-regulated dynamic actin filament arrays. Mol. Biol. Cell 2014, 25, 1111–1126. [Google Scholar] [CrossRef] [PubMed]
- Dabora, S.L.; Sheetz, M.F. The microtubule-dependent formation of a tubulovesicular network with characteristics of the ER from cultured cell extracts. Cell 1988, 54, 27–35. [Google Scholar] [CrossRef]
- Allan, V.J.; Vale, R.D. Cell cycle control of microtuble-based membrane transport and tubule formation in vitro. J. Cell Biol. 1991, 113, 347–359. [Google Scholar] [CrossRef] [Green Version]
- Allan, V.; Vale, R. Movement of membrane tubules along microtubules in vitro: Evidence for specialised sites of motor attachment. J. Cell Sci. 1994, 107, 1885–1897. [Google Scholar] [CrossRef]
- Dreier, L.; Rapoport, T.A. In vitro formation of the endoplasmic reticulum occurs independently of microtubules by a controlled fusion reaction. J. Cell Biol. 2000, 148, 883–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woźniak, M.J.; Allan, V.J. Cargo selection by specific kinesin light chain 1 isoforms. EMBO J. 2006, 25, 5457–5468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farías, G.G.; Fréal, A.; Tortosa, E.; Stucchi, R.; Pan, X.; Portegies, S.; Will, L.; Altelaar, M.; Hoogenraad, C.C. Feedback-Driven Mechanisms between Microtubules and the Endoplasmic Reticulum Instruct Neuronal Polarity. Neuron 2019, 102, 184–201.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sleigh, J.N.; Rossor, A.M.; Fellows, A.D.; Tosolini, A.P.; Schiavo, G. Axonal transport and neurological disease. Nat. Rev. Neurol. 2019, 15, 691–703. [Google Scholar] [CrossRef]
- Kumar, J.; Yu, H.; Sheetz, M.P. Kinectin, an essential anchor for kinesin-driven vesicle motility. Science 1995, 67, 1834–1837. [Google Scholar] [CrossRef]
- Ong, L.L.; Lim, A.P.C.; Er, C.P.N.; Kuznetsov, S.A.; Yu, H. Kinectin-kinesin binding domains and their effects on organelle motility. J. Biol. Chem. 2000, 275, 32854–32860. [Google Scholar] [CrossRef] [Green Version]
- Guadagno, N.A.; Margiotta, A.; Bjørnestad, S.A.; Haugen, L.H.; Kjos, I.; Xu, X.; Hu, X.; Bakke, O.; Margadant, F.; Progida, C. Rab18 regulates focal adhesion dynamics by interacting with kinectin-1 at the endoplasmic reticulum. J. Cell Biol. 2020, 219, e201809020. [Google Scholar] [CrossRef]
- Ng, I.C.; Pawijit, P.; Teo, L.Y.; Li, H.; Lee, S.Y.; Yu, H. Kinectin-dependent ER transport supports the focal complex maturation required for chemotaxis in shallow gradients. J. Cell Sci. 2016, 129, 2660–2672. [Google Scholar] [CrossRef] [Green Version]
- Plitz, T.; Pfeffer, K. Intact Lysosome Transport and Phagosome Function Despite Kinectin Deficiency. Mol. Cell. Biol. 2001, 21, 6044–6055. [Google Scholar] [CrossRef] [Green Version]
- Deng, C.Y.; Lei, W.L.; Xu, X.H.; Ju, X.C.; Liu, Y.; Luo, Z.G. JIP1 mediates anterograde transport of Rab10 cargos during neuronal polarization. J. Neurosci. 2014, 34, 1710–1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diefenbach, R.J.; Diefenbach, E.; Douglas, M.W.; Cunningham, A.L. The ribosome receptor, p180, interacts with kinesin heavy chain, KIF5B. Biochem. Biophys. Res. Commun. 2004, 319, 987–992. [Google Scholar] [CrossRef] [PubMed]
- Hein, M.Y.; Hubner, N.C.; Poser, I.; Cox, J.; Nagaraj, N.; Toyoda, Y.; Gak, I.A.; Weisswange, I.; Mansfeld, J.; Buchholz, F.; et al. A Human Interactome in Three Quantitative Dimensions Organized by Stoichiometries and Abundances. Cell 2015, 163, 712–723. [Google Scholar] [CrossRef] [Green Version]
- Raiborg, C.; Wenzel, E.M.; Pedersen, N.M.; Olsvik, H.; Schink, K.O.; Schultz, S.W.; Vietri, M.; Nisi, V.; Bucci, C.; Brech, A.; et al. Repeated ER-endosome contacts promote endosome translocation and neurite outgrowth. Nature 2015, 520, 234–238. [Google Scholar] [CrossRef]
- Pedersen, N.M.; Wenzel, E.M.; Wang, L.; Antoine, S.; Chavrier, P.; Stenmark, H.; Raiborg, C. Protrudin-mediated ER–endosome contact sites promote MT1-MMP exocytosis and cell invasion. J. Cell Biol. 2020, 219, e202003063. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, M.S.; Engelke, M.F.; Verhey, K.J.; Tsai, B. Exploiting the kinesin-1 molecular motor to generate a virus membrane penetration site. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Niclas, J.; Allan, V.J.; Vale, R.D. Cell cycle regulation of dynein association with membranes modulates microtubule-based organelle transport. J. Cell Biol. 1996, 133, 585–593. [Google Scholar] [CrossRef]
- Pelletier, J.F.; Field, C.M.; Fürthauer, S.; Sonnett, M.; Mitchison, T.J. Co-movement of astral microtubules, organelles and f-actin by dynein and actomyosin forces in frog egg cytoplasm. Elife 2020, 9, 1–30. [Google Scholar] [CrossRef]
- Reinsch, S.; Karsenti, E. Movement of nuclei along microtubules in Xenopus egg extracts. Curr. Biol. 1997, 7, 211–214. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, R.N.; Sallé, J.; Dmitrieff, S.; Nelson, K.M.; Oakey, J.; Minc, N.; Levy, D.L. The Perinuclear ER Scales Nuclear Size Independently of Cell Size in Early Embryos. Dev. Cell 2020, 54, 395–409.e7. [Google Scholar] [CrossRef]
- Wöllert, T.; Weiss, D.G.; Gerdes, H.H.; Kuznetsov, S.A. Activation of myosin V-based motility and F-actin-dependent network formation of endoplasmic reticulum during mitosis. J. Cell Biol. 2002, 159, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, J.I.; Jaureguiberry-Bravo, M.; Salas, D.A.; Ramírez, O.A.; Cornejo, V.H.; Lu, H.E.; Blanpied, T.A.; Couve, A. Transport along the dendritic endoplasmic reticulum mediates the trafficking of GABAB receptors. J. Cell Sci. 2014, 127, 3382–3395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannai, H.; Inoue, T.; Nakayama, T.; Hattori, M.; Mikoshiba, K. Kinesin dependent, rapid, bi-directional transport of ER sub-compartment in dendrites of hippocampal neurons. J. Cell Sci. 2004, 117, 163–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonçalves, J.C.; Quintremil, S.; Yi, J.; Vallee, R.B. Nesprin-2 Recruitment of BicD2 to the Nuclear Envelope Controls Dynein/Kinesin-Mediated Neuronal Migration In Vivo. Curr. Biol. 2020, 30, 3116–3129.e4. [Google Scholar] [CrossRef]
- Starr, D.A. A network of nuclear envelope proteins and cytoskeletal force generators mediates movements of and within nuclei throughout Caenorhabditis elegans development. Exp. Biol. Med. 2019, 244, 1323–1332. [Google Scholar] [CrossRef] [PubMed]
- Ungricht, R.; Kutay, U. Mechanisms and functions of nuclear envelope remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 229–245. [Google Scholar] [CrossRef]
- Splinter, D.; Tanenbaum, M.E.; Lindqvist, A.; Jaarsma, D.; Flotho, A.; Yu, K.L.; Grigoriev, I.; Engelsma, D.; Haasdijk, E.D.; Keijzer, N.; et al. Bicaudal D2, dynein, and kinesin-1 associate with nuclear pore complexes and regulate centrosome and nuclear positioning during mitotic entry. PLoS Biol. 2010, 8, e1000350. [Google Scholar] [CrossRef] [Green Version]
- Roux, K.J.; Crisp, M.L.; Liu, Q.; Kim, D.; Kozlov, S.; Stewart, C.L.; Burke, B. Nesprin 4 is an outer nuclear membrane protein that can induce kinesin-mediated cell polarization. Proc. Natl. Acad. Sci. USA 2009, 106, 2194–2199. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Van Tartwijk, F.W.; Lin, J.Q.; Nijenhuis, W.; Parutto, P.; Fantham, M.; Christensen, C.N.; Avezov, E.; Holt, C.E.; Tunnacliffe, A.; et al. The structure and global distribution of the endoplasmic reticulum network are actively regulated by lysosomes. Sci. Adv. 2020, 6, 1–16. [Google Scholar] [CrossRef]
- English, A.R.; Voeltz, G.K. Endoplasmic reticulum structure and interconnections with other organelles. Cold Spring Harb. Perspect. Biol. 2013, 5, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guimaraes, S.C.; Schuster, M.; Bielska, E.; Dagdas, G.; Kilaru, S.; Meadows, B.R.A.; Schrader, M.; Steinberg, G. Peroxisomes, lipid droplets, and endoplasmic reticulum “hitchhike” on motile early endosomes. J. Cell Biol. 2015, 211, 945–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salogiannis, J.; Reck-Peterson, S.L. Hitchhiking: A Non-Canonical Mode of Microtubule-Based Transport. Trends Cell Biol. 2017, 27, 141–150. [Google Scholar] [CrossRef] [Green Version]
- Flores-Rodriguez, N.; Rogers, S.S.; Kenwright, D.A.; Waigh, T.A.; Woodman, P.G.; Allan, V.J. Roles of dynein and dynactin in early endosome dynamics revealed using automated tracking and global analysis. PLoS ONE 2011, 6, e24479. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.; Lee, S.; Blackstone, C. Protrudin binds atlastins and endoplasmic reticulum-shaping proteins and regulates network formation. Proc. Natl. Acad. Sci. USA 2013, 110, 14954–14959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirane, M. Lipid Transfer–Dependent Endosome Maturation Mediated by Protrudin and PDZD8 in Neurons. Front. Cell Dev. Biol. 2020, 8, 1–10. [Google Scholar] [CrossRef]
- Matsuzaki, F.; Shirane, M.; Matsumoto, M.; Nakayama, K.I. Protrudin serves as an adaptor molecule that connects KIF5 and its cargoes in vesicular transport during process formation. Mol. Biol. Cell 2011, 22, 4602–4620. [Google Scholar] [CrossRef]
- Öztürk, Z.; O’Kane, C.J.; Pérez-Moreno, J.J. Axonal Endoplasmic Reticulum Dynamics and Its Roles in Neurodegeneration. Front. Neurosci. 2020, 14, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Palomo-Guerrero, M.; Fadó, R.; Casas, M.; Pérez-Montero, M.; Baena, M.; Helmer, P.O.; Domínguez, J.L.; Roig, A.; Serra, D.; Hayen, H.; et al. Sensing of nutrients by CPT1C regulates late endosome/lysosome anterograde transport and axon growth. Elife 2019, 8, 1–26. [Google Scholar] [CrossRef]
- Serra-Marques, A.; Martin, M.; Katrukha, E.A.; Grigoriev, I.; Peeters, C.A.E.; Liu, Q.; Hooikaas, P.J.; Yao, Y.; Solianova, V.; Smal, I.; et al. Concerted action of kinesins kif5b and kif13b promotes efficient secretory vesicle transport to microtubule plus ends. Elife 2020, 9, 1–37. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Guo, Y.; Xue, B.; Shi, P.; Chen, Y.; Su, Q.P.; Hao, H.; Zhao, S.; Wu, C.; Yu, L.; et al. ER-mitochondria contacts promote mtDNA nucleoids active transportation via mitochondrial dynamic tubulation. Nat. Commun. 2020, 11, 4471. [Google Scholar] [CrossRef] [PubMed]
- Mateus, D.; Marini, E.S.; Progida, C.; Bakke, O. Rab7a modulates ER stress and ER morphology. Biochim. Biophys. Acta-Mol. Cell Res. 2018, 1865, 781–793. [Google Scholar] [CrossRef]
- Waterman-Storer, C.M.; Gregory, J.; Parsons, S.F.; Salmon, E.D. Membrane/microtubule tip attachment complexes (TACs) allow the assembly dynamics of plus ends to push and pull membranes into tubulovesicular networks in interphase Xenopus egg extracts. J. Cell Biol. 1995, 130, 1161–1169. [Google Scholar] [CrossRef]
- Pavez, M.; Thompson, A.C.; Arnott, H.J.; Mitchell, C.B.; D’Atri, I.; Don, E.K.; Chilton, J.K.; Scott, E.K.; Lin, J.Y.; Young, K.M.; et al. STIM1 is required for remodeling of the endoplasmic reticulum and microtubule cytoskeleton in steering growth cones. J. Neurosci. 2019, 39, 5095–5114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Zhang, G.; Lin, S.; Shi, J.; Zhang, H.; Hu, J. Sec61β facilitates the maintenance of endoplasmic reticulum homeostasis by associating microtubules. Protein Cell 2018, 9, 616–628. [Google Scholar] [CrossRef] [Green Version]
- Upadhyaya, A.; Sheetz, M.P. Tension in Tubulovesicular Networks of Golgi and Endoplasmic Reticulum Membranes. Biophys. J. 2004, 86, 2923–2928. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Hussey, P.J. Interactions between plant endomembrane systems and the actin cytoskeleton. Front. Plant. Sci. 2015, 6, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Quader, H.; Schnepf, E. Endoplasmic reticulum and cytoplasmic streaming: Fluorescence microscopical observations in adaxial epidermis cells of onion bulb scales. Protoplasma 1986, 131, 250–252. [Google Scholar] [CrossRef]
- Shimmen, T.; Yokota, E. Cytoplasmic streaming in plants. Curr. Opin. Cell Biol. 2004, 16, 68–72. [Google Scholar] [CrossRef]
- Delgadillo, M.O.; Ruano, G.; Zouhar, J.; Sauer, M.; Shen, J.; Lazarova, A.; Sanmartín, M.; Faat Lai, L.T.; Deng, C.; Wang, P.; et al. MTV proteins unveil ER- And microtubule-associated compartments in the plant vacuolar trafficking pathway. Proc. Natl. Acad. Sci. USA 2020, 117, 9884–9895. [Google Scholar] [CrossRef]
- Hamada, T.; Tominaga, M.; Fukaya, T.; Nakamura, M.; Nakano, A.; Watanabe, Y.; Hashimoto, T.; Baskin, T.I. RNA processing bodies, peroxisomes, golgi bodies, mitochondria, and endoplasmic reticulum tubule junctions frequently pause at cortical microtubules. Plant. Cell Physiol. 2012, 53, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.; Kalra, S.; Jameson, L.E.; Guerrero, L.A.; Cain, N.E.; Bolivar, J.; Starr, D.A. The nesprin-1/-2 ortholog anc-1 regulates organelle positioning in c. Elegans independently from its kash or actin-binding domains. Elife 2021, 10, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Tabb, J.S.; Molyneaux, B.J.; Cohen, D.L.; Kuznetsov, S.A.; Langford, G.M. Transport of ER vesicles on actin filaments in neurons by myosin V. J. Cell Sci. 1998, 111, 3221–3234. [Google Scholar] [CrossRef] [PubMed]
- Lynch, C.D.; Gauthier, N.C.; Biais, N.; Lazar, A.M.; Roca-Cusachs, P.; Yu, C.H.; Sheetz, M.P. Filamin depletion blocks endoplasmic spreading and destabilizes force-bearing adhesions. Mol. Biol. Cell 2011, 22, 1263–1273. [Google Scholar] [CrossRef] [Green Version]
- Terasaki, M.; Reese, T.S. Interactions among endoplasmic reticulum, microtubules, and retrograde movements of the cell surface. Cell Motil. Cytoskeleton 1994, 29, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Speckner, K.; Stadler, L.; Weiss, M. Anomalous dynamics of the endoplasmic reticulum network. Phys. Rev. E 2018, 98, 012406. [Google Scholar] [CrossRef] [PubMed]
- Granek, R. From Semi-Flexible Polymers to Membranes: Anomalous Diffusion and Reptation. J. Phys. II 1997, 7, 1761–1788. [Google Scholar] [CrossRef]
- Macháň, R.; Hof, M. Lipid diffusion in planar membranes investigated by fluorescence correlation spectroscopy. Biochim. Biophys. Acta-Biomembr. 2010, 1798, 1377–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jan Akhunzada, M.; D’Autilia, F.; Chandramouli, B.; Bhattacharjee, N.; Catte, A.; Di Rienzo, R.; Cardarelli, F.; Brancato, G. Interplay between lipid lateral diffusion, dye concentration and membrane permeability unveiled by a combined spectroscopic and computational study of a model lipid bilayer. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef]
- Dietrich, C.; Yang, B.; Fujiwara, T.; Kusumi, A.; Jacobson, K. Relationship of lipid rafts to transient confinement zones detected by single particle tracking. Biophys. J. 2002, 82, 274–284. [Google Scholar] [CrossRef] [Green Version]
- Eggeling, C.; Ringemann, C.; Medda, R.; Schwarzmann, G.; Sandhoff, K.; Polyakova, S.; Belov, V.N.; Hein, B.; Von Middendorff, C.; Schönle, A.; et al. Direct observation of the nanoscale dynamics of membrane lipids in a living cell. Nature 2009, 457, 1159–1162. [Google Scholar] [CrossRef]
- Peters, R.; Cherry, R.J. Lateral and rotational diffusion of bacteriorhodopsin in lipid bilayers: Experimental test of the Saffman-Delbruck equations. Proc. Natl. Acad. Sci. USA 1982, 79, 4317–4321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domański, J.; Marrink, S.J.; Schäfer, L.V. Transmembrane helices can induce domain formation in crowded model membranes. Biochim. Biophys. Acta-Biomembr. 2012, 1818, 984–994. [Google Scholar] [CrossRef]
- Malchus, N.; Weiss, M. Anomalous diffusion reports on the interaction of misfolded proteins with the quality control machinery in the endoplasmic reticulum. Biophys. J. 2010, 99, 1321–1328. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, T.; Ritchie, K.; Murakoshi, H.; Jacobson, K.; Kusumi, A. Phospholipids undergo hop diffusion in compartmentalized cell membrane. J. Cell Biol. 2002, 157, 1071–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Rienzo, C.; Gratton, E.; Beltram, F.; Cardarelli, F. Fast spatiotemporal correlation spectroscopy to determine protein lateral diffusion laws in live cell membranes. Proc. Natl. Acad. Sci. USA 2013, 110, 12307–12312. [Google Scholar] [CrossRef] [Green Version]
- Runions, J.; Brach, T.; Kühner, S.; Hawes, C. Photoactivation of GFP reveals protein dynamics within the endoplasmic reticulum membrane. J. Exp. Bot. 2006, 57, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.S.; Warshaviak, D.T.; Chachisvilis, M. Effect of membrane tension on the physical properties of DOPC lipid bilayer membrane. Biochim. Biophys. Acta-Biomembr. 2012, 1818, 2271–2281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Essmann, U.; Berkowitz, M.L. Dynamical properties of phospholipid bilayers from computer simulation. Biophys. J. 1999, 76, 2081–2089. [Google Scholar] [CrossRef] [Green Version]
- Dayel, M.J.; Horn, E.F.Y.; Verkman, A.S. Diffusion of green fluorescent protein in the aqueous-phase lumen of endoplasmic reticulum. Biophys. J. 1999, 76, 2843–2851. [Google Scholar] [CrossRef] [Green Version]
- Snapp, E.L.; Sharma, A.; Lippincott-Schwartz, J.; Hegde, R.S. Monitoring chaperone engagement of substrates in the endoplasmic reticulum of live cells. Proc. Natl. Acad. Sci. USA 2006, 103, 6536–6541. [Google Scholar] [CrossRef] [Green Version]
- Nagaya, H.; Tamura, T.; Higa-Nishiyama, A.; Ohashi, K.; Takeuchi, M.; Hashimoto, H.; Hatsuzawa, K.; Kinjo, M.; Okada, T.; Wada, I. Regulated motion of glycoproteins revealed by direct visualization of a single cargo in the endoplasmic reticulum. J. Cell Biol. 2008, 180, 129–143. [Google Scholar] [CrossRef] [Green Version]
- Kernes, J.; Levine, A.J. Equilibrium fluctuations of a semiflexible filament cross linked into a network. Phys. Rev. E 2020, 101, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Broedersz, C.P.; Mackintosh, F.C. Modeling semiflexible polymer networks. Rev. Mod. Phys. 2014, 86, 995–1036. [Google Scholar] [CrossRef] [Green Version]
- Kernes, J.; Levine, A.J. Dynamics of undulatory fluctuations of semiflexible filaments in a network. Phys. Rev. E 2020, 102, 1–19. [Google Scholar] [CrossRef]
- Müller, K.W.; Cyron, C.J.; Wall, W.A. Computational analysis of morphologies and phase transitions of cross-linked, semi-flexible polymer networks. Proc. R. Soc. A Math. Phys. Eng. Sci. 2015, 471, 20150332. [Google Scholar] [CrossRef]
- Gladrow, J.; Fakhri, N.; MacKintosh, F.C.; Schmidt, C.F.; Broedersz, C.P. Broken Detailed Balance of Filament Dynamics in Active Networks. Phys. Rev. Lett. 2016, 116, 248301. [Google Scholar] [CrossRef] [Green Version]
- Joo, S.; Durang, X.; Lee, O.C.; Jeon, J.H. Anomalous diffusion of active Brownian particles cross-linked to a networked polymer: Langevin dynamics simulation and theory. Soft Matter 2020, 16, 9188–9201. [Google Scholar] [CrossRef] [PubMed]
- Gov, N.S. Phases of membrane tubules pulled by molecular motors. Soft Matter 2009, 5, 2431–2437. [Google Scholar] [CrossRef]
- Gong, B.; Lin, J.; Wei, X.; Qian, J.; Lin, Y. Cross-linked biopolymer networks with active motors: Mechanical response and intra-network transport. J. Mech. Phys. Solids 2019, 127, 80–93. [Google Scholar] [CrossRef]
- Lin, C.; Zhang, Y.; Sparkes, I.; Ashwin, P. Structure and dynamics of ER: Minimal networks and biophysical constraints. Biophys. J. 2014, 107, 763–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.; White, R.R.; Sparkes, I.; Ashwin, P. Modeling Endoplasmic Reticulum Network Maintenance in a Plant Cell. Biophys. J. 2017, 113, 214–222. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Lemarchand, L.; Euler, R.; Sparkes, I. Modeling the Geometry and Dynamics of the Endoplasmic Reticulum Network. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018, 15, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.I.; Westrate, L.M.; Koslover, E.F. Impact of global structure on diffusive exploration of organelle networks. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Bassot, A.; Giuliani, F.; Simmen, T. Amyotrophic Lateral Sclerosis (ALS): Stressed by Dysfunctional Mitochondria-Endoplasmic Reticulum Contacts (MERCs). Cells 2021, 10, 1789. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.A.; Song, C.H. Insights Into the Role of Endoplasmic Reticulum Stress in Infectious Diseases. Front. Immunol. 2020, 10, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safiedeen, Z.; Andriantsitohaina, R.; Martinez, M.C. Dialogue between endoplasmic reticulum and mitochondria as a key actor of vascular dysfunction associated to metabolic disorders. Int. J. Biochem. Cell Biol. 2016, 77, 10–14. [Google Scholar] [CrossRef]
- Ren, J.; Bi, Y.; Sowers, J.R.; Hetz, C.; Zhang, Y. Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases. Nat. Rev. Cardiol. 2021, 18, 499–521. [Google Scholar] [CrossRef]
- Hazan, J.; Fonknechten, N.; Mavel, D.; Paternotte, C.; Samson, D.; Artiguenave, F.; Davoine, C.S.; Cruaud, C.; Dürr, A.; Wincker, P.; et al. Spastin, a new AAA protein, is altered in the most frequent form of autosomal dominant spastic paraplegia. Nat. Genet. 1999, 23, 296–303. [Google Scholar] [CrossRef]
- Mannan, A.U.; Krawen, P.; Sauter, S.M.; Boehm, J.; Chronowska, A.; Paulus, W.; Neesen, J.; Engel, W. ZFYVE27 (SPG33), a novel spastin-binding protein, is mutated in hereditary spastic paraplegia. Am. J. Hum. Genet. 2006, 79, 351–357. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, Y.; Shirane, M.; Matsuzaki, F.; Saita, S.; Ohnishi, T.; Nakayama, K.I. Protrudin regulates endoplasmic reticulum morphology and function associated with the pathogenesis of hereditary spastic paraplegia. J. Biol. Chem. 2014, 289, 12946–12961. [Google Scholar] [CrossRef] [Green Version]
- Windpassinger, C.; Auer-Grumbach, M.; Irobi, J.; Patel, H.; Petek, E.; Hörl, G.; Malli, R.; Reed, J.A.; Dierick, I.; Verpoorten, N.; et al. Heterozygous missense mutations in BSCL2 are associated with distal hereditary motor neuropathy and Silver syndrome. Nat. Genet. 2004, 36, 271–276. [Google Scholar] [CrossRef]
- Szymanski, K.M.; Binns, D.; Bartz, R.; Grishin, N.V.; Li, W.P.; Agarwal, A.K.; Garg, A.; Anderson, R.G.W.; Goodman, J.M. The lipodystrophy protein seipin is found at endoplasmic reticulum lipid droplet junctions and is important for droplet morphology. Proc. Natl. Acad. Sci. USA 2007, 104, 20890–20895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, K.; Stone, M.C.; Weiner, A.T.; Gheres, K.W.; Zhou, C.; Deitcher, D.L.; Levitan, E.S.; Rolls, M.M. Spastin, atlastin, and ER relocalization are involved in axon but not dendrite regeneration. Mol. Biol. Cell 2016, 27, 3245–3256. [Google Scholar] [CrossRef]
- Zhao, X.; Alvarado, D.; Rainier, S.; Lemons, R.; Hedera, P.; Weber, C.H.; Tukel, T.; Apak, M.; Heiman-Patterson, T.; Ming, L.; et al. Mutations in a newly identified GTPase gene cause autosomal dominant hereditary spastic paraplegia. Nat. Genet. 2001, 29, 326–331. [Google Scholar] [CrossRef]
- Züchner, S.; Wang, G.; Tran-Viet, K.N.; Nance, M.A.; Gaskell, P.C.; Vance, J.M.; Ashley-Koch, A.E.; Pericak-Vance, M.A. Mutations in the novel mitochondrial protein REEP1 cause hereditary spastic paraplegia type 31. Am. J. Hum. Genet. 2006, 79, 365–369. [Google Scholar] [CrossRef] [Green Version]
- Montenegro, G.; Rebelo, A.P.; Connell, J.; Allison, R.; Babalini, C.; D’Aloia, M.; Montieri, P.; Schüle, R.; Ishiura, H.; Price, J.; et al. Mutations in the ER-shaping protein reticulon 2 cause the axon-degenerative disorder hereditary spastic paraplegia type 12. J. Clin. Invest. 2012, 122, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Esteves, T.; Durr, A.; Mundwiller, E.; Loureiro, J.L.; Boutry, M.; Gonzalez, M.A.; Gauthier, J.; El-Hachimi, K.H.; Depienne, C.; Muriel, M.P.; et al. Loss of association of REEP2 with membranes leads to hereditary spastic paraplegia. Am. J. Hum. Genet. 2014, 94, 268–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, A.L.; Mitne-Neto, M.; Silva, H.C.A.; Richieri-Costa, A.; Middleton, S.; Cascio, D.; Kok, F.; Oliveira, J.R.M.; Gillingwater, T.; Webb, J.; et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet. 2004, 75, 822–831. [Google Scholar] [CrossRef] [Green Version]
- Murphy, S.E.; Levine, T.P. VAP, a Versatile Access Point for the Endoplasmic Reticulum: Review and analysis of FFAT-like motifs in the VAPome. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2016, 1861, 952–961. [Google Scholar] [CrossRef]
- Borgese, N.; Iacomino, N.; Colombo, S.F.; Navone, F. The Link between VAPB Loss of Function and Amyotrophic Lateral Sclerosis. Cells 2021, 10, 1865. [Google Scholar] [CrossRef]
- Teuling, E.; Ahmed, S.; Haasdijk, E.; Demmers, J.; Steinmetz, M.O.; Akhmanova, A.; Jaarsma, D.; Hoogenraad, C.C. Motor neuron disease-associated mutant vesicle-associated membrane protein-associated protein (VAP) B recruits wild-type VAPs into endoplasmic reticulum-derived tubular aggregates. J. Neurosci. 2007, 27, 9801–9815. [Google Scholar] [CrossRef] [Green Version]
- Tsuda, H.; Han, S.M.; Yang, Y.; Tong, C.; Lin, Y.Q.; Mohan, K.; Haueter, C.; Zoghbi, A.; Harati, Y.; Kwan, J.; et al. The Amyotrophic Lateral Sclerosis 8 Protein VAPB is Cleaved, Secreted, and Acts as a Ligand for Eph Receptors. Cell 2008, 133, 963–977. [Google Scholar] [CrossRef] [Green Version]
- Fasana, E.; Fossati, M.; Ruggiano, A.; Brambillasca, S.; Hoogenraad, C.C.; Navone, F.; Francolini, M.; Borgese, N. A VAPB mutant linked to amyotrophic lateral sclerosis generates a novel form of organized smooth endoplasmic reticulum. FASEB J. 2010, 24, 1419–1430. [Google Scholar] [CrossRef]
- Papiani, G.; Ruggiano, A.; Fossati, M.; Raimondi, A.; Bertoni, G.; Francolini, M.; Benfante, R.; Navone, F.; Borgese, N. Restructured endoplasmic reticulum generated by mutant amyotrophic lateral sclerosis-linked VAPB is cleared by the proteasome. J. Cell Sci. 2012, 125, 3601–3611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De vos, K.J.; Mórotz, G.M.; Stoica, R.; Tudor, E.L.; Lau, K.F.; Ackerley, S.; Warley, A.; Shaw, C.E.; Miller, C.C.J. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet. 2012, 21, 1299–1311. [Google Scholar] [CrossRef] [Green Version]
- Paillusson, S.; Stoica, R.; Gomez-Suaga, P.; Lau, D.H.W.; Mueller, S.; Miller, T.; Miller, C.C.J. There’s Something Wrong with my MAM; the ER-Mitochondria Axis and Neurodegenerative Diseases. Trends Neurosci. 2016, 39, 146–157. [Google Scholar] [CrossRef] [Green Version]
- Moustaqim-barrette, A.; Lin, Y.Q.; Pradhan, S.; Neely, G.G.; Bellen, H.J.; Tsuda, H. The amyotrophic lateral sclerosis 8 protein, VAP, is required for ER protein quality control. Hum. Mol. Genet. 2014, 23, 1975–1989. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.S.; Harel, N.Y.; Strittmatter, S.M. Reticulon-4A (Nogo-A) redistributes protein disulfide isomerase to protect mice from SOD1-dependent amyotrophic lateral sclerosis. J. Neurosci. 2009, 29, 13850–13859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Saif, A.; Al-Mohanna, F.; Bohlega, S. A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis. Ann. Neurol. 2011, 70, 913–919. [Google Scholar] [CrossRef]
- Hayashi, T.; Su, T.P. Sigma-1 Receptor Chaperones at the ER- Mitochondrion Interface Regulate Ca2+ Signaling and Cell Survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, S.; Ilieva, H.; Tamada, H.; Nomura, H.; Komine, O.; Endo, F.; Jin, S.; Mancias, P.; Kiyama, H.; Yamanaka, K. Mitochondria-associated membrane collapse is a common pathomechanism in SIGMAR 1- and SOD 1-linked ALS. EMBO Mol. Med. 2016, 8, 1421–1437. [Google Scholar] [CrossRef] [PubMed]
- Züchner, S.; Noureddine, M.; Kennerson, M.; Verhoeven, K.; Claeys, K.; De Jonghe, P.; Merory, J.; Oliveira, S.A.; Speer, M.C.; Stenger, J.E.; et al. Mutations in the pleckstrin homology domain of dynamin 2 cause dominant intermediate Charcot-Marie-Tooth disease. Nat. Genet. 2005, 37, 289–294. [Google Scholar] [CrossRef]
- Lee, J.E.; Westrate, L.M.; Wu, H.; Page, C.; Voeltz, G.K. Multiple dynamin family members collaborate to drive mitochondrial division. Nature 2016, 540, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, T.B.; Sánchez-Guerrero, Á.; Milosevic, I.; Raimundo, N. Mitochondrial fission requires DRP1 but not dynamins. Nature 2019, 570, e34–e42. [Google Scholar] [CrossRef]
- Züchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J.; et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004, 36, 449–451. [Google Scholar] [CrossRef] [PubMed]
- Bernard-Marissal, N.; Van Hameren, G.; Juneja, M.; Pellegrino, C.; Louhivuori, L.; Bartesaghi, L.; Rochat, C.; El Mansour, O.; Médard, J.J.; Croisier, M.; et al. Altered interplay between endoplasmic reticulum and mitochondria in Charcot-Marie-Tooth type 2A neuropathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2328–2337. [Google Scholar] [CrossRef] [Green Version]
- Bach, D.; Naon, D.; Pich, S.; Soriano, F.X.; Vega, N.; Rieusset, J.; Laville, M.; Guillet, C.; Boirie, Y.; Wallberg-Henriksson, H.; et al. Expression of Mfn2, the Charcot-Marie-Tooth neuropathy type 2A gene, in human skeletal muscle: Effects of type 2 diabetes, obesity, weight loss, and the regulatory role of tumor necrosis factor α and interleukin-6. Diabetes 2005, 54, 2685–2693. [Google Scholar] [CrossRef] [Green Version]
- Sebastián, D.; Hernández-Alvarez, M.I.; Segalés, J.; Sorianello, E.; Muñoz, J.P.; Sala, D.; Waget, A.; Liesa, M.; Paz, J.C.; Gopalacharyulu, P.; et al. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 5523–5528. [Google Scholar] [CrossRef] [Green Version]
- Tubbs, E.; Theurey, P.; Vial, G.; Bendridi, N.; Bravard, A.; Chauvin, M.A.; Ji-Cao, J.; Zoulim, F.; Bartosch, B.; Ovize, M.; et al. Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 2014, 63, 3279–3294. [Google Scholar] [CrossRef] [Green Version]
- Böhm, J.; Chevessier, F.; De Paula, A.M.; Koch, C.; Attarian, S.; Feger, C.; Hantaï, D.; Laforêt, P.; Ghorab, K.; Vallat, J.M.; et al. Constitutive activation of the calcium sensor STIM1 causes tubular-aggregate myopathy. Am. J. Hum. Genet. 2013, 92, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Hedberg, C.; Niceta, M.; Fattori, F.; Lindvall, B.; Ciolfi, A.; D’Amico, A.; Tasca, G.; Petrini, S.; Tulinius, M.; Tartaglia, M.; et al. Childhood onset tubular aggregate myopathy associated with de novo STIM1 mutations. J. Neurol. 2014, 261, 870–876. [Google Scholar] [CrossRef]
- Morin, G.; Bruechle, N.O.; Singh, A.R.; Knopp, C.; Jedraszak, G.; Elbracht, M.; Brémond-Gignac, D.; Hartmann, K.; Sevestre, H.; Deutz, P.; et al. Gain-of-Function Mutation in STIM1 (P.R304W) is Associated with Stormorken Syndrome. Hum. Mutat. 2014, 35, 1221–1232. [Google Scholar] [CrossRef]
- Misceo, D.; Holmgren, A.; Louch, W.E.; Holme, P.A.; Mizobuchi, M.; Morales, R.J.; De Paula, A.M.; Stray-Pedersen, A.; Lyle, R.; Dalhus, B.; et al. A Dominant STIM1 Mutation Causes Stormorken Syndrome. Hum. Mutat. 2014, 35, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Nesin, V.; Wiley, G.; Kousi, M.; Ong, E.C.; Lehmann, T.; Nicholl, D.J.; Suri, M.; Shahrizaila, N.; Katsanis, N.; Gaffney, P.M.; et al. Activating mutations in STIM1 and ORAI1 cause overlapping syndromes of tubular myopathy and congenital miosis. Proc. Natl. Acad. Sci. USA 2014, 111, 4197–4202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luik, R.M.; Wu, M.M.; Buchanan, J.A.; Lewis, R.S. The elementary unit of store-operated Ca2+ entry: Local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J. Cell Biol. 2006, 174, 815–825. [Google Scholar] [CrossRef]
- Xu, P.; Lu, J.; Li, Z.; Yu, X.; Chen, L.; Xu, T. Aggregation of STIM1 underneath the plasma membrane induces clustering of Orai1. Biochem. Biophys. Res. Commun. 2006, 350, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.H.; Shaltouki, A.; Gonzalez, A.E.; Bettencourt da Cruz, A.; Burbulla, L.F.; St. Lawrence, E.; Schüle, B.; Krainc, D.; Palmer, T.D.; Wang, X. Functional Impairment in Miro Degradation and Mitophagy is a Shared Feature in Familial and Sporadic Parkinson’s Disease. Cell Stem Cell 2016, 19, 709–724. [Google Scholar] [CrossRef] [Green Version]
- Modi, S.; López-Doménech, G.; Halff, E.F.; Covill-Cooke, C.; Ivankovic, D.; Melandri, D.; Arancibia-Cárcamo, I.L.; Burden, J.J.; Lowe, A.R.; Kittler, J.T. Miro clusters regulate ER-mitochondria contact sites and link cristae organization to the mitochondrial transport machinery. Nat. Commun. 2019, 10, 17–19. [Google Scholar] [CrossRef] [Green Version]
- Berenguer-Escuder, C.; Grossmann, D.; Antony, P.; Arena, G.; Wasner, K.; Massart, F.; Jarazo, J.; Walter, J.; Schwamborn, J.C.; Grünewald, A.; et al. Impaired mitochondrial-endoplasmic reticulum interaction and mitophagy in Miro1-mutant neurons in Parkinson’s disease. Hum. Mol. Genet. 2020, 29, 1353–1364. [Google Scholar] [CrossRef]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Calì, T.; Ottolini, D.; Negro, A.; Brini, M. Enhanced parkin levels favor ER-mitochondria crosstalk and guarantee Ca2+ transfer to sustain cell bioenergetics. Biochim. Biophys. Acta-Mol. Basis Dis. 2013, 1832, 495–508. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; Squitieri, F.; Krieger, E.; Vanacore, N.; van Swieten, J.C.; Brice, A.; van Duijn, C.M.; Oostra, B.; Meco, G.; et al. DJ-1 (PARK7), a novel gene for autosomal recessive, early onset parkinsonism. Neurol. Sci. 2003, 24, 159–160. [Google Scholar] [CrossRef] [Green Version]
- Ottolini, D.; Calì, T.; Negro, A.; Brini, M. The Parkinson disease-related protein DJ-1 counteracts mitochondrial impairment induced by the tumour suppressor protein p53 by enhancing endoplasmic reticulum-mitochondria tethering. Hum. Mol. Genet. 2013, 22, 2152–2168. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [Green Version]
- Guardia-Laguarta, C.; Area-Gomez, E.; Rüb, C.; Liu, Y.; Magrané, J.; Becker, D.; Voos, W.; Schon, E.A.; Przedborski, S. α-synuclein is localized to mitochondria-associated ER membranes. J. Neurosci. 2014, 34, 249–259. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelmetti, V.; De Rosa, P.; Torosantucci, L.; Marini, E.S.; Romagnoli, A.; Di Rienzo, M.; Arena, G.; Vignone, D.; Fimia, G.M.; Valente, E.M. PINK1 and BECN1 relocalize at mitochondria-associated membranes during mitophagy and promote ER-mitochondria tethering and autophagosome formation. Autophagy 2017, 13, 654–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyofuku, T.; Okamoto, Y.; Ishikawa, T.; Sasawatari, S.; Kumanogoh, A. LRRK 2 regulates endoplasmic reticulum–mitochondrial tethering through the PERK-mediated ubiquitination pathway. EMBO J. 2020, 39, 1–19. [Google Scholar] [CrossRef]
- Scheuner, D.; Eckman, C.; Jensen, M.; Song, X.; Citron, M.; Suzuki, N.; Bird, T.D.; Hardy, J.; Hutton, M.; Kukull, W.; et al. Secreted amyloid β–protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat. Med. 1996, 2, 864–870. [Google Scholar] [CrossRef]
- Tu, H.; Nelson, O.; Bezprozvanny, A.; Wang, Z.; Lee, S.F.; Hao, Y.H.; Serneels, L.; De Strooper, B.; Yu, G.; Bezprozvanny, I. Presenilins Form ER Ca2+ Leak Channels, a Function Disrupted by Familial Alzheimer’s Disease-Linked Mutations. Cell 2006, 126, 981–993. [Google Scholar] [CrossRef] [Green Version]
- Ferreiro, E.; Oliveira, C.R.; Pereira, C.M.F. Involvement of endoplasmic reticulum Ca2+ release through ryanodine and inositol 1,4,5-triphosphate receptors in the neurotoxic effects induced by the amyloid-β peptide. J. Neurosci. Res. 2004, 76, 872–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasri, N.N.; Kocks, S.L.; Verbert, L.; Hébert, S.S.; Callewaert, G.; Parys, J.B.; Missiaen, L.; De Smedt, H. Up-regulation of inositol 1,4,5-trisphosphate receptor type 1 is responsible for a decreased endoplasmic-reticulum Ca2+ content in presenilin double knock-out cells. Cell Calcium 2006, 40, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Zatti, G.; Burgo, A.; Giacomello, M.; Barbiero, L.; Ghidoni, R.; Sinigaglia, G.; Florean, C.; Bagnoli, S.; Binetti, G.; Sorbi, S.; et al. Presenilin mutations linked to familial Alzheimer’s disease reduce endoplasmic reticulum and Golgi apparatus calcium levels. Cell Calcium 2006, 39, 539–550. [Google Scholar] [CrossRef]
- He, W.; Lu, Y.; Qahwash, I.; Hu, X.Y.; Chang, A.; Yan, R. Reticulon family members modulate BACE1 activity and amyloid-β peptide generation. Nat. Med. 2004, 10, 959–965. [Google Scholar] [CrossRef]
- Wakana, Y.; Koyama, S.; Nakajima, K.I.; Hatsuzawa, K.; Nagahama, M.; Tani, K.; Hauri, H.P.; Melançon, P.; Tagaya, M. Reticulon 3 is involved in membrane trafficking between the endoplasmic reticulum and Golgi. Biochem. Biophys. Res. Commun. 2005, 334, 1198–1205. [Google Scholar] [CrossRef]
- Shi, Q.; Prior, M.; He, W.; Tang, X.; Hu, X.; Yan, R. Reduced amyloid deposition in mice overexpressing RTN3 is adversely affected by preformed dystrophic neurites. J. Neurosci. 2009, 29, 9163–9173. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Shi, Q.; Zhou, X.; He, W.; Yi, H.; Yin, X.; Gearing, M.; Levey, A.; Yan, R. Transgenic mice overexpressing reticulon 3 develop neuritic abnormalities. EMBO J. 2007, 26, 2755–2767. [Google Scholar] [CrossRef] [Green Version]
- Waterham, H.R.; Koster, J.; van Roermund, C.W.T.; Mooyer, P.A.W.; Wanders, R.J.A.; Leonard, J.V. A Lethal Defect of Mitochondrial and Peroxisomal Fission. N. Engl. J. Med. 2007, 356, 1736–1741. [Google Scholar] [CrossRef]
- Abu-Safieh, L.; Alrashed, M.; Anazi, S.; Alkuraya, H.; Khan, A.O.; Al-Owain, M.; Al-Zahrani, J.; Al-Abdi, L.; Hashem, M.; Al-Tarimi, S.; et al. Autozygome-guided exome sequencing in retinal dystrophy patients reveals pathogenetic mutations and novel candidate disease genes. Genome Res. 2013, 23, 236–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, R.; Cheng, D.; Coyaud, É.; Freeman, S.; Di Pietro, E.; Wang, Y.; Vissa, A.; Yip, C.M.; Fairn, G.D.; Braverman, N.; et al. VAPs and ACBD5 tether peroxisomes to the ER for peroxisome maintenance and lipid homeostasis. J. Cell Biol. 2017, 216, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Ozeki, S.; Cheng, J.; Tauchi-Sato, K.; Hatano, N.; Taniguchi, H.; Fujimoto, T. Rab18 localizes to lipid droplets and induces their close apposition to the endoplasmic reticulum-derived membrane. J. Cell Sci. 2005, 118, 2601–2611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takáts, S.; Lévay, L.; Boda, A.; Tóth, S.; Simon-Vecsei, Z.; Rubics, A.; Varga, Á.; Lippai, M.; Lőrincz, P.; Glatz, G.; et al. The Warburg Micro Syndrome-associated Rab3GAP-Rab18 module promotes autolysosome maturation through the Vps34 Complex I. FEBS J. 2021, 288, 190–211. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Giorgi, C.; Oparka, M.; Duszynski, J.; Wieckowski, M.R.; Pinton, P. Oncogenic and oncosuppressive signal transduction at mitochondria-associated endoplasmic reticulum membranes. Mol. Cell. Oncol. 2014, 1. [Google Scholar] [CrossRef] [Green Version]
- Fischer, D.; Schabhüttl, M.; Wieland, T.; Windhager, R.; Strom, T.M.; Auer-Grumbach, M. A novel missense mutation confirms ATL3 as a gene for hereditary sensory neuropathy type 1. Brain 2014, 137, 91–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornak, U.; Mademan, I.; Schinke, M.; Voigt, M.; Krawitz, P.; Hecht, J.; Barvencik, F.; Schinke, T.; Gießelmann, S.; Beil, F.T.; et al. Sensory neuropathy with bone destruction due to a mutation in the membrane-shaping atlastin GTPase 3. Brain 2014, 137, 683–692. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Wu, F.; Sun, S.; Yu, W.; Hu, J. Human atlastin GTPases mediate differentiated fusion of endoplasmic reticulum membranes. Protein Cell 2015, 6, 307–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krols, M.; Detry, S.; Asselbergh, B.; Almeida-Souza, L.; Kremer, A.; Lippens, S.; De Rycke, R.; De Winter, V.; Müller, F.J.; Kurth, I.; et al. Sensory-Neuropathy-Causing Mutations in ATL3 Cause Aberrant ER Membrane Tethering. Cell Rep. 2018, 23, 2026–2038. [Google Scholar] [CrossRef]
- Pulst, S.; Nechiporuk, A.; Nechiporuk, T.; Gispert, S.; Chen, X.; Lopes-Cendes, I.; Pearlman, S.; Starkman, S.; Orozco-Diaz, G.; Lunkes, A.; et al. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat. Genet. 1996, 14, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Steiner, B.; Swart, A.L.; Welin, A.; Weber, S.; Personnic, N.; Kaech, A.; Freyre, C.; Ziegler, U.; Klemm, R.W.; Hilbi, H. ER remodeling by the large GTPase atlastin promotes vacuolar growth of Legionella pneumophila. EMBO Rep. 2017, 18, 1817–1836. [Google Scholar] [CrossRef]
- Haenssler, E.; Ramabhadran, V.; Murphy, C.S.; Heidtman, M.I.; Isberg, R.R. Endoplasmic reticulum tubule protein reticulon 4 associates with the Legionella pneumophila vacuole and with translocated substrate Ceg9. Infect. Immun. 2015, 83, 3479–3489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanhope, R.; Flora, E.; Bayne, C.; Derré, I. IncV, a FFAT motif-containing Chlamydia protein, tethers the endoplasmic reticulum to the pathogen-containing vacuole. Proc. Natl. Acad. Sci. USA 2017, 114, 12039–12044. [Google Scholar] [CrossRef] [Green Version]
- Derré, I.; Swiss, R.; Agaisse, H. The lipid transfer protein CERT interacts with the Chlamydia inclusion protein IncD and participates to ER-Chlamydia inclusion membrane contact sites. PLoS Pathog. 2011, 7, e1002092. [Google Scholar] [CrossRef] [PubMed]
- Agaisse, H.; Derré, I. STIM1 is a novel component of ER-Chlamydia trachomatis inclusion membrane contact sites. PLoS ONE 2015, 10, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz, A.; Wang, X.; Ahlquist, P. Membrane-shaping host reticulon proteins play crucial roles in viral RNA replication compartment formation and function. Proc. Natl. Acad. Sci. USA 2010, 107, 16291–16296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.F.; Yang, S.Y.; Wu, B.W.; Jheng, J.R.; Chen, Y.L.; Shih, C.H.; Lin, K.H.; Lai, H.C.; Tang, P.; Horng, J.T. Reticulon 3 binds the 2C protein of enterovirus 71 and is required for viral replication. J. Biol. Chem. 2007, 282, 5888–5898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neufeldt, C.J.; Cortese, M.; Scaturro, P.; Cerikan, B.; Wideman, J.G.; Tabata, K.; Moraes, T.; Oleksiuk, O.; Pichlmair, A.; Bartenschlager, R. ER-shaping atlastin proteins act as central hubs to promote flavivirus replication and virion assembly. Nat. Microbiol. 2019, 4, 2416–2429. [Google Scholar] [CrossRef] [PubMed]
- Mookherjee, D.; Majumder, P.; Mukherjee, R.; Chatterjee, D.; Kaul, Z.; Das, S.; Sougrat, R.; Chakrabarti, S.; Chakrabarti, O. Cytosolic aggregates in presence of non-translocated proteins perturb endoplasmic reticulum structure and dynamics. Traffic 2019, 20, 943–960. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Li, Y.; Wu, L.; Li, Y.; Zhao, D.; Yu, J.; Huang, T.; Ferguson, C.; Parton, R.G.; Yang, H.; et al. Rab18 promotes lipid droplet (LD) growth by tethering the ER to LDs through SNARE and NRZ interactions. J. Cell Biol. 2018, 217, 975–995. [Google Scholar] [CrossRef] [Green Version]
- Khachaturian, Z.S. Calcium hypothesis of Alzheimer’s disease and brain aging. Ann. N. Y. Acad. Sci. 1994, 747, 1–11. [Google Scholar] [CrossRef]
- Campbell, P.D.; Shen, K.; Sapio, M.R.; Glenn, T.D.; Talbot, W.S.; Marlow, F.L. Unique function of Kinesin Kif5A in localization of mitochondria in axons. J. Neurosci. 2014, 34, 14717–14732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etcheberrigaray, R.; Hirashima, N.; Nee, L.; Prince, J.; Govoni, S.; Racchi, M.; Tanzi, R.E.; Alkon, D.L. Calcium responses in fibroblasts from asymptomatic members of Alzheimer’s disease families. Neurobiol. Dis. 1998, 5, 37–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero-Brey, I.; Bartenschlager, R. Endoplasmic reticulum: The favorite intracellular niche for viral replication and assembly. Viruses 2016, 8, 160. [Google Scholar] [CrossRef] [Green Version]
- Martinez, E.; Siadous, F.A.; Bonazzi, M. Tiny architects: Biogenesis of intracellular replicative niches by bacterial pathogens. FEMS Microbiol. Rev. 2018, 42, 425–447. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.J.; Ke, P.Y.; Hsu, J.T.A.; Yeh, C.T.; Horng, J.T. Reticulon 3 interacts with NS4B of the hepatitis C virus and negatively regulates viral replication by disrupting NS4B self-interaction. Cell. Microbiol. 2014, 16, 1603–1618. [Google Scholar] [CrossRef]
- Gouttenoire, J.; Roingeard, P.; Penin, F.; Moradpour, D. Amphipathic α-Helix AH2 is a Major Determinant for the Oligomerization of Hepatitis C Virus Nonstructural Protein 4B. J. Virol. 2010, 84, 12529–12537. [Google Scholar] [CrossRef] [Green Version]
- Geiger, R.; Andritschke, D.; Friebe, S.; Herzog, F.; Luisoni, S.; Heger, T.; Helenius, A. BAP31 and BiP are essential for dislocation of SV40 from the endoplasmic reticulum to the cytosol. Nat. Cell Biol. 2011, 13, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
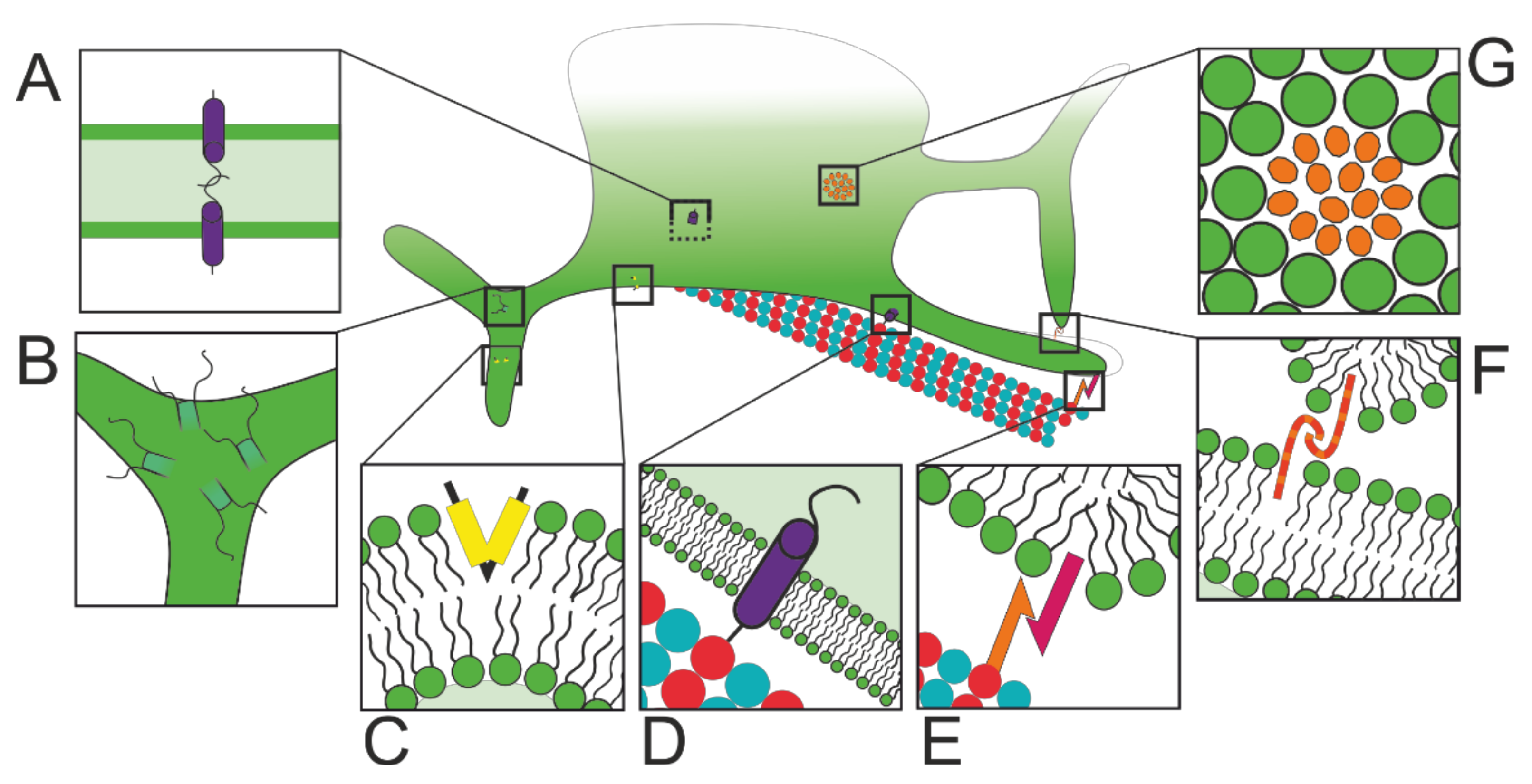
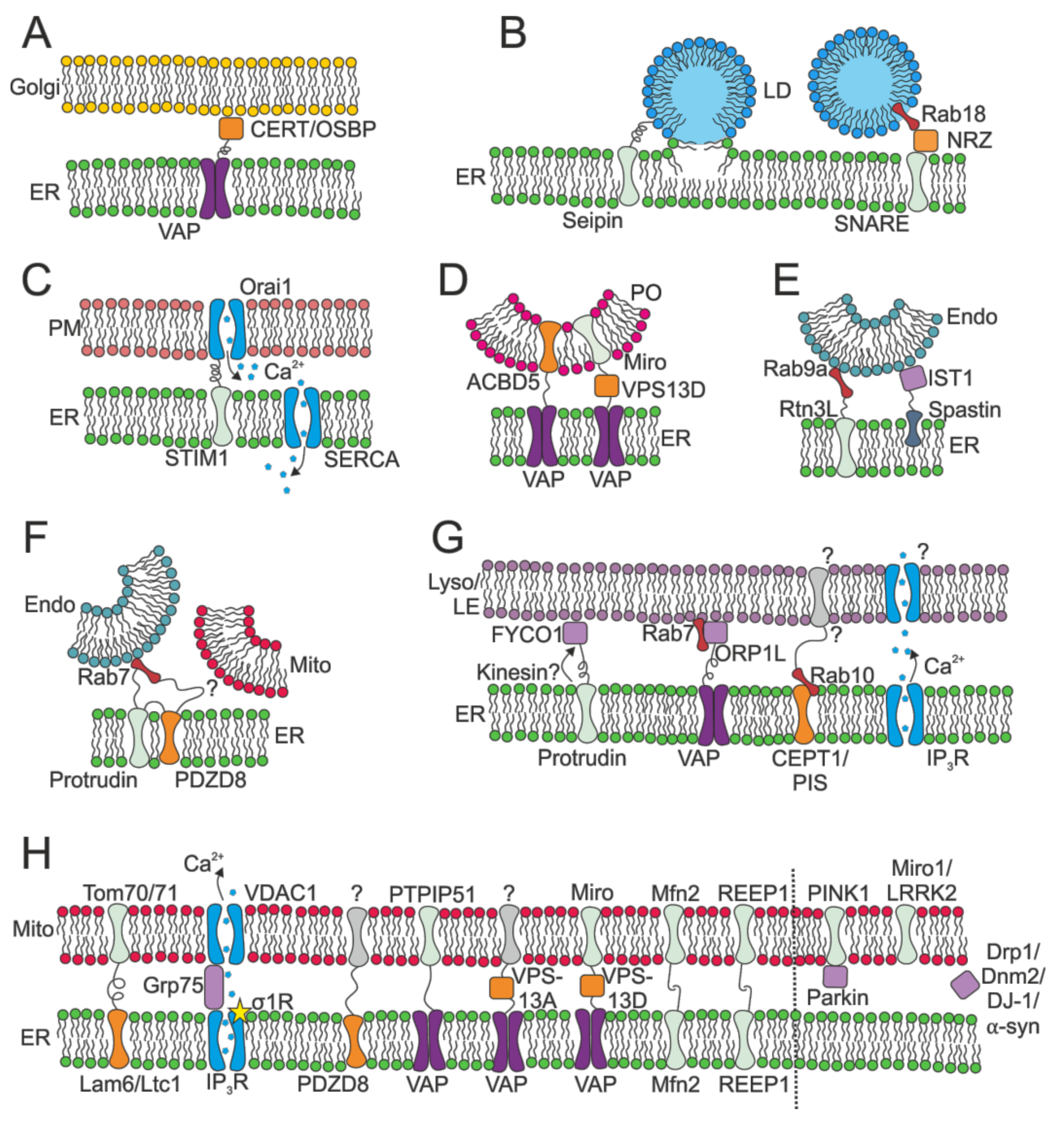

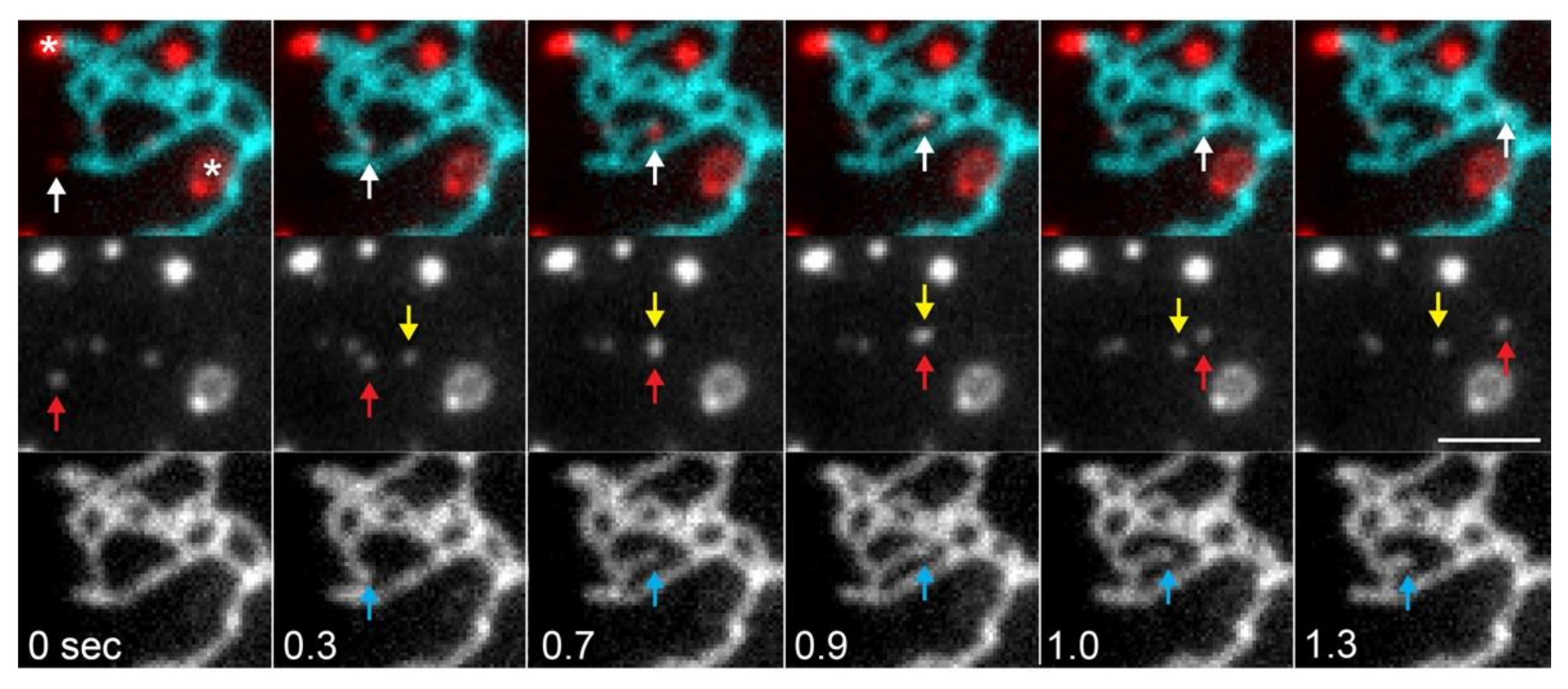

{kind=link}
{kind=link}
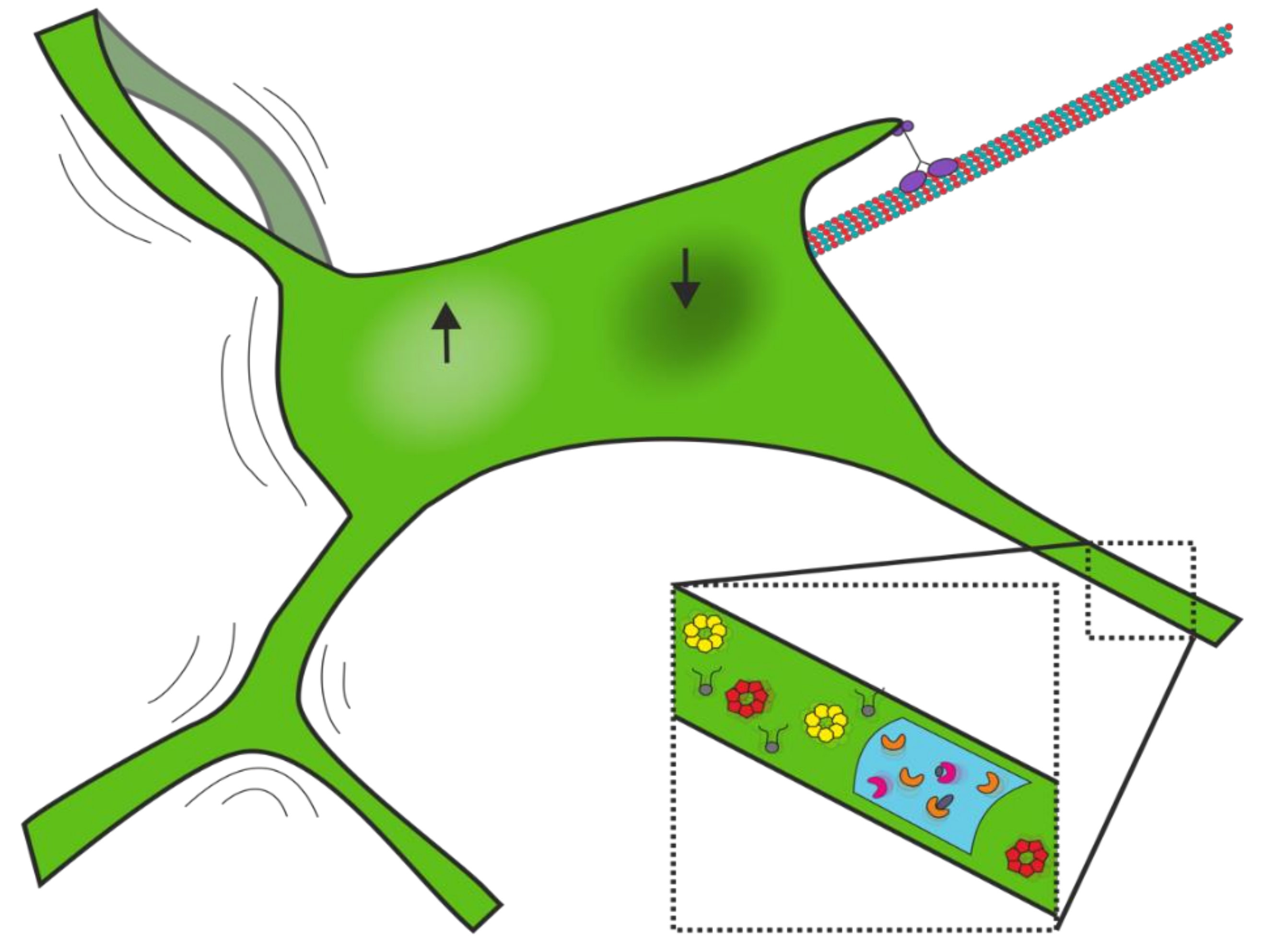{kind=link}
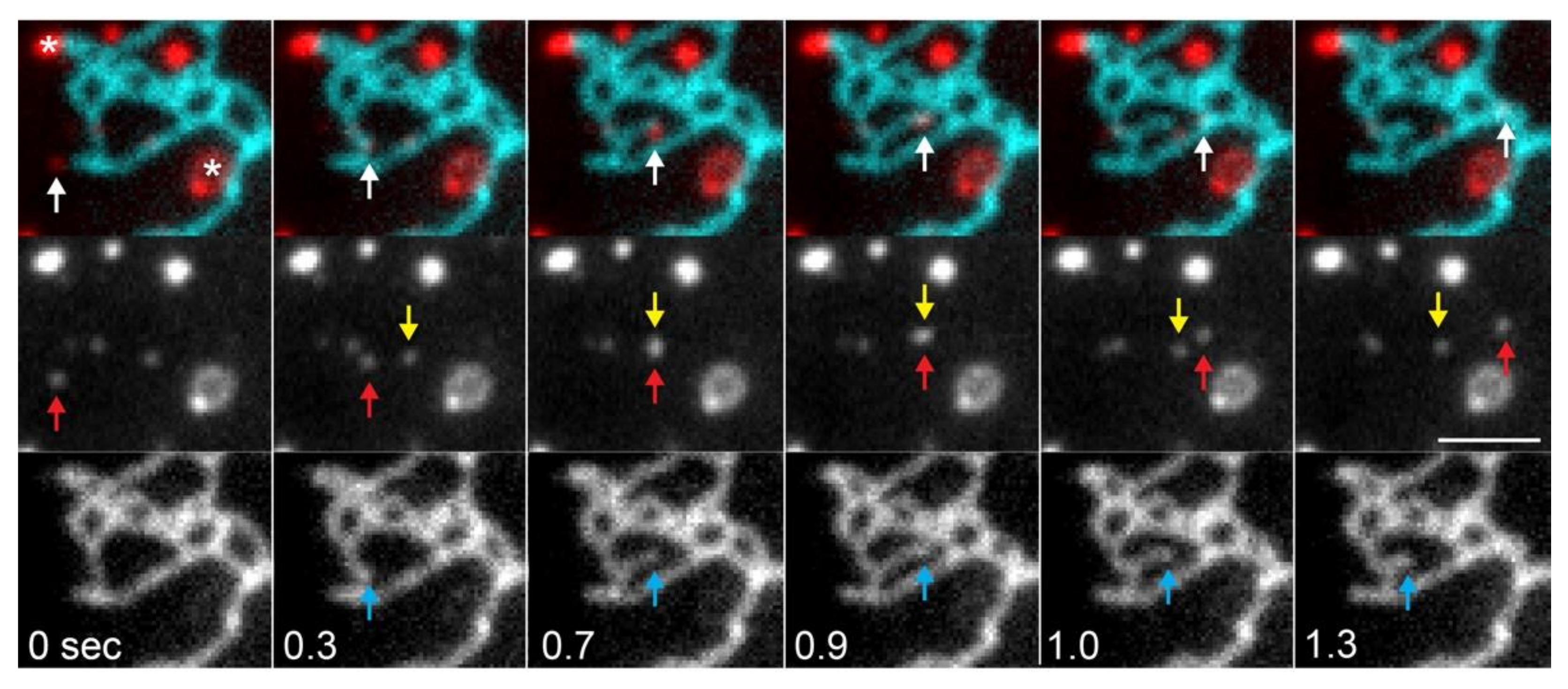{kind=link}
| Disease | Protein Implicated | Protein Role in Healthy ER | Function Affected in Disease |
|---|---|---|---|
| Hereditary Spastic Paraplegias | Spastin [279] | ER tubule regulation [17], ER-endosome MCS [177] | Endosomal fission fails at MCS [177] |
| Protrudin [280] | ER membrane curvature and tubule fission [281], ER-endosome MCS [204], ER MCS-dependent endosome maturation [125] | ER morphology and function dysregulated [225,281] | |
| Seipin [282] | ER-lipid droplet MCS [283] | Axon regeneration impaired [284] | |
| Atlastin-1 [285] | ER membrane fusion [5] | ER morphology dysregulated [17] | |
| REEP1 [286] | ER membrane curvature [2], ER-mitochondria MCS [106] | ER-mitochondria interactions disrupted [106] | |
| REEP2 [287] | ER membrane curvature regulation [2] | ER morphology disrupted [288] | |
| KIF5A [195] | ER dynamics via kinesin-1 [195] | Impaired axonal transport [195] | |
| Amyotrophic Lateral Sclerosis | VAPB [289] | MCS between ER and many organelles (reviewed [290,291]) | ER morphology dysregulated [292,293,294,295], ER-mitochondria MCS [296,297],interactions between VAPB and oxysterol binding protein (OSBP) perturbed [298] |
| Reticulon 4 [299] | ER membrane curvature [2] | Chaperone protein disulfide isomerase (PDI) distribution altered [299] | |
| Sigma-1 Receptor [300] | ER-mitochondria MCS [301], ER Ca2+ homeostasis [301] | Disrupts ER-mitochondria MCS [302] | |
| KIF5A [195] | ER dynamics via kinesin-1 [195] | Impaired axonal transport [195] | |
| Charcot Marie Tooth | Dynamin 2 [303] | Possibly involved in mitochondrial fission at ER-mitochondria MCS [304,305] | Unknown |
| Mitofusin 2 [306] | ER-mitochondria MCS [105] | Reduction in ER-mitochondria MCS [307] | |
| KIF5A [195] | ER dynamics via kinesin-1 [195] | Impaired axonal transport [195] | |
| Diabetes | Mitofusin 2 [308] | ER-mitochondria MCS [105] | Insulin signaling impaired [309] |
| VDAC-1/grp75/IP3R-1 [310] | ER-mitochondria MCS [165], ER Ca2+ homeostasis [165] | Diminished ER-mitochondria interaction impairs insulin signaling [310] | |
| Tubular Aggregate Myopathy/Stormorken Syndrome | STIM1 [311,312,313] | SOCE [168], ER-plasma membrane MCS [170], TACs [21] | Ca2+ homeostasis dysregulated [311,313,314] |
| Orai1 [315] | SOCE and ER-plasma membrane MCS [316,317] | Ca2+ homeostasis dysregulated [315] | |
| Parkinson’s Disease | Miro1 [318] | ER-mitochondria MCS [319] | Altered ER-mitochondria Ca2+ transfer [320] |
| Parkin [321] | ER-mitochondria MCS [322] | Altered ER-mitochondria Ca2+ transfer [322] | |
| DJ-1 [323] | ER-mitochondria MCS [324] | Disrupts ER-mitochondria MCS and Ca2+ transfer [324] | |
| α-synuclein [325] | ER-mitochondria MCS [326] | Fewer ER-mitochondria MCS [326] | |
| PINK1 [327] | ER-mitochondria MCS [328] | Altered ER-mitochondria Ca2+ transfer [328] | |
| LRRK2 [329] | ER-mitochondria MCS [330] | Disrupts ER-mitochondria MCS [330] | |
| Alzheimer’s Disease | Presenilins [331] | ER Ca2+ homeostasis [332] | Ca2+ homeostasis dysregulated [333,334,335] |
| Reticulon 3 [336] | ER morphology regulation, Golgi to ER trafficking [337] | Amyloid plaque formation altered [336,338], cognitive dysfunction due to Rtn3 aggregates in AD brains [339] | |
| Encephalopathy | Dynamin-related protein 1 [340] | ER-mitochondria MCS [73] | Defective mitochondrial fission [340] |
| Retinal Dystrophy | ACBD5 [341] | ER-peroxisome MCS [136,342] | Unknown |
| Warburg Micro Syndrome | Rab18 [29] | ER-lipid droplet MCS [343], ER morphology and dynamics [8,198] | Perturbed autophagy [344] |
| Cancer | Various ER-resident and ER-mitochondria MCS proteins (reviewed [345]) | ER-mitochondria MCS and Ca2+ dynamics implicated | Various |
| Hereditary Sensory and Autonomic Neuropathy | Atlastin-3 [346,347] | ER tubule fusion [348] | Aberrant bundling of ER tubules [349] |
| Spinocerebellar Ataxia Type 2 | Ataxin-2 [350] | ER morphology and dynamics [28] | ER morphology collapse, ER dynamics impaired [28] |
| Legionnaires’ Disease | Atlastin-3 [351] | ER tubule fusion [348] | Atlastin-3-mediated ER remodeling promotes bacterial replication [351] |
| Reticulon 4 [352] | ER membrane curvature [2] | Connects ER to pathogen by binding Ceg9 [352] to promote bacterial replication [351] | |
| Chlamydia | VAPA & VAPB [353] | MCS between ER and many organelles (reviewed [84]) | Chlamydia protein IncV binds VAP to form ER-bacterial inclusion MCS [353] |
| CERT [354] | ER-Golgi ceramide transfer [117] | Chlamydia protein IncD binds to CERT to form ER-bacterial inclusion MCS [354] | |
| STIM1 [355] | SOCE [168], ER-plasma membrane MCS [170], TACs [21] | STIM1 localises to ER-bacterial inclusion MCS [355] | |
| Brome mosaic virus | Reticulons [356] | ER membrane curvature [2] | Reticulons bind viral protein 1a to promote viral replication [356] |
| Enterovirus 71 | Reticulon 3 [357] | ER morphology regulation, Golgi to ER trafficking [337] | Enterovirus protein 2C binds Rtn3 to promote viral replication [357] |
| Flaviviruses (Dengue, Zika, West Nile) | Atlastin-2 and -3 [358] | ER tubule fusion [348] | Atlastins promote viral replication via distinct mechanisms [358] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perkins, H.T.; Allan, V. Intertwined and Finely Balanced: Endoplasmic Reticulum Morphology, Dynamics, Function, and Diseases. Cells 2021, 10, 2341. https://doi.org/10.3390/cells10092341
Perkins HT, Allan V. Intertwined and Finely Balanced: Endoplasmic Reticulum Morphology, Dynamics, Function, and Diseases. Cells. 2021; 10(9):2341. https://doi.org/10.3390/cells10092341
Chicago/Turabian StylePerkins, Hannah T., and Viki Allan. 2021. "Intertwined and Finely Balanced: Endoplasmic Reticulum Morphology, Dynamics, Function, and Diseases" Cells 10, no. 9: 2341. https://doi.org/10.3390/cells10092341
APA StylePerkins, H. T., & Allan, V. (2021). Intertwined and Finely Balanced: Endoplasmic Reticulum Morphology, Dynamics, Function, and Diseases. Cells, 10(9), 2341. https://doi.org/10.3390/cells10092341