
Aging and Cancer: The Waning of Community Bonds


Abstract

{kind=link}
{kind=link}
{kind=link}
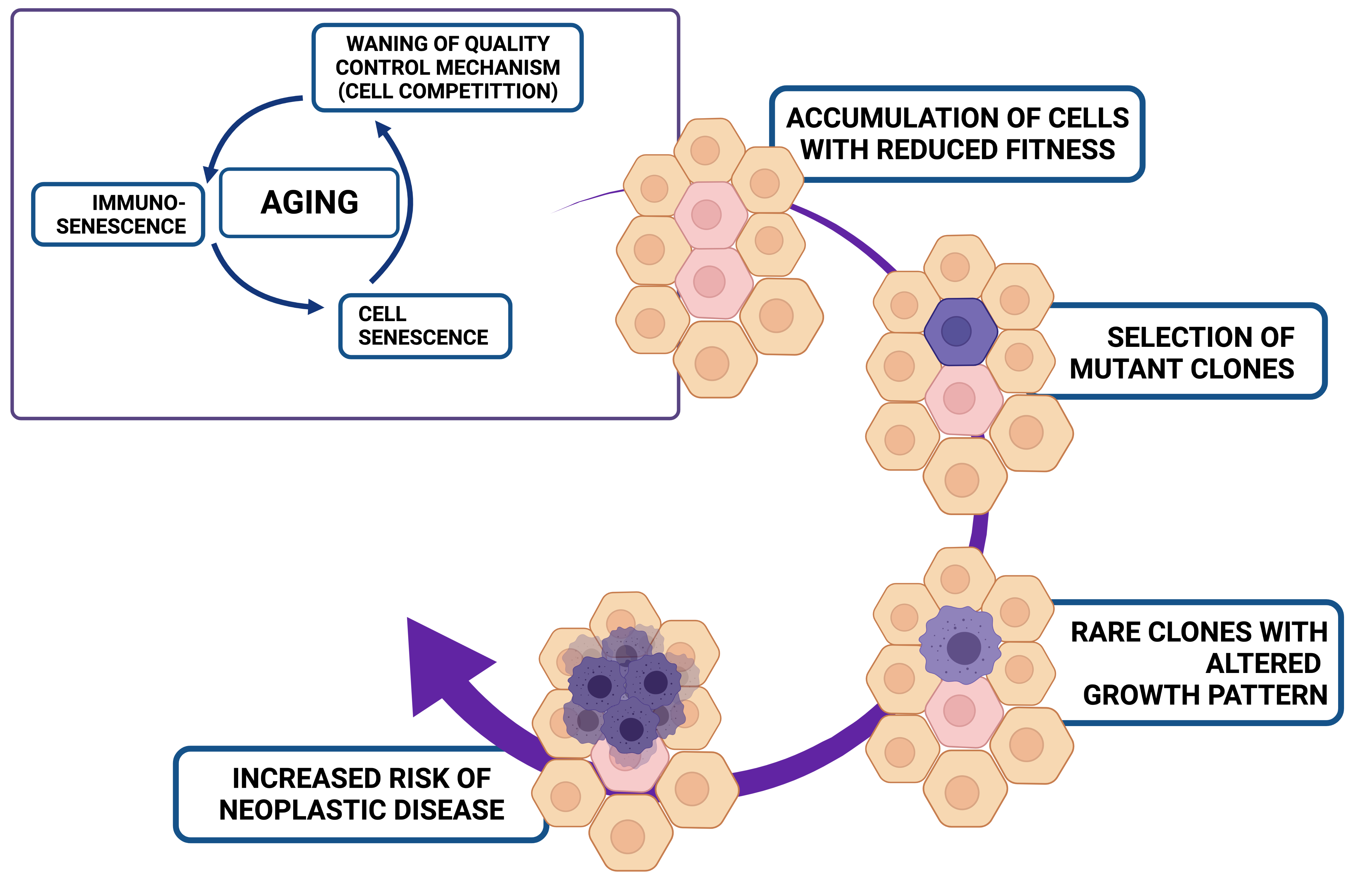
1. Aging and Cancer: The Essential Facts
2. How Do We Avoid Cancer Early in Life
2.1. Cell Competition and the Concept of Relative Fitness
2.2. Cell Senescence
2.3. Immune Surveillance
3. Aging and Cancer: How Does It Happen
3.1. A Cell-Oriented View
3.2. A Tissue-Oriented View
3.3. An Ecological View
4. Aging and Cancer: How to Loosen the Link
4.1. Cancer Risk Factors and Aging
4.2. Dietary Interventions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fane, M.; Weeraratna, A.T. How the Ageing Microenvironment Influences Tumour Progression. Nat. Rev. Cancer 2020, 20, 89–106. [Google Scholar] [CrossRef] [PubMed]
- Cancer of Any Site—Cancer Stat Facts. Available online: https://seer.cancer.gov/statfacts/html/all.html (accessed on 11 June 2021).
- West, S.A.; Fisher, R.M.; Gardner, A.; Kiers, E.T. Major Evolutionary Transitions in Individuality. Proc. Natl. Acad. Sci. USA 2015, 112, 10112–10119. [Google Scholar] [CrossRef] [PubMed]
- Queller, D.C.; Strassmann, J.E. Beyond Society: The Evolution of Organismality. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2009, 364, 3143–3155. [Google Scholar] [CrossRef]
- Laconi, E.; Marongiu, F.; DeGregori, J. Cancer as a Disease of Old Age: Changing Mutational and Microenvironmental Landscapes. Br. J. Cancer 2020, 122, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Morata, G.; Ripoll, P. Minutes: Mutants of Drosophila Autonomously Affecting Cell Division Rate. Dev. Biol. 1975, 42, 211–221. [Google Scholar] [CrossRef]
- Kim, W.; Jain, R. Picking Winners and Losers: Cell Competition in Tissue Development and Homeostasis. Trends Genet. 2020, 36, 490–498. [Google Scholar] [CrossRef]
- Baker, N.E. Emerging Mechanisms of Cell Competition. Nat. Rev. Genet. 2020, 21, 683–697. [Google Scholar] [CrossRef]
- Bowling, S.; Lawlor, K.; Rodríguez, T.A. Cell Competition: The Winners and Losers of Fitness Selection. Dev. Camb. Engl. 2019, 146. [Google Scholar] [CrossRef]
- Clavería, C.; Giovinazzo, G.; Sierra, R.; Torres, M. Myc-Driven Endogenous Cell Competition in the Early Mammalian Embryo. Nature 2013, 500, 39–44. [Google Scholar] [CrossRef]
- Sancho, M.; Di-Gregorio, A.; George, N.; Pozzi, S.; Sánchez, J.M.; Pernaute, B.; Rodríguez, T.A. Competitive Interactions Eliminate Unfit Embryonic Stem Cells at the Onset of Differentiation. Dev. Cell 2013, 26, 19–30. [Google Scholar] [CrossRef]
- Moreno, E.; Basler, K. DMyc Transforms Cells into Super-Competitors. Cell 2004, 117, 117–129. [Google Scholar] [CrossRef]
- Rhiner, C.; López-Gay, J.M.; Soldini, D.; Casas-Tinto, S.; Martín, F.A.; Lombardía, L.; Moreno, E. Flower Forms an Extracellular Code That Reveals the Fitness of a Cell to Its Neighbors in Drosophila. Dev. Cell 2010, 18, 985–998. [Google Scholar] [CrossRef]
- Madan, E.; Pelham, C.J.; Nagane, M.; Parker, T.M.; Canas-Marques, R.; Fazio, K.; Shaik, K.; Yuan, Y.; Henriques, V.; Galzerano, A.; et al. Flower Isoforms Promote Competitive Growth in Cancer. Nature 2019, 572, 260–264. [Google Scholar] [CrossRef]
- Ji, Z.; Chuen, J.; Kiparaki, M.; Baker, N. Cell Competition Removes Segmental Aneuploid Cells from Drosophila Imaginal Disc-Derived Tissues Based on Ribosomal Protein Gene Dose. eLife 2021, 10, e61172. [Google Scholar] [CrossRef] [PubMed]
- Kanda, H.; Igaki, T. Mechanism of Tumor-Suppressive Cell Competition in Flies. Cancer Sci. 2020, 111, 3409–3415. [Google Scholar] [CrossRef]
- Kon, S.; Fujita, Y. Cell Competition-Induced Apical Elimination of Transformed Cells, EDAC, Orchestrates the Cellular Homeostasis. Dev. Biol. 2021, 476, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Norman, M.; Wisniewska, K.A.; Lawrenson, K.; Garcia-Miranda, P.; Tada, M.; Kajita, M.; Mano, H.; Ishikawa, S.; Ikegawa, M.; Shimada, T.; et al. Loss of Scribble Causes Cell Competition in Mammalian Cells. J. Cell Sci. 2012, 125, 59–66. [Google Scholar] [CrossRef]
- Liu, N.; Matsumura, H.; Kato, T.; Ichinose, S.; Takada, A.; Namiki, T.; Asakawa, K.; Morinaga, H.; Mohri, Y.; De Arcangelis, A.; et al. Stem Cell Competition Orchestrates Skin Homeostasis and Ageing. Nature 2019, 568, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Marongiu, F.; Laconi, E. Cell Competition in Liver Carcinogenesis. World J. Hepatol. 2020, 12, 475–484. [Google Scholar] [CrossRef]
- Gutierrez-Martinez, P.; Hogdal, L.; Nagai, M.; Kruta, M.; Singh, R.; Sarosiek, K.; Nussenzweig, A.; Beerman, I.; Letai, A.; Rossi, D.J. Diminished Apoptotic Priming and ATM Signalling Confer a Survival Advantage onto Aged Haematopoietic Stem Cells in Response to DNA Damage. Nat. Cell Biol. 2018, 20, 413–421. [Google Scholar] [CrossRef]
- Campisi, J.; Robert, L. Cell Senescence: Role in Aging and Age-Related Diseases. Interdiscip. Top. Gerontol. 2014, 39, 45–61. [Google Scholar] [CrossRef]
- Braig, M.; Lee, S.; Loddenkemper, C.; Rudolph, C.; Peters, A.H.F.M.; Schlegelberger, B.; Stein, H.; Dörken, B.; Jenuwein, T.; Schmitt, C.A. Oncogene-Induced Senescence as an Initial Barrier in Lymphoma Development. Nature 2005, 436, 660–665. [Google Scholar] [CrossRef]
- Fumagalli, M.; d’Adda di Fagagna, F. SASPense and DDRama in Cancer and Ageing. Nat. Cell Biol. 2009, 11, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.-W.; Lasitschka, F.; Andrulis, M.; et al. A Complex Secretory Program Orchestrated by the Inflammasome Controls Paracrine Senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Childs, B.G.; Baker, D.J.; Kirkland, J.L.; Campisi, J.; van Deursen, J.M. Senescence and Apoptosis: Dueling or Complementary Cell Fates? EMBO Rep. 2014, 15, 1139–1153. [Google Scholar] [CrossRef]
- Ritschka, B.; Storer, M.; Mas, A.; Heinzmann, F.; Ortells, M.C.; Morton, J.P.; Sansom, O.J.; Zender, L.; Keyes, W.M. The Senescence-Associated Secretory Phenotype Induces Cellular Plasticity and Tissue Regeneration. Genes Dev. 2017, 31, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Prata, L.G.P.L.; Ovsyannikova, I.G.; Tchkonia, T.; Kirkland, J.L. Senescent Cell Clearance by the Immune System: Emerging Therapeutic Opportunities. Semin. Immunol. 2018, 40, 101275. [Google Scholar] [CrossRef]
- Song, P.; An, J.; Zou, M.-H. Immune Clearance of Senescent Cells to Combat Ageing and Chronic Diseases. Cells 2020, 9, 671. [Google Scholar] [CrossRef] [PubMed]
- Tauber, A.I. Reconceiving Autoimmunity: An Overview. J. Theor. Biol. 2015, 375, 52–60. [Google Scholar] [CrossRef]
- Pradeu, T.; Vivier, E. The Discontinuity Theory of Immunity. Sci. Immunol. 2016, 1. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.-W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence Surveillance of Pre-Malignant Hepatocytes Limits Liver Cancer Development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef]
- Sagiv, A.; Burton, D.G.A.; Moshayev, Z.; Vadai, E.; Wensveen, F.; Ben-Dor, S.; Golani, O.; Polic, B.; Krizhanovsky, V. NKG2D Ligands Mediate Immunosurveillance of Senescent Cells. Aging 2016, 8, 328–344. [Google Scholar] [CrossRef]
- Mittal, D.; Gubin, M.M.; Schreiber, R.D.; Smyth, M.J. New Insights into Cancer Immunoediting and Its Three Component Phases--Elimination, Equilibrium and Escape. Curr. Opin. Immunol. 2014, 27, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Mittelbrunn, M.; Kroemer, G. Hallmarks of T Cell Aging. Nat. Immunol. 2021, 22, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, G.R.; Almeida, P.P.; de Oliveira Santos, L.; Rodrigues, L.P.; de Carvalho, J.L.; Boroni, M. Hallmarks of Aging in Macrophages: Consequences to Skin Inflammaging. Cells 2021, 10, 1323. [Google Scholar] [CrossRef] [PubMed]
- van Beek, A.A.; Van den Bossche, J.; Mastroberardino, P.G.; de Winther, M.P.J.; Leenen, P.J.M. Metabolic Alterations in Aging Macrophages: Ingredients for Inflammaging? Trends Immunol. 2019, 40, 113–127. [Google Scholar] [CrossRef]
- Almanzar, N.; Antony, J.; Baghel, A.S.; Bakerman, I.; Bansal, I.; Barres, B.A.; Beachy, P.A.; Berdnik, D.; Bilen, B.; Brownfield, D.; et al. A Single-Cell Transcriptomic Atlas Characterizes Ageing Tissues in the Mouse. Nature 2020, 583, 590–595. [Google Scholar] [CrossRef]
- Chambers, E.S.; Vukmanovic-Stejic, M. Skin Barrier Immunity and Ageing. Immunology 2020, 160, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Risques, R.A.; Kennedy, S.R. Aging and the Rise of Somatic Cancer-Associated Mutations in Normal Tissues. PLoS Genet. 2018, 14, e1007108. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Vogelstein, B. Variation in Cancer Risk among Tissues Can Be Explained by the Number of Stem Cell Divisions. Science 2015, 347, 78–81. [Google Scholar] [CrossRef]
- Kim, S.K.; Park, Y.-K. Ewing Sarcoma: A Chronicle of Molecular Pathogenesis. Hum. Pathol. 2016, 55, 91–100. [Google Scholar] [CrossRef]
- Rosell, R.; Karachaliou, N. Relationship between Gene Mutation and Lung Cancer Metastasis. Cancer Metastasis Rev. 2015, 34, 243–248. [Google Scholar] [CrossRef]
- Gibbons, D.L.; Byers, L.A.; Kurie, J.M. Smoking, P53 Mutation, and Lung Cancer. Mol. Cancer Res. MCR 2014, 12, 3–13. [Google Scholar] [CrossRef]
- Luo, J. KRAS Mutation in Pancreatic Cancer. Semin. Oncol. 2021. [Google Scholar] [CrossRef]
- Al Zouabi, L.; Bardin, A.J. Stem Cell DNA Damage and Genome Mutation in the Context of Aging and Cancer Initiation. Cold Spring Harb. Perspect. Biol. 2020, 12. [Google Scholar] [CrossRef]
- Petr, M.A.; Tulika, T.; Carmona-Marin, L.M.; Scheibye-Knudsen, M. Protecting the Aging Genome. Trends Cell Biol. 2020, 30, 117–132. [Google Scholar] [CrossRef] [PubMed]
- McNeely, T.; Leone, M.; Yanai, H.; Beerman, I. DNA Damage in Aging, the Stem Cell Perspective. Hum. Genet. 2020, 139, 309–331. [Google Scholar] [CrossRef] [PubMed]
- Beerman, I. Accumulation of DNA Damage in the Aged Hematopoietic Stem Cell Compartment. Semin. Hematol. 2017, 54, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Fiala, C.; Diamandis, E.P. Mutations in Normal Tissues-Some Diagnostic and Clinical Implications. BMC Med. 2020, 18, 283. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.A. Gerontology as Oncology. Research on Aging as the Key to the Understanding of Cancer. Cancer 1991, 68, 2496–2501. [Google Scholar] [CrossRef]
- Grist, S.A.; McCarron, M.; Kutlaca, A.; Turner, D.R.; Morley, A.A. In Vivo Human Somatic Mutation: Frequency and Spectrum with Age. Mutat. Res. 1992, 266, 189–196. [Google Scholar] [CrossRef]
- Loeb, L.A. Human Cancers Express a Mutator Phenotype: Hypothesis, Origin, and Consequences. Cancer Res. 2016, 76, 2057–2059. [Google Scholar] [CrossRef] [PubMed]
- Laconi, E. The Evolving Concept of Tumor Microenvironments. BioEssays News Rev. Mol. Cell. Dev. Biol. 2007, 29, 738–744. [Google Scholar] [CrossRef] [PubMed]
- McCullough, K.D.; Coleman, W.B.; Smith, G.J.; Grisham, J.W. Age-Dependent Induction of Hepatic Tumor Regression by the Tissue Microenvironment after Transplantation of Neoplastically Transformed Rat Liver Epithelial Cells into the Liver. Cancer Res. 1997, 57, 1807–1813. [Google Scholar]
- Petr, M.A.; Alfaras, I.; Krawcyzk, M.; Bair, W.-N.; Mitchell, S.J.; Morrell, C.H.; Studenski, S.A.; Price, N.L.; Fishbein, K.W.; Spencer, R.G.; et al. A Cross-Sectional Study of Functional and Metabolic Changes during Aging through the Lifespan in Male Mice. eLife 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Bissell, M.J.; Radisky, D. Putting Tumours in Context. Nat. Rev. Cancer 2001, 1, 46–54. [Google Scholar] [CrossRef]
- Greco, S.J. Breast Cancer Risk in a Rapidly Aging Population: Advances and Approaches to Study the Aging Tissue Microenvironment. Breast Cancer Dove Med. Press 2019, 11, 111–113. [Google Scholar] [CrossRef]
- Guo, Y.; Ayers, J.L.; Carter, K.T.; Wang, T.; Maden, S.K.; Edmond, D.; Newcomb, P.P.; Li, C.; Ulrich, C.; Yu, M.; et al. Senescence-Associated Tissue Microenvironment Promotes Colon Cancer Formation through the Secretory Factor GDF15. Aging Cell 2019, 18, e13013. [Google Scholar] [CrossRef]
- Laconi, S.; Pani, P.; Pillai, S.; Pasciu, D.; Sarma, D.S.R.; Laconi, E. A Growth-Constrained Environment Drives Tumor Progression in Vivo. Proc. Natl. Acad. Sci. USA 2001, 98, 7806–7811. [Google Scholar] [CrossRef]
- McCullough, K.D.; Coleman, W.B.; Smith, G.J.; Grishan, J.W. Age-Dependent Regulation of the Tumorigenic Potential of Neoplastically Transformed Rat Liver Epithelial Cells by the Liver Microenvironment. Cancer Res. 1994, 54, 3668–3671. [Google Scholar]
- Pasciu, D.; Montisci, S.; Greco, M.; Doratiotto, S.; Pitzalis, S.; Pani, P.; Laconi, S.; Laconi, E. Aging Is Associated with Increased Clonogenic Potential in Rat Liver in Vivo. Aging Cell 2006, 5, 373–377. [Google Scholar] [CrossRef]
- Marongiu, F.; Serra, M.P.; Doratiotto, S.; Sini, M.; Fanti, M.; Cadoni, E.; Serra, M.; Laconi, E. Aging Promotes Neoplastic Disease through Effects on the Tissue Microenvironment. Aging 2016, 8, 3390–3399. [Google Scholar] [CrossRef]
- Henry, C.J.; Marusyk, A.; Zaberezhnyy, V.; Adane, B.; DeGregori, J. Declining Lymphoid Progenitor Fitness Promotes Aging-Associated Leukemogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 21713–21718. [Google Scholar] [CrossRef]
- Schneider, J.L.; Rowe, J.H.; Garcia-de-Alba, C.; Kim, C.F.; Sharpe, A.H.; Haigis, M.C. The Aging Lung: Physiology, Disease, and Immunity. Cell 2021, 184, 1990–2019. [Google Scholar] [CrossRef]
- Worrede, A.; Douglass, S.M.; Weeraratna, A.T. The Dark Side of Daylight: Photoaging and the Tumor Microenvironment in Melanoma Progression. J. Clin. Invest. 2021, 131. [Google Scholar] [CrossRef]
- Santos, S.A.A.; Camargo, A.C.; Constantino, F.B.; Colombelli, K.T.; Mani, F.; Rinaldi, J.C.; Franco, S.; Portela, L.M.F.; Duran, B.O.S.; Scarano, W.R.; et al. Maternal Low-Protein Diet Impairs Prostate Growth in Young Rat Offspring and Induces Prostate Carcinogenesis With Aging. J. Gerontol. A. Biol. Sci. Med. Sci. 2019, 74, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Landry, D.A.; Vaishnav, H.T.; Vanderhyden, B.C. The Significance of Ovarian Fibrosis. Oncotarget 2020, 11, 4366–4370. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A New Immune-Metabolic Viewpoint for Age-Related Diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef]
- Tibbs, T.N.; Lopez, L.R.; Arthur, J.C. The Influence of the Microbiota on Immune Development, Chronic Inflammation, and Cancer in the Context of Aging. Microb. Cell Graz Austria 2019, 6, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Henry, C.J.; Casás-Selves, M.; Kim, J.; Zaberezhnyy, V.; Aghili, L.; Daniel, A.E.; Jimenez, L.; Azam, T.; McNamee, E.N.; Clambey, E.T.; et al. Aging-Associated Inflammation Promotes Selection for Adaptive Oncogenic Events in B Cell Progenitors. J. Clin. Invest. 2015, 125, 4666–4680. [Google Scholar] [CrossRef] [PubMed]
- Parikh, N.; Shuck, R.L.; Gagea, M.; Shen, L.; Donehower, L.A. Enhanced Inflammation and Attenuated Tumor Suppressor Pathways Are Associated with Oncogene-Induced Lung Tumors in Aged Mice. Aging Cell 2018, 17. [Google Scholar] [CrossRef]
- Alter, N.M. Mechanical Irritation as Etiologic Factor of Cancer. Am. J. Pathol. 1925, 1, 511–518.3. [Google Scholar]
- Kuraishy, A.; Karin, M.; Grivennikov, S.I. Tumor Promotion via Injury- and Death-Induced Inflammation. Immunity 2011, 35, 467–477. [Google Scholar] [CrossRef]
- Bottazzi, B.; Riboli, E.; Mantovani, A. Aging, Inflammation and Cancer. Semin. Immunol. 2018, 40, 74–82. [Google Scholar] [CrossRef]
- Li, C.M.-C.; Shapiro, H.; Tsiobikas, C.; Selfors, L.M.; Chen, H.; Rosenbluth, J.; Moore, K.; Gupta, K.P.; Gray, G.K.; Oren, Y.; et al. Aging-Associated Alterations in Mammary Epithelia and Stroma Revealed by Single-Cell RNA Sequencing. Cell Rep. 2020, 33, 108566. [Google Scholar] [CrossRef] [PubMed]
- Langevin, H.M.; Keely, P.; Mao, J.; Hodge, L.M.; Schleip, R.; Deng, G.; Hinz, B.; Swartz, M.A.; de Valois, B.A.; Zick, S.; et al. Connecting (T)Issues: How Research in Fascia Biology Can Impact Integrative Oncology. Cancer Res. 2016, 76, 6159–6162. [Google Scholar] [CrossRef]
- Vogel, V. Unraveling the Mechanobiology of Extracellular Matrix. Annu. Rev. Physiol. 2018, 80, 353–387. [Google Scholar] [CrossRef]
- Mueller, M.M.; Fusenig, N.E. Friends or Foes—Bipolar Effects of the Tumour Stroma in Cancer. Nat. Rev. Cancer 2004, 4, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Ingber, D.E. Can Cancer Be Reversed by Engineering the Tumor Microenvironment? Semin. Cancer Biol. 2008, 18, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Adaptive Oncogenesis—James DeGregori. Available online: https://www.hup.harvard.edu/catalog.php?isbn=9780674545397 (accessed on 27 May 2021).
- Haddow, A. Cellular Inhibition and Origin of Cancer. Acta Unio. Int. Contra. Cancrum. 1938, 3, 342–353. [Google Scholar]
- Solt, D.; Farber, E. New Principle for the Analysis of Chemical Carcinogenesis. Nature 1976, 263, 701–703. [Google Scholar] [CrossRef]
- Barcellos-Hoff, M.H.; Park, C.; Wright, E.G. Radiation and the Microenvironment—Tumorigenesis and Therapy. Nat. Rev. Cancer 2005, 5, 867–875. [Google Scholar] [CrossRef] [PubMed]
- Laconi, E.; Pani, P.; Farber, E. The Resistance Phenotype in the Development and Treatment of Cancer. Lancet Oncol. 2000, 1, 235–241. [Google Scholar] [CrossRef]
- Lynch, M. Rate, Molecular Spectrum, and Consequences of Human Mutation. Proc. Natl. Acad. Sci. USA 2010, 107, 961–968. [Google Scholar] [CrossRef]
- Milholland, B.; Suh, Y.; Vijg, J. Mutation and Catastrophe in the Aging Genome. Exp. Gerontol. 2017, 94, 34–40. [Google Scholar] [CrossRef]
- Timchenko, N.A. Aging and Liver Regeneration. Trends Endocrinol. Metab. TEM 2009, 20, 171–176. [Google Scholar] [CrossRef]
- Ambrosi, T.H.; Scialdone, A.; Graja, A.; Gohlke, S.; Jank, A.-M.; Bocian, C.; Woelk, L.; Fan, H.; Logan, D.W.; Schürmann, A.; et al. Adipocyte Accumulation in the Bone Marrow during Obesity and Aging Impairs Stem Cell-Based Hematopoietic and Bone Regeneration. Cell Stem Cell 2017, 20, 771–784.e6. [Google Scholar] [CrossRef]
- Saito, Y.; Morine, Y.; Shimada, M. Mechanism of Impairment on Liver Regeneration in Elderly Patients: Role of Hepatic Stellate Cell Function. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2017, 47, 505–513. [Google Scholar] [CrossRef]
- Serra, M.P.; Marongiu, F.; Marongiu, M.; Contini, A.; Laconi, E. Cell-Autonomous Decrease in Proliferative Competitiveness of the Aged Hepatocyte. J. Hepatol. 2015, 62, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Marongiu, F.; Doratiotto, S.; Montisci, S.; Pani, P.; Laconi, E. Liver Repopulation and Carcinogenesis: Two Sides of the Same Coin? Am. J. Pathol. 2008, 172, 857–864. [Google Scholar] [CrossRef]
- Yoshida, K.; Gowers, K.H.C.; Lee-Six, H.; Chandrasekharan, D.P.; Coorens, T.; Maughan, E.F.; Beal, K.; Menzies, A.; Millar, F.R.; Anderson, E.; et al. Tobacco Smoking and Somatic Mutations in Human Bronchial Epithelium. Nature 2020, 578, 266–272. [Google Scholar] [CrossRef]
- Forsberg, L.A.; Gisselsson, D.; Dumanski, J.P. Mosaicism in Health and Disease—Clones Picking up Speed. Nat. Rev. Genet. 2017, 18, 128–142. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef]
- Martincorena, I.; Roshan, A.; Gerstung, M.; Ellis, P.; Van Loo, P.; McLaren, S.; Wedge, D.C.; Fullam, A.; Alexandrov, L.B.; Tubio, J.M.; et al. Tumor Evolution. High Burden and Pervasive Positive Selection of Somatic Mutations in Normal Human Skin. Science 2015, 348, 880–886. [Google Scholar] [CrossRef]
- Yokoyama, A.; Kakiuchi, N.; Yoshizato, T.; Nannya, Y.; Suzuki, H.; Takeuchi, Y.; Shiozawa, Y.; Sato, Y.; Aoki, K.; Kim, S.K.; et al. Age-Related Remodelling of Oesophageal Epithelia by Mutated Cancer Drivers. Nature 2019, 565, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Lu, T.; Jia, Y.; Luo, X.; Gopal, P.; Li, L.; Odewole, M.; Renteria, V.; Singal, A.G.; Jang, Y.; et al. Somatic Mutations Increase Hepatic Clonal Fitness and Regeneration in Chronic Liver Disease. Cell 2019, 177, 608–621.e12. [Google Scholar] [CrossRef]
- Watson, C.J.; Papula, A.L.; Poon, G.Y.P.; Wong, W.H.; Young, A.L.; Druley, T.E.; Fisher, D.S.; Blundell, J.R. The Evolutionary Dynamics and Fitness Landscape of Clonal Hematopoiesis. Science 2020, 367, 1449–1454. [Google Scholar] [CrossRef] [PubMed]
- Martincorena, I.; Fowler, J.C.; Wabik, A.; Lawson, A.R.J.; Abascal, F.; Hall, M.W.J.; Cagan, A.; Murai, K.; Mahbubani, K.; Stratton, M.R.; et al. Somatic Mutant Clones Colonize the Human Esophagus with Age. Science 2018, 362, 911–917. [Google Scholar] [CrossRef]
- Oertel, M.; Menthena, A.; Dabeva, M.D.; Shafritz, D.A. Cell Competition Leads to a High Level of Normal Liver Reconstitution by Transplanted Fetal Liver Stem/Progenitor Cells. Gastroenterology 2006, 130, 507–520, quiz 590. [Google Scholar] [CrossRef]
- Marongiu, F.; Serra, M.; Laconi, E. Development versus Evolution in Cancer Biology. Trends Cancer 2018, 4, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Doratiotto, S.; Marongiu, F.; Faedda, S.; Pani, P.; Laconi, E. Altered Growth Pattern, Not Altered Growth per Se, Is the Hallmark of Early Lesions Preceding Cancer Development. Histol. Histopathol. 2009, 24, 101–106. [Google Scholar] [CrossRef]
- Doratiotto, S.; Krause, P.; Serra, M.P.; Marongiu, F.; Sini, M.; Koenig, S.; Laconi, E. The Growth Pattern of Transplanted Normal and Nodular Hepatocytes. Histochem. Cell Biol. 2011, 135, 581–591. [Google Scholar] [CrossRef][Green Version]
- Marongiu, F.; Doratiotto, S.; Sini, M.; Serra, M.P.; Laconi, E. Cancer as a Disease of Tissue Pattern Formation. Prog. Histochem. Cytochem. 2012, 47, 175–207. [Google Scholar] [CrossRef]
- Ruiz i Altaba, A.; Mas, C.; Stecca, B. The Gli Code: An Information Nexus Regulating Cell Fate, Stemness and Cancer. Trends Cell Biol. 2007, 17, 438–447. [Google Scholar] [CrossRef]
- Zhao, Y.L.; Piao, C.Q.; Hall, E.J.; Hei, T.K. Mechanisms of Radiation-Induced Neoplastic Transformation of Human Bronchial Epithelial Cells. Radiat. Res. 2001, 155, 230–234. [Google Scholar] [CrossRef]
- Fernandez-Antoran, D.; Piedrafita, G.; Murai, K.; Ong, S.H.; Herms, A.; Frezza, C.; Jones, P.H. Outcompeting P53-Mutant Cells in the Normal Esophagus by Redox Manipulation. Cell Stem Cell 2019, 25, 329–341.e6. [Google Scholar] [CrossRef]
- Roshan, A.; Jones, P.H. Chronic Low Dose UV Exposure and P53 Mutation: Tilting the Odds in Early Epidermal Preneoplasia? Int. J. Radiat. Biol. 2012, 88, 682–687. [Google Scholar] [CrossRef]
- Nguyen, R.H.; Vater, L.B.; Timsina, L.R.; Durm, G.A.; Rupp, K.; Wright, K.; Spitznagle, M.H.; Paul, B.; Jalal, S.I.; Carter-Harris, L.; et al. Impact of Smoke-Free Ordinance Strength on Smoking Prevalence and Lung Cancer Incidence. PLoS ONE 2021, 16, e0250285. [Google Scholar] [CrossRef] [PubMed]
- Marongiu, F.; Laconi, E. Dietary Patterns and the Neoplastic-Prone Tissue Landscape of Old Age. Aging Cancer 2020, 1, 45–57. [Google Scholar] [CrossRef]
- Colman, R.J.; Anderson, R.M.; Johnson, S.C.; Kastman, E.K.; Kosmatka, K.J.; Beasley, T.M.; Allison, D.B.; Cruzen, C.; Simmons, H.A.; Kemnitz, J.W.; et al. Caloric Restriction Delays Disease Onset and Mortality in Rhesus Monkeys. Science 2009, 325, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Mattison, J.A.; Roth, G.S.; Beasley, T.M.; Tilmont, E.M.; Handy, A.H.; Herbert, R.L.; Longo, D.L.; Allison, D.B.; Young, J.E.; Bryant, M.; et al. Impact of Caloric Restriction on Health and Survival in Rhesus Monkeys: The NIA Study. Nature 2012, 489. [Google Scholar] [CrossRef]
- Madeo, F.; Carmona-Gutierrez, D.; Hofer, S.J.; Kroemer, G. Caloric Restriction Mimetics against Age-Associated Disease: Targets, Mechanisms, and Therapeutic Potential. Cell Metab. 2019, 29, 592–610. [Google Scholar] [CrossRef]
- Dirks, A.J.; Leeuwenburgh, C. Caloric Restriction in Humans: Potential Pitfalls and Health Concerns. Mech. Ageing Dev. 2006, 127, 1–7. [Google Scholar] [CrossRef]
- Serra, M.; Marongiu, F.; Pisu, M.G.; Serra, M.; Laconi, E. Time-Restricted Feeding Delays the Emergence of the Age-Associated, Neoplastic-Prone Tissue Landscape. Aging 2019, 11, 3851–3863. [Google Scholar] [CrossRef]
- Cadoni, E.; Marongiu, F.; Fanti, M.; Serra, M.; Laconi, E. Caloric Restriction Delays Early Phases of Carcinogenesis via Effects on the Tissue Microenvironment. Oncotarget 2017, 8, 36020–36032. [Google Scholar] [CrossRef]
- Brandhorst, S.; Choi, I.Y.; Wei, M.; Cheng, C.W.; Sedrakyan, S.; Navarrete, G.; Dubeau, L.; Yap, L.P.; Park, R.; Vinciguerra, M.; et al. A Periodic Diet That Mimics Fasting Promotes Multi-System Regeneration, Enhanced Cognitive Performance, and Healthspan. Cell Metab. 2015, 22, 86–99. [Google Scholar] [CrossRef]
- Descamps, O.; Riondel, J.; Ducros, V.; Roussel, A.-M. Mitochondrial Production of Reactive Oxygen Species and Incidence of Age-Associated Lymphoma in OF1 Mice: Effect of Alternate-Day Fasting. Mech. Ageing Dev. 2005, 126, 1185–1191. [Google Scholar] [CrossRef]
- Caffa, I.; Spagnolo, V.; Vernieri, C.; Valdemarin, F.; Becherini, P.; Wei, M.; Brandhorst, S.; Zucal, C.; Driehuis, E.; Ferrando, L.; et al. Fasting-Mimicking Diet and Hormone Therapy Induce Breast Cancer Regression. Nature 2020, 583, 620–624. [Google Scholar] [CrossRef]
- Ling, G.; Persson, Å.; Berne, B.; Uhlén, M.; Lundeberg, J.; Ponten, F. Persistent P53 Mutations in Single Cells from Normal Human Skin. Am. J. Pathol. 2001, 159, 1247–1253. [Google Scholar] [CrossRef]
- McLellan, J.S.; Zheng, X.; Hauk, G.; Ghirlando, R.; Beachy, P.A.; Leahy, D.J. The Mode of Hedgehog Binding to Ihog Homologues Is Not Conserved across Different Phyla. Nature 2008, 455, 979–983. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laconi, E.; Cheri, S.; Fanti, M.; Marongiu, F. Aging and Cancer: The Waning of Community Bonds. Cells 2021, 10, 2269. https://doi.org/10.3390/cells10092269
Laconi E, Cheri S, Fanti M, Marongiu F. Aging and Cancer: The Waning of Community Bonds. Cells. 2021; 10(9):2269. https://doi.org/10.3390/cells10092269
Chicago/Turabian StyleLaconi, Ezio, Samuele Cheri, Maura Fanti, and Fabio Marongiu. 2021. "Aging and Cancer: The Waning of Community Bonds" Cells 10, no. 9: 2269. https://doi.org/10.3390/cells10092269
APA StyleLaconi, E., Cheri, S., Fanti, M., & Marongiu, F. (2021). Aging and Cancer: The Waning of Community Bonds. Cells, 10(9), 2269. https://doi.org/10.3390/cells10092269