
Comprehensive Characterization of Multitissue Expression Landscape, Co-Expression Networks and Positive Selection in Pikeperch

 , , , ,
, , , ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Tissue Sampling, Library Preparation, RNA-Sequencing
2.2. De Novo Transcriptome Assembly, Functional Annotation
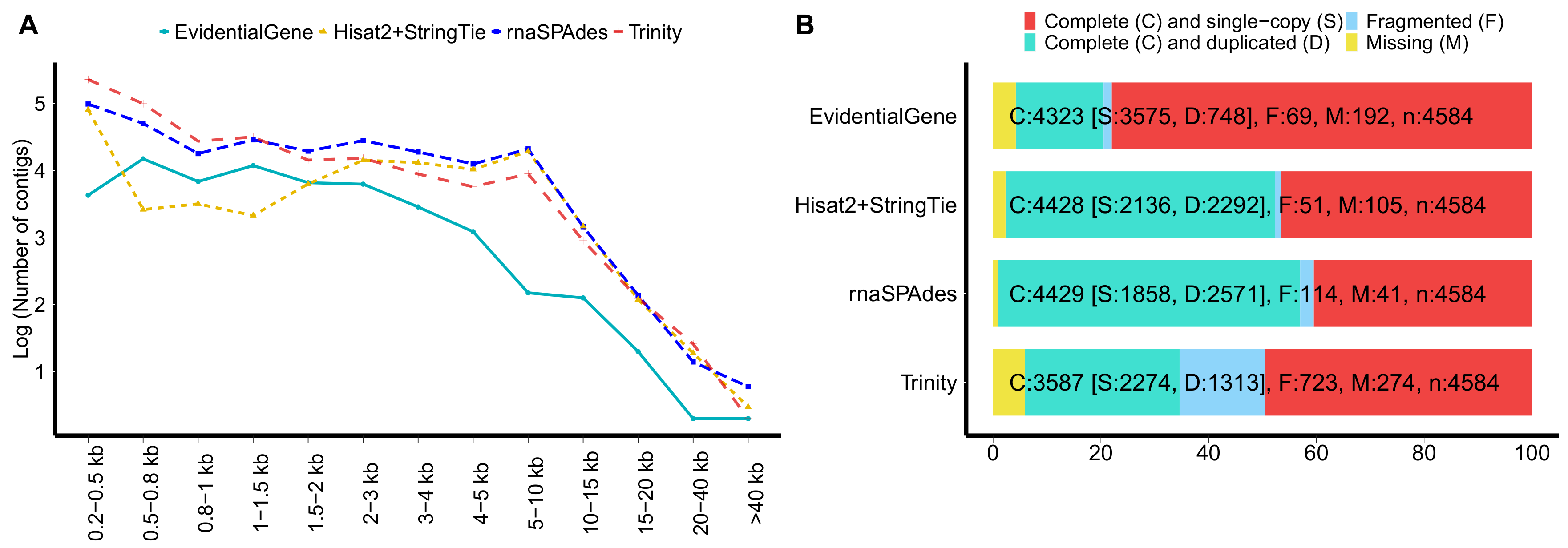
2.3. Quality Assessment of the Multitissues Transcriptome Assembly
2.4. Quantification of Tissues Expression Profiles
2.5. Tissue Specificity Index, Differential Expression Analysis
2.6. Tissue-Specific Co-Expression and Network Modules Analysis
2.7. Positive Selection Analysis
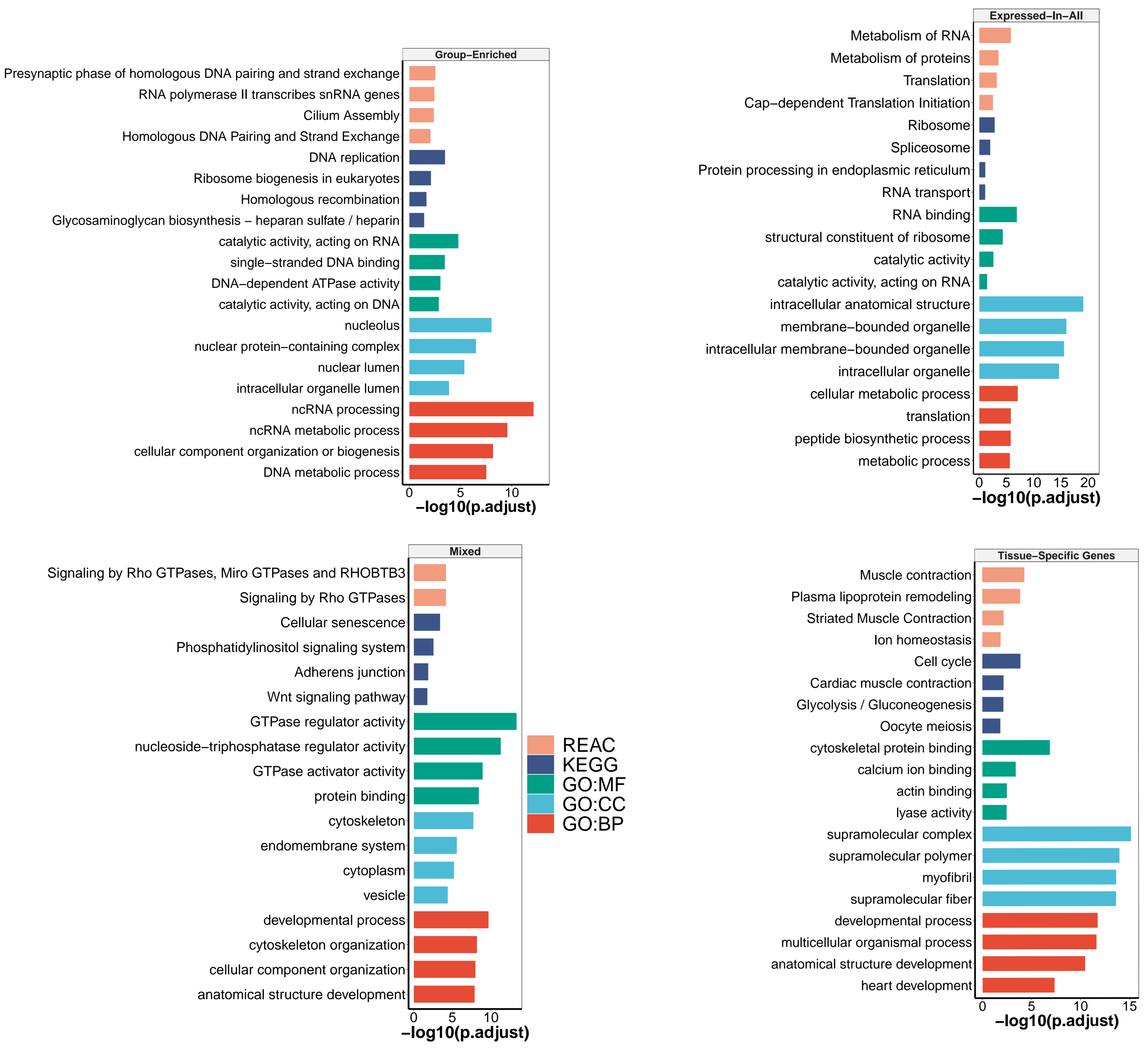
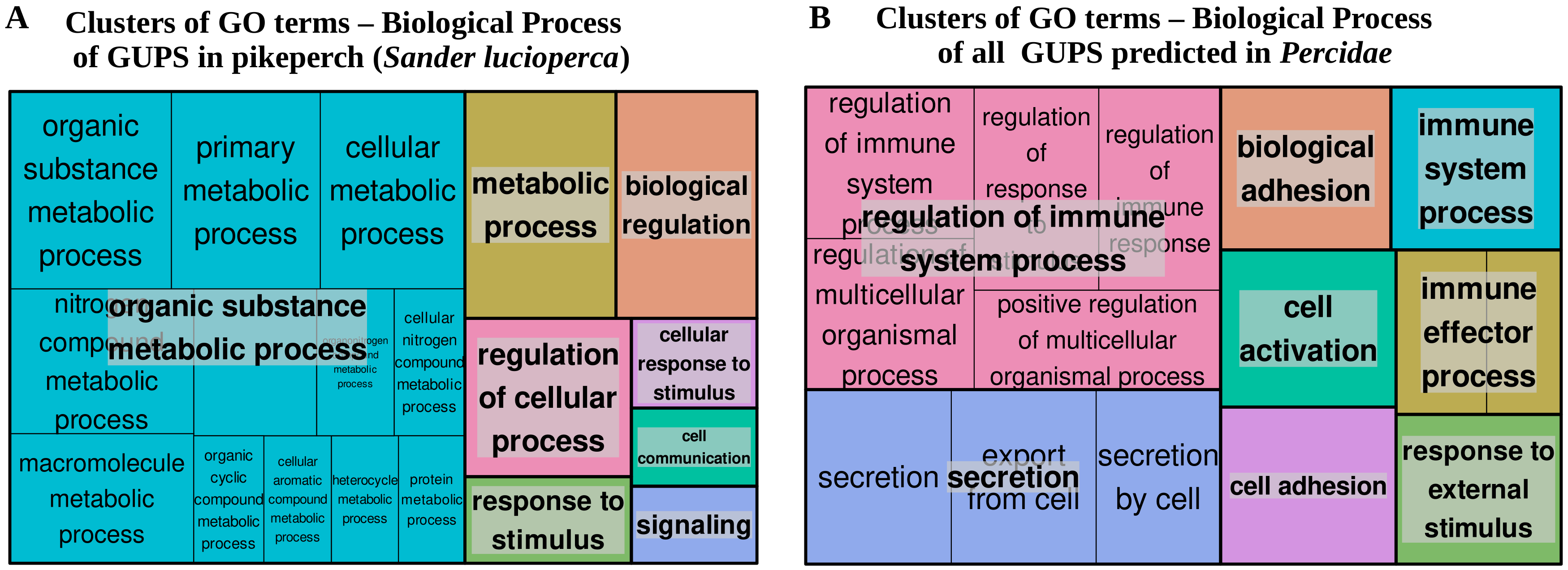
2.8. Functional Enrichment Analyses
3. Results
3.1. RNA-Seq, Assembly and Functional Annotation
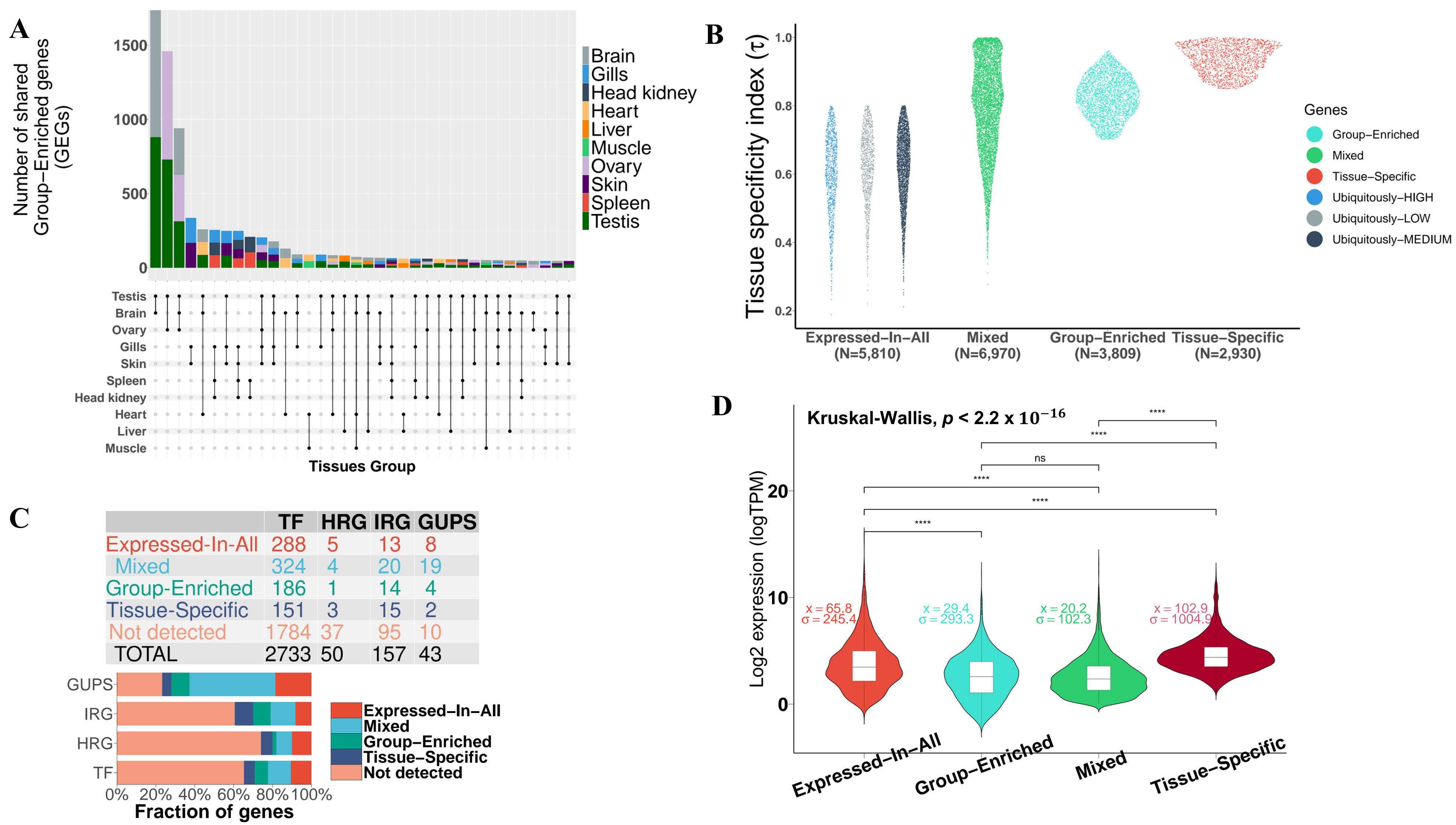
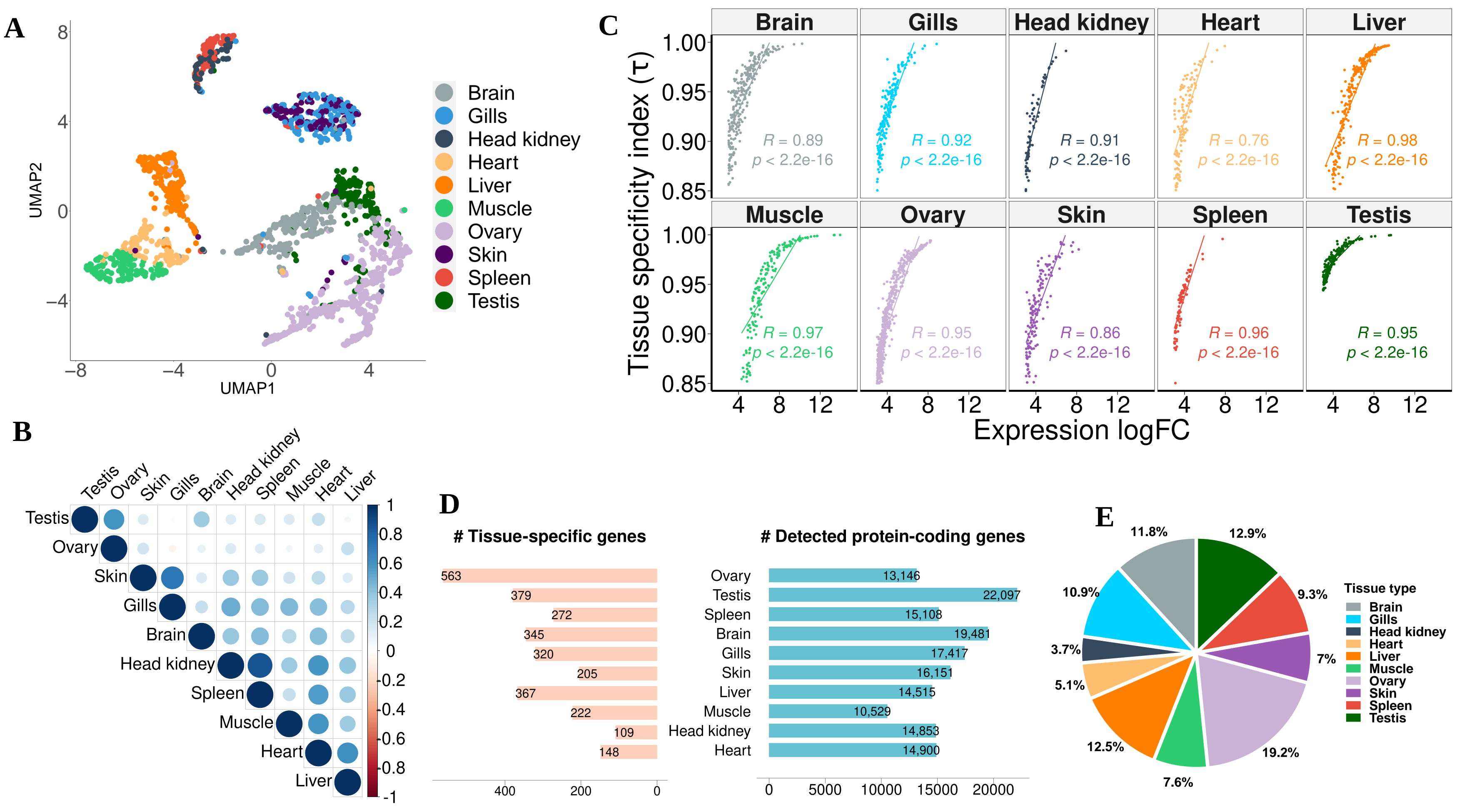
3.2. Expression Atlas of Pikeperch Protein-Coding Genes
3.2.1. Mixed-Expressed Genes
3.2.2. Expressed-in-All Genes
3.2.3. Group-Enriched Genes
3.2.4. Tissue-Specific Genes
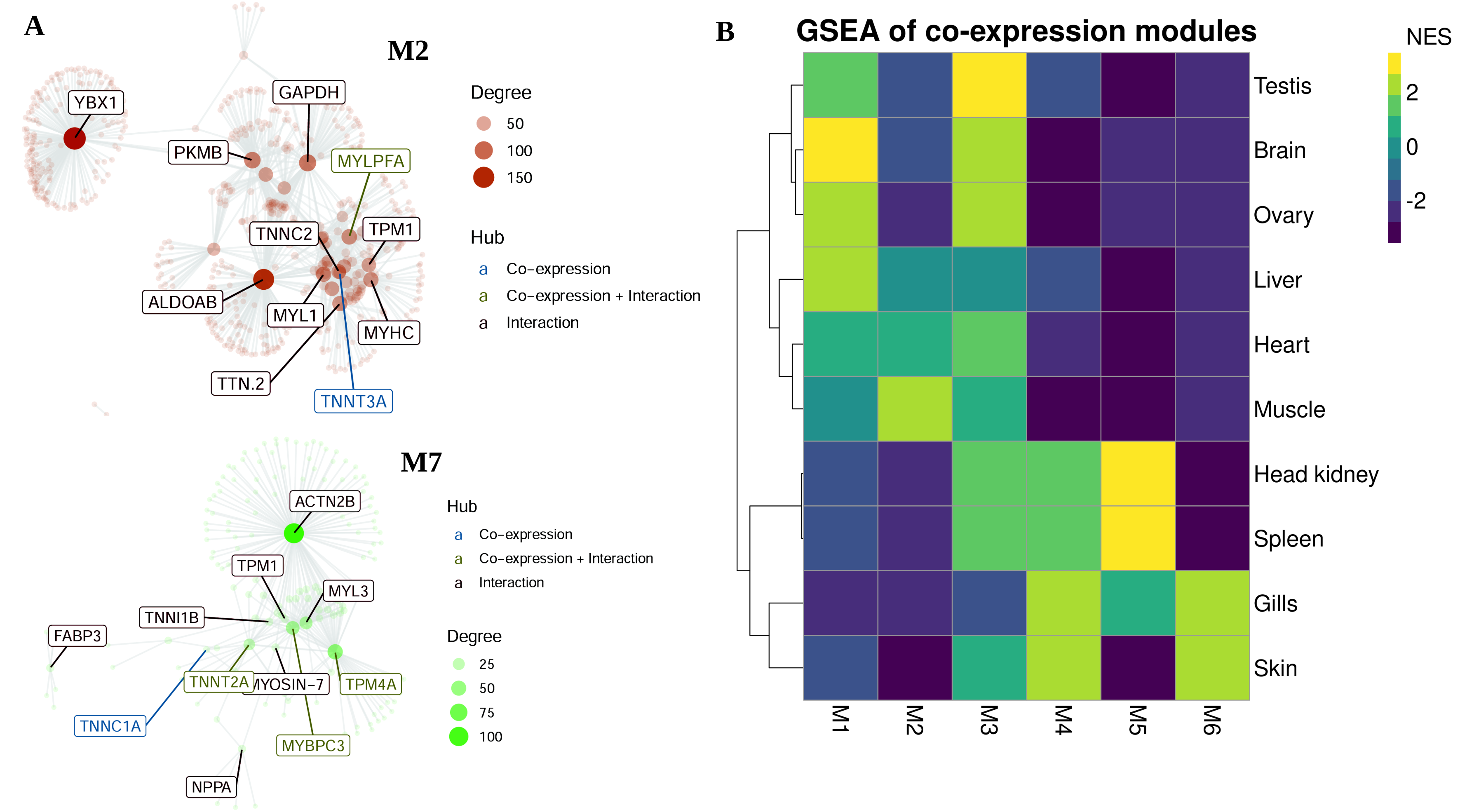
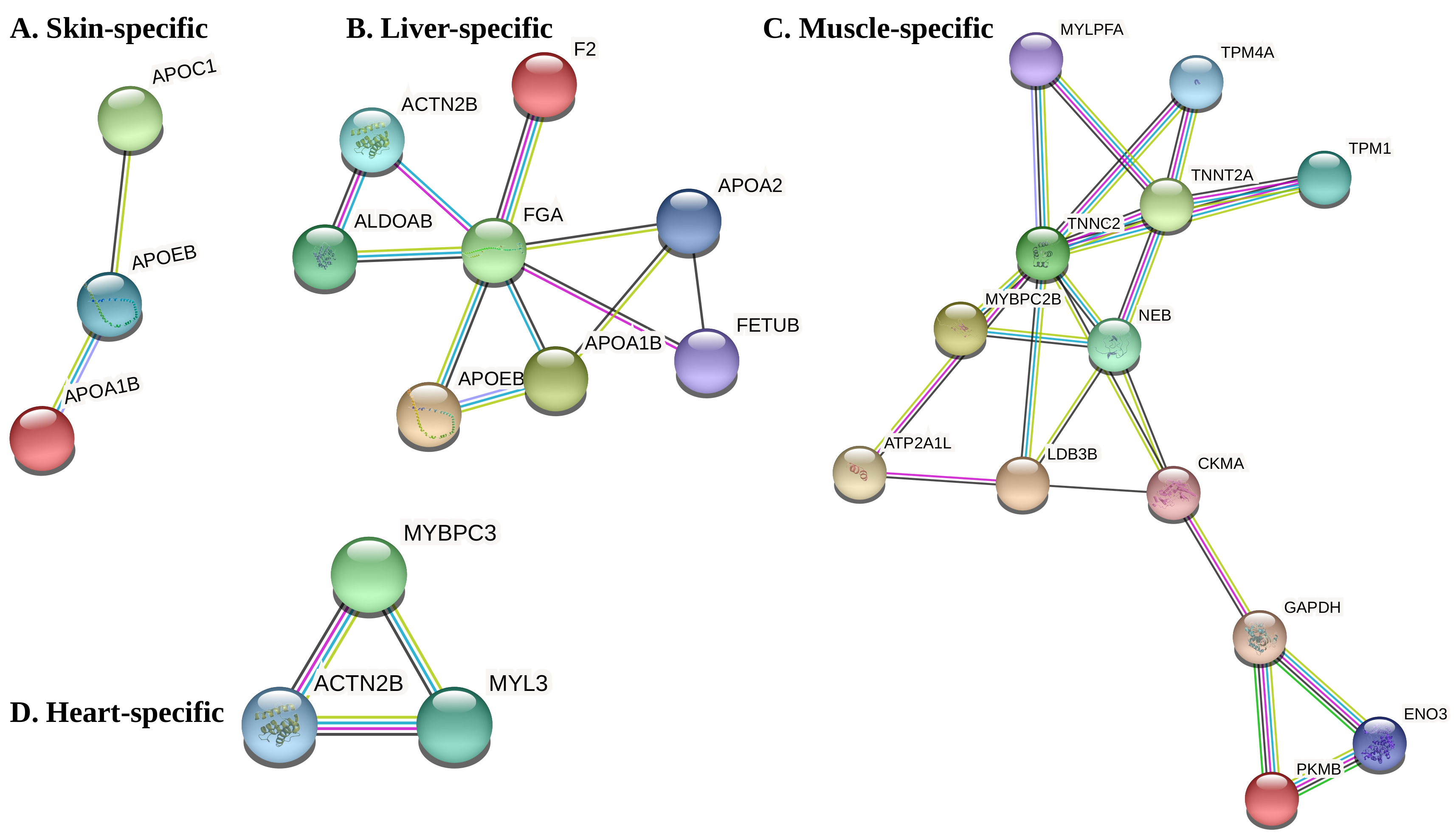
3.3. Co-Expression Modules, Hubs and Tissue-Specific Networks
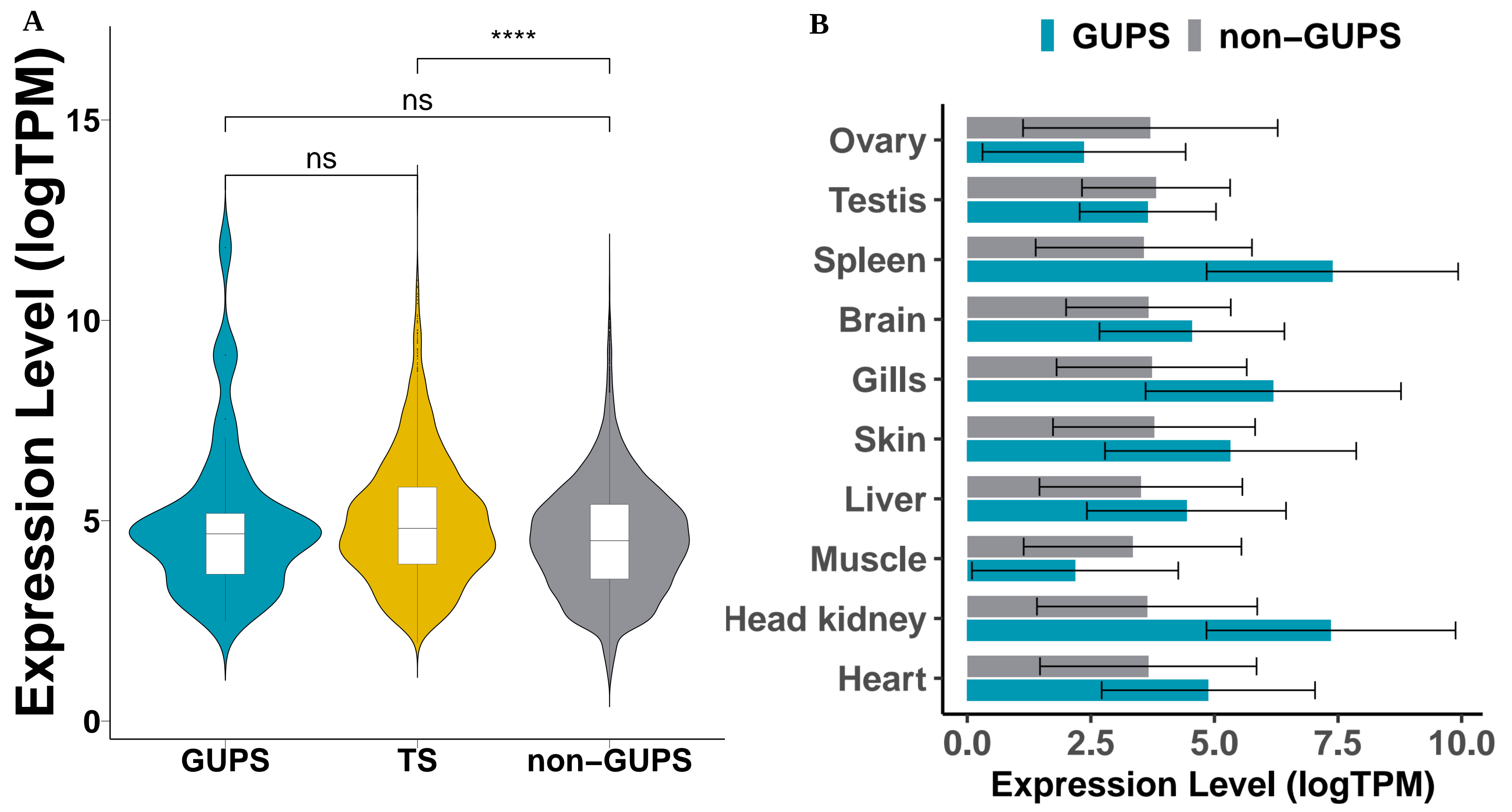
3.4. Positive Selection Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mylonas, C.; Robles, R. DIVERSIFY-Exploring the Biological and Socioeconomic Potential of New/Emerging Fish Species for the Expansion of the European Aquaculture Industry 2017. Aquaculture Europe. 2017. Available online: https://www.diversifyfish.eu/uploads/1/4/2/0/14206280/mylonas_diversifyoct1810.30.pdf (accessed on 30 July 2021).
- Rufchaei, R.; Nedaei, S.; Hoseinifar, S.H.; Hassanpour, S.; Golshan, M.; Bourani, M.S. Improved growth performance, serum and mucosal immunity, haematology and antioxidant capacity in pikeperch (Sander lucioperca) using dietary water hyacinth (Eichhornia crassipes) leaf powder. Aquac. Res. 2020, 52, 2194–2204. [Google Scholar] [CrossRef]
- Alexi, N.; Byrne, D.V.; Nanou, E.; Grigorakis, K. Investigation of sensory profiles and hedonic drivers of emerging aquaculture fish species. J. Sci. Food Agric. 2018, 98, 1179–1187. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Xu, X.; Kestemont, P. Effect of temperature and feeding frequency on growth performances, feed efficiency and body composition of pikeperch juveniles (Sander lucioperca). Aquaculture 2009, 289, 70–73. [Google Scholar] [CrossRef]
- Bercsényi, M.; Urbányi, B.; Bódis, M.; Müller, T. Comparison of Growth in Pike-Perch (Sander lucioperca) and Hybrids of Pike-Perch (S. lucioperca) × Volga Pike-Perch (S. volgensis). Isr. J. Aquac.-Bamidgeh 2011, 63. [Google Scholar] [CrossRef]
- Kestemont, P.; Xueliang, X.; Hamza, N.; Maboudou, J.; Toko, I.I. Effect of weaning age and diet on pikeperch larviculture. Aquaculture 2007, 264, 197–204. [Google Scholar] [CrossRef]
- Baekelandt, S.; Redivo, B.; Mandiki, S.N.; Bournonville, T.; Houndji, A.; Bernard, B.; Kertaoui, N.E.; Schmitz, M.; Fontaine, P.; Gardeur, J.N.; et al. Multifactorial analyses revealed optimal aquaculture modalities improving husbandry fitness without clear effect on stress and immune status of pikeperch Sander lucioperca. Gen. Comp. Endocrinol. 2018, 258, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Szkudlarek, M.; Zakęś, Z. Effect of stocking density on survival and growth performance of pikeperch, Sander lucioperca (L.), larvae under controlled conditions. Aquac. Int. 2007, 15, 67–81. [Google Scholar] [CrossRef]
- Policar, T.; Blecha, M.; Křišťan, J.; Mráz, J.; Velíšek, J.; Stará, A.; Stejskal, V.; Malinovskyi, O.; Svačina, P.; Samarin, A.M. Comparison of production efficiency and quality of differently cultured pikeperch (Sander lucioperca L.) juveniles as a valuable product for ongrowing culture. Aquac. Int. 2016, 24, 1607–1626. [Google Scholar] [CrossRef]
- Żarski, D.; Le Cam, A.; Nynca, J.; Klopp, C.; Ciesielski, S.; Sarosiek, B.; Montfort, J.; Król, J.; Fontaine, P.; Ciereszko, A.; et al. Domestication modulates the expression of genes involved in neurogenesis in high-quality eggs of Sander lucioperca. Mol. Reprod. Dev. 2020, 87, 934–951. [Google Scholar] [CrossRef]
- Han, X.; Ling, Q.; Li, C.; Wang, G.; Xu, Z.; Lu, G. Characterization of pikeperch (Sander lucioperca) transcriptome and development of SSR markers. Biochem. Syst. Ecol. 2016, 66, 188–195. [Google Scholar] [CrossRef]
- Guo, J.; Li, C.; Teng, T.; Shen, F.; Chen, Y.; Wang, Y.; Pan, C.; Ling, Q. Construction of the first high-density genetic linkage map of pikeperch (Sander lucioperca) using specific length amplified fragment (SLAF) sequencing and QTL analysis of growth-related traits. Aquaculture 2018, 497, 299–305. [Google Scholar] [CrossRef]
- de los Ríos-Pérez, L.; Nguinkal, J.A.; Verleih, M.; Rebl, A.; Brunner, R.M.; Klosa, J.; Schäfer, N.; Stüeken, M.; Goldammer, T.; Wittenburg, D. An ultra-high density SNP-based linkage map for enhancing the pikeperch (Sander lucioperca) genome assembly to chromosome-scale. Sci. Rep. 2020, 10, 22335. [Google Scholar] [CrossRef] [PubMed]
- Nguinkal, J.A.; Brunner, R.M.; Verleih, M.; Rebl, A.; de los Rios-Perez, L.; Schafer, N.; Hadlich, F.; Stueken, M.; Wittenburg, D.; Goldammer, T. The First Highly Contiguous Genome Assembly of Pikeperch (Sander lucioperca), an Emerging Aquaculture Species in Europe. Genes 2019, 10, 708. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yu, X.; Zack, D.J.; Zhu, H.; Qian, J. TiGER: A database for tissue-specific gene expression and regulation. BMC Bioinform. 2008, 9, 271. [Google Scholar] [CrossRef] [PubMed]
- Chikina, M.D.; Huttenhower, C.; Murphy, C.T.; Troyanskaya, O.G. Global prediction of tissue-specific gene expression and context-dependent gene networks in Caenorhabditis elegans. PLoS Comput. Biol. 2009, 5, e1000417. [Google Scholar] [CrossRef]
- Bentz, A.B.; Dossey, E.K.; Rosvall, K.A. Tissue-specific gene regulation corresponds with seasonal plasticity in female testosterone. Gen. Comp. Endocrinol. 2019, 270, 26–34. [Google Scholar] [CrossRef]
- Liao, X.; Cheng, L.; Xu, P.; Lu, G.; Wachholtz, M.; Sun, X.; Chen, S. Transcriptome Analysis of Crucian Carp (Carassius auratus), an Important Aquaculture and Hypoxia-Tolerant Species. PLoS ONE 2013, 8, e62308. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.R.; King, H.; Evans, B.; Reverter, A.; Kijas, J.W. Multi-Tissue Transcriptome Profiling of North American Derived Atlantic Salmon. Front. Genet. 2018, 9, 369. [Google Scholar] [CrossRef]
- Salem, M.; Paneru, B.; Al-Tobasei, R.; Abdouni, F.; Thorgaard, G.H.; Rexroad, C.E.; Yao, J. Transcriptome assembly, gene annotation and tissue gene expression atlas of the rainbow trout. PLoS ONE 2015, 10, e0121778. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xia, S.; Arand, B.; Zhu, H.; Machiraju, R.; Huang, K.; Ji, H.; Qian, J. Single-Cell Co-expression Analysis Reveals Distinct Functional Modules, Co-regulation Mechanisms and Clinical Outcomes. PLoS Comput. Biol. 2016, 12, e1004892. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Bushmanova, E.; Antipov, D.; Lapidus, A.; Prjibelski, A.D. rrnaSPAdes: A de novo transcriptome assembler and its application to RNA-Seq data. GigaScience 2019, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 2019, 20, 278. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Nakasugi, K.; Crowhurst, R.; Bally, J.; Waterhouse, P. Combining transcriptome assemblies from multiple de novo assemblers in the allo-tetraploid plant Nicotiana benthamiana. PLoS ONE 2014, 9, e91776. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, D. Gene-omes built from mRNA seq not genome DNA. F1090Research 2013, 5, 1695. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2018, 47, D309–D314. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Pertea, G.; Pertea, M. GFF Utilities: GffRead and GffCompare. F1000Research 2020, 9, 304. [Google Scholar] [CrossRef]
- Waterhouse, R.M.; Seppey, M.; Sim?o, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Vera Alvarez, R.; Pongor, L.S.; Mariño-Ramírez, L.; Landsman, D. TPMCalculator: One-step software to quantify mRNA abundance of genomic features. Bioinformatics 2019, 35, 1960–1962. [Google Scholar] [CrossRef] [PubMed]
- Yanai, I.; Benjamin, H.; Shmoish, M.; Chalifa-Caspi, V.; Shklar, M.; Ophir, R.; Bar-Even, A.; Horn-Saban, S.; Safran, M.; Domany, E.; et al. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics 2005, 21, 650–659. [Google Scholar] [CrossRef]
- Mank, J.E.; Hultin-Rosenberg, L.; Zwahlen, M.; Ellegren, H. Pleiotropic constraint hampers the resolution of sexual antagonism in vertebrate gene expression. Am. Nat. 2008, 171, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Kryuchkova-Mostacci, N.; Robinson-Rechavi, M. A benchmark of gene expression tissue-specificity metrics. Brief Bioinform. 2017, 18, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Liao, B.Y.; Zhang, J. Low rates of expression profile divergence in highly expressed genes and tissue-specific genes during mammalian evolution. Mol. Biol. Evol. 2006, 23, 1119–1128. [Google Scholar] [CrossRef]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef]
- Jain, A.; Tuteja, G. TissueEnrich: Tissue-specific gene enrichment analysis. Bioinformatics 2019, 35, 1966–1967. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics: Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Russo, P.S.T.; Ferreira, G.R.; Cardozo, L.E.; Bürger, M.C.; Arias-Carrasco, R.; Maruyama, S.R.; Hirata, T.D.C.; Lima, D.S.; Passos, F.M.; Fukutani, K.F.; et al. CEMiTool: A Bioconductor package for performing comprehensive modular co-expression analyses. BMC Bioinform. 2018, 19, 56. [Google Scholar] [CrossRef] [PubMed]
- Feron, R.; Zahm, M.; Cabau, C.; Klopp, C.; Roques, C.; Bouchez, O.; Eche, C.; Valière, S.; Donnadieu, C.; Haffray, P.; et al. Characterization of a Y-specific duplication/insertion of the anti-Mullerian hormone type II receptor gene based on a chromosome-scale genome assembly of yellow perch, Perca flavescens. Mol. Ecol. Resour. 2020, 20, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Moran, R.L.; Catchen, J.M.; Fuller, R.C. Genomic Resources for Darters (Percidae: Etheostominae) Provide Insight into Postzygotic Barriers Implicated in Speciation. Mol. Biol. Evol. 2020, 37, 711–729. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Sahm, A.; Bens, M.; Platzer, M.; Szafranski, K. PosiGene: Automated and easy-to-use pipeline for genome-wide detection of positively selected genes. Nucleic Acids Res. 2017, 45, e100. [Google Scholar] [CrossRef] [PubMed]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef]
- Huang, X.; Chen, X.G.; Armbruster, P.A. Comparative performance of transcriptome assembly methods for non-model organisms. BMC Genom. 2016, 17, 523. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Miao, Y.R.; Jia, L.H.; Yu, Q.Y.; Zhang, Q.; Guo, A.Y. AnimalTFDB 3.0: A comprehensive resource for annotation and prediction of animal transcription factors. Nucleic Acids Res. 2019, 47, D33–D38. [Google Scholar] [CrossRef]
- Houston, R.D.; Bean, T.P.; Macqueen, D.J.; Gundappa, M.K.; Jin, Y.H.; Jenkins, T.L.; Selly, S.L.C.; Martin, S.A.M.; Stevens, J.R.; Santos, E.M.; et al. Harnessing genomics to fast-track genetic improvement in aquaculture. Nat. Rev. Genet. 2020, 21, 389–409. [Google Scholar] [CrossRef]
- Cerveau, N.; Jackson, D.J. Combining independent de novo assemblies optimizes the coding transcriptome for nonconventional model eukaryotic organisms. BMC Bioinform. 2016, 17, 525. [Google Scholar] [CrossRef]
- Sadat-Hosseini, M.; Bakhtiarizadeh, M.R.; Boroomand, N.; Tohidfar, M.; Vahdati, K. Combining independent de novo assemblies to optimize leaf transcriptome of Persian walnut. PLoS ONE 2020, 15, e0232005. [Google Scholar] [CrossRef]
- Ferraz, A.L.; Ojeda, A.; López-Béjar, M.; Fernandes, L.T.; Castelló, A.; Folch, J.M.; Pérez-Enciso, M. Transcriptome architecture across tissues in the pig. BMC Genom. 2008, 9, 173. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Fuscoe, J.C.; Zhao, C.; Guo, C.; Jia, M.; Qing, T.; Bannon, D.I.; Lancashire, L.; Bao, W.; Du, T.; et al. A rat RNA-Seq transcriptomic BodyMap across 11 organs and 4 developmental stages. Nat. Commun. 2014, 5, 3230. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Qing, T.; Zhu, J.; Wen, Z.; Yu, Y.; Fukumura, R.; Zheng, Y.; Gondo, Y.; Shi, L. A Comprehensive Mouse Transcriptomic BodyMap across 17 Tissues by RNA-seq. Sci. Rep. 2017, 7, 4200. [Google Scholar] [CrossRef] [PubMed]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Sonawane, A.R.; Platig, J.; Fagny, M.; Chen, C.Y.; Paulson, J.N.; Lopes-Ramos, C.M.; DeMeo, D.L.; Quackenbush, J.; Glass, K.; Kuijjer, M.L. Understanding Tissue-Specific Gene Regulation. Cell Rep. 2017, 21, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Chen, G.; Zhu, S.; Li, S.; Wen, Z.; Li, B.; Zheng, Y.; Shi, L. Identification of Tissue-Specific Protein-Coding and Noncoding Transcripts across 14 Human Tissues Using RNA-seq. Sci. Rep. 2016, 6, 28400. [Google Scholar] [CrossRef]
- Saju, J.M.; Hossain, M.S.; Liew, W.C.; Pradhan, A.; Thevasagayam, N.M.; Tan, L.S.E.; Anand, A.; Olsson, P.E.; Orbán, L. Heat Shock Factor 5 Is Essential for Spermatogenesis in Zebrafish. Cell Rep. 2018, 25, 3252–3261. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Wang, B.; Du, H. A review onsoxgenes in fish. Rev. Aquac. 2021. [Google Scholar] [CrossRef]
- To, K.K.; Huang, L.E. Suppression of hypoxia-inducible factor 1alpha (HIF-1alpha) transcriptional activity by the HIF prolyl hydroxylase EGLN1. J. Biol. Chem. 2005, 280, 38102–38107. [Google Scholar] [CrossRef]
- Pescador, N.; Cuevas, Y.; Naranjo, S.; Alcaide, M.; Villar, D.; Landázuri, M.O.; Del Peso, L. Identification of a functional hypoxia-responsive element that regulates the expression of the egl nine homologue 3 (egln3/phd3) gene. Biochem. J. 2005, 390, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, C.K.; Mazumder, B.; Fox, P.L.; Fox, P.L. Role of hypoxia-inducible factor-1 in transcriptional activation of ceruloplasmin by iron deficiency. J. Biol. Chem. 2000, 275, 21048–21054. [Google Scholar] [CrossRef]
- Liu, S.; Brown, J.D.; Stanya, K.J.; Homan, E.; Leidl, M.; Inouye, K.; Bhargava, P.; Gangl, M.R.; Dai, L.; Hatano, B.; et al. A diurnal serum lipid integrates hepatic lipogenesis and peripheral fatty acid use. Nature 2013, 502, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Solaro, R.J. Calcium, thin filaments, and the integrative biology of cardiac contractility. Annu. Rev. Physiol. 2005, 67, 39–67. [Google Scholar] [CrossRef]
- Handley-Goldstone, H.M.; Grow, M.W.; Stegeman, J.J. Cardiovascular gene expression profiles of dioxin exposure in zebrafish embryos. Toxicol. Sci. 2005, 85, 683–693. [Google Scholar] [CrossRef]
- Jagadeeswaran, P.; Kulkarni, V.; Carrillo, M.; Kim, S. Zebrafish: From hematology to hydrology. J. Thromb. Haemost. 2007, 5 (Suppl. S1), 300–304. [Google Scholar] [CrossRef]
- Fish, R.J.; Vorjohann, S.; Béna, F.; Fort, A.; Neerman-Arbez, M. Developmental expression and organisation of fibrinogen genes in the zebrafish. Thromb. Haemost. 2012, 107, 158–166. [Google Scholar] [PubMed]
- Axelsson, E.; Hultin-Rosenberg, L.; Brandström, M.; Zwahlén, M.; Clayton, D.F.; Ellegren, H. Natural selection in avian protein-coding genes expressed in brain. Mol. Ecol. 2008, 17, 3008–3017. [Google Scholar] [CrossRef]
- Ekblom, R.; French, L.; Slate, J.; Burke, T. Evolutionary analysis and expression profiling of zebra finch immune genes. Genome Biol. Evol. 2010, 2, 781–790. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Libraries | No. of Raw Reads | Q30 Raw Reads (%) | No. of Clean Reads | Q30 Clean Reads (%) |
|---|---|---|---|---|
| Heart-1 | 33,908,652 | 93.98 | 30,984,679 | 97.42 |
| Heart-2 | 35,031,697 | 94.16 | 33,180,437 | 97.49 |
| Head kidney-1 | 33,002,587 | 93.95 | 30,015,644 | 97.51 |
| Head kidney-2 | 39,938,681 | 94.16 | 36,623,609 | 97.50 |
| Muscle-1 | 35,047,416 | 94.74 | 32,410,797 | 97.52 |
| Muscle-2 | 39,426,896 | 94.32 | 36,506,458 | 97.53 |
| Liver-1 | 32,566,471 | 94.20 | 30,209,964 | 97.42 |
| Liver-2 | 35,071,990 | 94.11 | 32,007,540 | 97.65 |
| Brain-1 | 40,422,234 | 93.58 | 37,106,005 | 97.40 |
| Brain-2 | 35,567,608 | 93.46 | 36,458,085 | 97.41 |
| Skin-1 | 37,989,173 | 94.21 | 35,172,646 | 97.42 |
| Skin-2 | 40,633,032 | 94.34 | 37,687,356 | 97.47 |
| Gills-1 | 38,586,131 | 93.97 | 35,630,505 | 97.42 |
| Gills-2 | 39,427,046 | 94.51 | 36,458,085 | 97.41 |
| Spleen-1 | 45,127,155 | 94.00 | 41,790,579 | 97.36 |
| Spleen-2 | 33,329,198 | 94.27 | 30,504,153 | 97.44 |
| Ovary | 37,553,020 | 94.38 | 34,742,133 | 97.51 |
| Testis | 48,694,199 | 93.90 | 44,903,971 | 97.37 |
| Average | 37,851,288 | 94.12 | 35,132,924 | 97.45 |
| Total | 681,323,186 | — | 632,392,646 | — |
| Trinity | rnaSPAdes | Hisat2 + StringTie2 | EvidentialGene | |
|---|---|---|---|---|
| Number of contigs | 438,462 | 295,387 | 79,936 | 56,302 |
| Cumulative contigs length (Mb) | 399.28 | 502.46 | 299.28 | 85.73 |
| Mean contigs length (bp) | 910.65 | 1701.05 | 3744.49 | 1522.81 |
| N50 contigs length (bp) | 1340 | 3436 | 4934 | 1977 |
| Largest contig (bp) | 70,079 | 80,089 | 78,909 | 79,815 |
| ∑ contigs > 1 Kb (%) | 57.11 | 83.16 | 98.31 | 80.51 |
| % of FL transcripts | 60.57 | 72.84 | 89.52 | 86.73 |
| % of transcripts with ORFs | 76.73 | 80.53 | 88.84 | 85.07 |
| % of BUSCO complete | 80.27 | 96.58 | 96.62 | 96.87 |
| % of transcripts with NCBI NR hits | 72.83 | 78.04 | 86.27 | 88.35 |
| % of transcripts with Swiss-Prot hits | 55.76 | 60.23 | 75.86 | 78.57 |
| Mapping rate RNA-Seq reads (%) | 83.92 | 84.75 | 90.86 | 88.15 |
| Category | No. of Genes | Fraction of Detected Genes (%) |
|---|---|---|
| Tissue-Specific | 2930 | 15.00 |
| Group-Enriched | 3809 | 19.50 |
| Expressed-in-All | 5810 | 29.80 |
| Mixed | 6970 | 35.70 |
| Total detected | 19,541 | 100 |
| Module | No. Genes | Tisssue-Specific Upregulation | Hubs (Gene Symbol) |
|---|---|---|---|
| M1 | 55 | Liver | C3, AFP4, C1QTNF3 |
| M2 | 53 | Muscle | PYGM, TNNT3A, TRIM21, MYLPFA |
| M3 | 27 | Ovary, Testis | SERPINA12, ALOX12B, LOC116046623 |
| M4 | 21 | Skin | RPS7, RPS3A, RPL5, RPL13A, RPL7A |
| M5 | 19 | Head kidney, Spleen | HBZ, NPRL3, AQP8A, HBB2 |
| M6 | 15 | Gills, Skin | LOC116046623, ZG16B, MPO |
| M7 | 14 | Heart | TNNT2A, MYBPC3, TNNC1A, TNNI1, TPM4A |
| Branch | No of. CDS | No. of GUPS | Mean () | Avg No. of Sites |
|---|---|---|---|---|
| Sander lucioperca | 56,899 | 43 | 5.11 | 6.63 |
| Sander vitreus | 34,187 | 63 | 4.08 | 9.16 |
| Perca flavescens | 43,150 | 137 | 3.41 | 8.41 |
| Perca fluviatilis | 50,212 | 154 | 5.97 | 7.80 |
| Etheostoma spectabile | 45,699 | 152 | 4.07 | 9.10 |
| Etheostoma cragini | 45,199 | 124 | 3.24 | 9.22 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguinkal, J.A.; Verleih, M.; de los Ríos-Pérez, L.; Brunner, R.M.; Sahm, A.; Bej, S.; Rebl, A.; Goldammer, T. Comprehensive Characterization of Multitissue Expression Landscape, Co-Expression Networks and Positive Selection in Pikeperch. Cells 2021, 10, 2289. https://doi.org/10.3390/cells10092289
Nguinkal JA, Verleih M, de los Ríos-Pérez L, Brunner RM, Sahm A, Bej S, Rebl A, Goldammer T. Comprehensive Characterization of Multitissue Expression Landscape, Co-Expression Networks and Positive Selection in Pikeperch. Cells. 2021; 10(9):2289. https://doi.org/10.3390/cells10092289
Chicago/Turabian StyleNguinkal, Julien Alban, Marieke Verleih, Lidia de los Ríos-Pérez, Ronald Marco Brunner, Arne Sahm, Saptarshi Bej, Alexander Rebl, and Tom Goldammer. 2021. "Comprehensive Characterization of Multitissue Expression Landscape, Co-Expression Networks and Positive Selection in Pikeperch" Cells 10, no. 9: 2289. https://doi.org/10.3390/cells10092289
APA StyleNguinkal, J. A., Verleih, M., de los Ríos-Pérez, L., Brunner, R. M., Sahm, A., Bej, S., Rebl, A., & Goldammer, T. (2021). Comprehensive Characterization of Multitissue Expression Landscape, Co-Expression Networks and Positive Selection in Pikeperch. Cells, 10(9), 2289. https://doi.org/10.3390/cells10092289