
Dynamic, Transient, and Robust Increase in the Innervation of the Inflamed Mucosa in Inflammatory Bowel Diseases

 , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mouse Housing and Husbandry
2.2. Experimental Models of Murine Colitis
2.3. RNA Extraction
2.4. Library Preparation and mRNA Sequencing
2.5. Single-Cell RNA-Seq-Assisted Generation of the Neuroglial Identifier Lists
2.6. Analyses of Publicly Available Datasets
2.7. Statistical Analysis of Differential Gene Expression
2.8. Immunohistological Staining and Analyses
3. Results
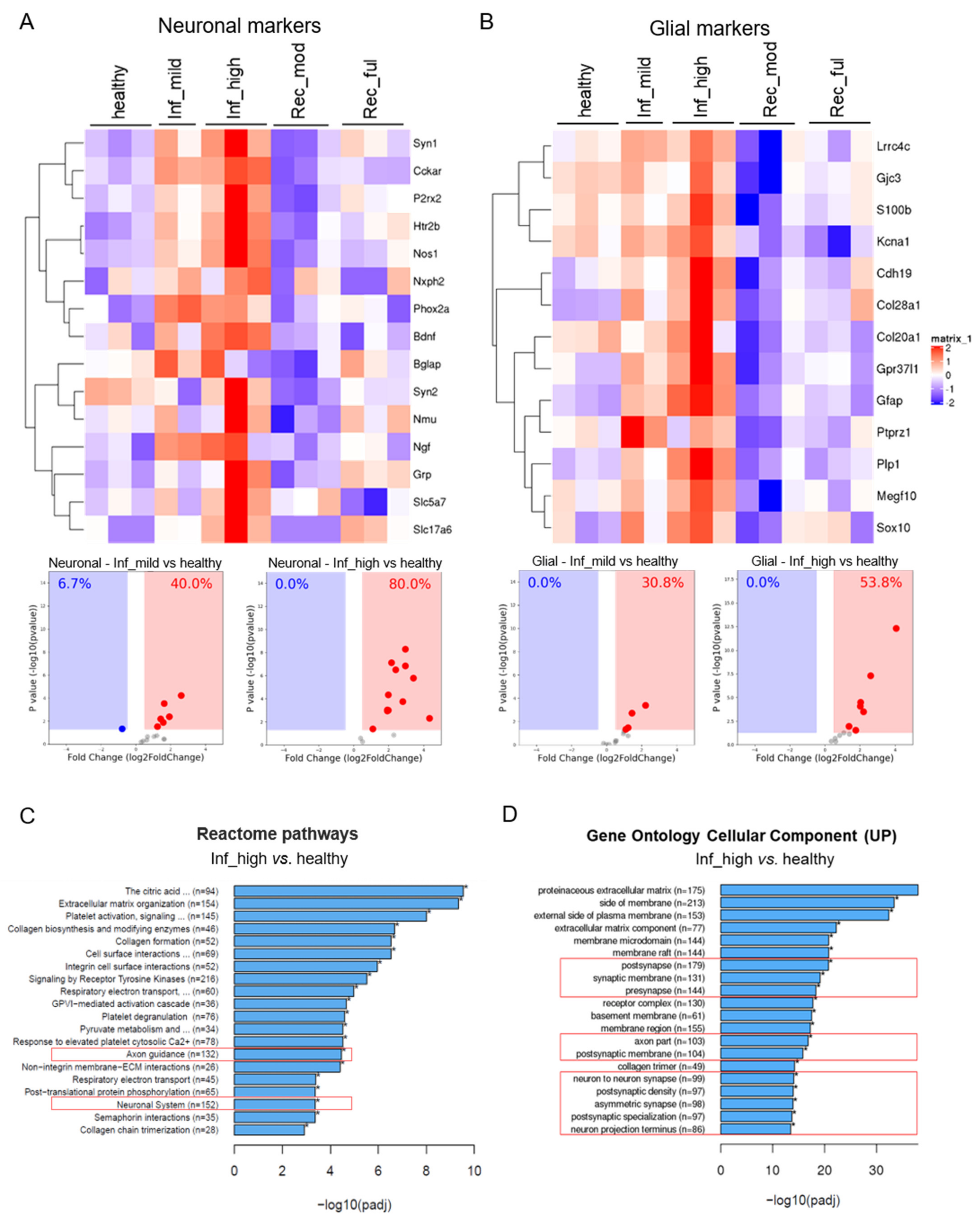
3.1. Experimental Colitis Causes an Increased Neuroglial Repertoire
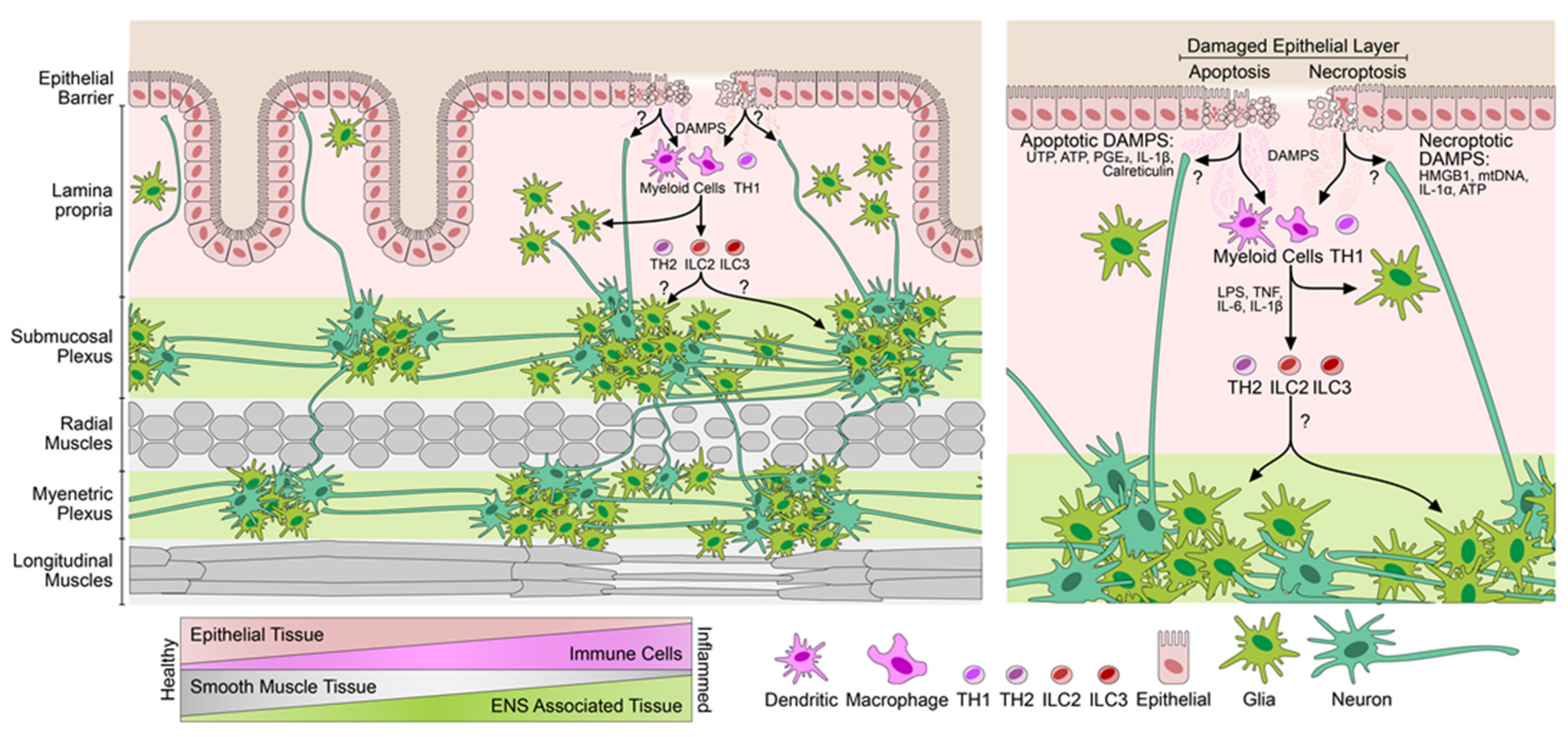
3.2. Increase in the Neuroglial Repertoire Is Accompanied by Heightened Innervation of the Inflamed Mucosa and Higher Barrier Disruption
3.3. The Neuroglial Repertoire Is Increased in the Inflamed Tissues of IBD Patients
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yarandi, S.S.; Kulkarni, S.; Saha, M.; Sylvia, K.E.; Sears, C.L.; Pasricha, P.J. Intestinal Bacteria Maintain Adult Enteric Nervous System and Nitrergic Neurons via Toll-like Receptor 2-induced Neurogenesis in Mice. Gastroenterology 2020, 159, 200–213.e8. [Google Scholar] [CrossRef]
- Obata, Y.; Castano, A.; Boeing, S.; Bon-Frauches, A.C.; Fung, C.; Fallesen, T.; de Aguero, M.G.; Yilmaz, B.; Lopes, R.; Huseynova, A.; et al. Neuronal programming by microbiota regulates intestinal physiology. Nature 2020, 578, 284–289. [Google Scholar] [CrossRef]
- Yoo, B.B.; Mazmanian, S.K. The Enteric Network: Interactions between the Immune and Nervous Systems of the Gut. Immunity 2017, 46, 910–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassotti, G.; Villanacci, V.; Nascimbeni, R.; Cadei, M.; Fisogni, S.; Antonelli, E.; Corazzi, N.; Salerni, B. Enteric neuroglial apoptosis in inflammatory bowel diseases. J. Crohns. Colitis. 2009, 3, 264–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vadder, F.; Grasset, E.; Manneras Holm, L.; Karsenty, G.; Macpherson, A.J.; Olofsson, L.E.; Backhed, F. Gut microbiota regulates maturation of the adult enteric nervous system via enteric serotonin networks. Proc. Natl. Acad. Sci. USA 2018, 115, 6458–6463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klose, C.S.N.; Mahlakoiv, T.; Moeller, J.B.; Rankin, L.C.; Flamar, A.L.; Kabata, H.; Monticelli, L.A.; Moriyama, S.; Putzel, G.G.; Rakhilin, N.; et al. The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nature 2017, 549, 282–286. [Google Scholar] [CrossRef]
- Wallrapp, A.; Riesenfeld, S.J.; Burkett, P.R.; Abdulnour, R.E.; Nyman, J.; Dionne, D.; Hofree, M.; Cuoco, M.S.; Rodman, C.; Farouq, D.; et al. Erratum: The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature 2017, 551, 658. [Google Scholar] [CrossRef] [Green Version]
- Boyapati, R.K.; Rossi, A.G.; Satsangi, J.; Ho, G.T. Gut mucosal DAMPs in IBD: From mechanisms to therapeutic implications. Mucosal. Immunol. 2016, 9, 567–582. [Google Scholar] [CrossRef] [Green Version]
- Patankar, J.V.; Becker, C. Cell death in the gut epithelium and implications for chronic inflammation. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 543–556. [Google Scholar] [CrossRef]
- Drokhlyansky, E.; Smillie, C.S.; Van Wittenberghe, N.; Ericsson, M.; Griffin, G.K.; Eraslan, G.; Dionne, D.; Cuoco, M.S.; Goder-Reiser, M.N.; Sharova, T.; et al. The Human and Mouse Enteric Nervous System at Single-Cell Resolution. Cell 2020, 182, 1606–1622.e23. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.M.; Schneider, S.; Smith-Edwards, K.M.; Mafra, F.; Leembruggen, A.J.L.; Gonzalez, M.V.; Kothakapa, D.R.; Anderson, J.B.; Maguire, B.A.; Gao, T.; et al. scRNA-Seq Reveals New Enteric Nervous System Roles for GDNF, NRTN, and TBX3. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 1548–1592.e1. [Google Scholar] [CrossRef]
- Zeisel, A.; Hochgerner, H.; Lonnerberg, P.; Johnsson, A.; Memic, F.; van der Zwan, J.; Haring, M.; Braun, E.; Borm, L.E.; La Manno, G.; et al. Molecular Architecture of the Mouse Nervous System. Cell 2018, 174, 999–1014.e22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morarach, K.; Mikhailova, A.; Knoflach, V.; Memic, F.; Kumar, R.; Li, W.; Ernfors, P.; Marklund, U. Diversification of molecularly defined myenteric neuron classes revealed by single-cell RNA sequencing. Nat. Neurosci. 2021, 24, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Belkind-Gerson, J.; Graham, H.K.; Reynolds, J.; Hotta, R.; Nagy, N.; Cheng, L.; Kamionek, M.; Shi, H.N.; Aherne, C.M.; Goldstein, A.M.; et al. Colitis promotes neuronal differentiation of Sox2+ and PLP1+ enteric cells. Sci. Rep. 2017, 7, 2525. [Google Scholar] [CrossRef] [PubMed]
- Belkind-Gerson, J.; Hotta, R.; Nagy, N.; Thomas, A.R.; Graham, H.; Cheng, L. Colitis induces enteric neurogenesis through a 5-HT4-dependent mechanism. Inflamm. Bowel. Dis. 2015, 21, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Sanovic, S.; Lamb, D.P.; Blennerhassett, M.G. Damage to the enteric nervous system in experimental colitis. Am. J. Pathol. 1999, 155, 1051–1057. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, S.; Micci, M.A.; Leser, J.; Shin, C.; Tang, S.C.; Fu, Y.Y. Adult enteric nervous system in health is maintained by a dynamic balance between neuronal apoptosis and neurogenesis. Proc. Natl. Acad. Sci. USA 2017, 114, E3709–E3718. [Google Scholar] [CrossRef] [Green Version]
- Joseph, N.M.; He, S.; Quintana, E.; Kim, Y.G.; Nunez, G.; Morrison, S.J. Enteric glia are multipotent in culture but primarily form glia in the adult rodent gut. J. Clin. Invest. 2011, 121, 3398–3411. [Google Scholar] [CrossRef]
- Patankar, J.V.; Muller, T.M.; Kantham, S.; Acera, M.G.; Mascia, F.; Scheibe, K.; Mahapatro, M.; Heichler, C.; Yu, Y.; Li, W.; et al. E-type prostanoid receptor 4 drives resolution of intestinal inflammation by blocking epithelial necroptosis. Nat. Cell. Biol. 2021, 23, 796–807. [Google Scholar] [CrossRef]
- Gonzalez Acera, M.; Patankar, J.V.; Diemand, L.; Siegmund, B.; Neurath, M.F.; Wirtz, S.; Becker, C. Comparative Transcriptomics of IBD Patients Indicates Induction of Type 2 Immunity Irrespective of the Disease Ideotype. Front Med. 2021, 8, 664045. [Google Scholar] [CrossRef]
- Eden, K. Adoptive Transfer Colitis. Methods Mol. Biol. 2019, 1960, 207–214. [Google Scholar]
- Wirtz, S.; Popp, V.; Kindermann, M.; Gerlach, K.; Weigmann, B.; Fichtner-Feigl, S.; Neurath, M.F. Chemically induced mouse models of acute and chronic intestinal inflammation. Nat. Protoc. 2017, 12, 1295–1309. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Haber, A.L.; Biton, M.; Rogel, N.; Herbst, R.H.; Shekhar, K.; Smillie, C.; Burgin, G.; Delorey, T.M.; Howitt, M.R.; Katz, Y.; et al. A single-cell survey of the small intestinal epithelium. Nature 2017, 551, 333–339. [Google Scholar] [CrossRef]
- Smillie, C.S.; Biton, M.; Ordovas-Montanes, J.; Sullivan, K.M.; Burgin, G.; Graham, D.B.; Herbst, R.H.; Rogel, N.; Slyper, M.; Waldman, J.; et al. Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis. Cell 2019, 178, 714–730.e22. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.H.; Swathi, Y.; Tan, S.; Goh, J.; Seishima, R.; Murakami, K.; Oshima, M.; Tsuji, T.; Phuah, P.; Tan, L.T.; et al. AQP5 enriches for stem cells and cancer origins in the distal stomach. Nature 2020, 578, 437–443. [Google Scholar] [CrossRef]
- Gaudino, S.J.; Beaupre, M.; Lin, X.; Joshi, P.; Rathi, S.; McLaughlin, P.A.; Kempen, C.; Mehta, N.; Eskiocak, O.; Yueh, B.; et al. IL-22 receptor signaling in Paneth cells is critical for their maturation, microbiota colonization, Th17-related immune responses, and anti-Salmonella immunity. Mucosal. Immunol. 2021, 14, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.D.; Thacker, M.; Castelucci, P.; Bagyanszki, M.; Epstein, M.L.; Furness, J.B. Immunohistochemical analysis of neuron types in the mouse small intestine. Cell. Tissue. Res. 2008, 334, 147–161. [Google Scholar] [CrossRef]
- Ochoa-Cortes, F.; Turco, F.; Linan-Rico, A.; Soghomonyan, S.; Whitaker, E.; Wehner, S.; Cuomo, R.; Christofi, F.L. Enteric Glial Cells: A New Frontier in Neurogastroenterology and Clinical Target for Inflammatory Bowel Diseases. Inflamm. Bowel. Dis. 2016, 22, 433–449. [Google Scholar] [CrossRef]
- Heanue, T.A.; Pachnis, V. Expression profiling the developing mammalian enteric nervous system identifies marker and candidate Hirschsprung disease genes. Proc. Natl. Acad. Sci. USA. 2006, 103, 6919–6924. [Google Scholar] [CrossRef] [Green Version]
- Rao, M.; Nelms, B.D.; Dong, L.; Salinas-Rios, V.; Rutlin, M.; Gershon, M.D.; Corfas, G. Enteric glia express proteolipid protein 1 and are a transcriptionally unique population of glia in the mammalian nervous system. Glia 2015, 63, 2040–2057. [Google Scholar] [CrossRef] [PubMed]
- Haberman, Y.; Karns, R.; Dexheimer, P.J.; Schirmer, M.; Somekh, J.; Jurickova, I.; Braun, T.; Novak, E.; Bauman, L.; Collins, M.H.; et al. Ulcerative colitis mucosal transcriptomes reveal mitochondriopathy and personalized mechanisms underlying disease severity and treatment response. Nat. Commun. 2019, 10, 38. [Google Scholar] [CrossRef] [Green Version]
- Fenton, C.G.; Taman, H.; Florholmen, J.; Sorbye, S.W.; Paulssen, R.H. Transcriptional Signatures That Define Ulcerative Colitis in Remission. Inflamm. Bowel. Dis. 2021, 27, 94–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome. Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, M.; Rastelli, D.; Dong, L.; Chiu, S.; Setlik, W.; Gershon, M.D.; Corfas, G. Enteric Glia Regulate Gastrointestinal Motility but Are Not Required for Maintenance of the Epithelium in Mice. Gastroenterology 2017, 153, 1068–1081.e7. [Google Scholar] [CrossRef]
- Bush, T.G.; Savidge, T.C.; Freeman, T.C.; Cox, H.J.; Campbell, E.A.; Mucke, L.; Johnson, M.H.; Sofroniew, M.V. Fulminant jejuno-ileitis following ablation of enteric glia in adult transgenic mice. Cell 1998, 93, 189–201. [Google Scholar] [CrossRef] [Green Version]
- Ibiza, S.; Garcia-Cassani, B.; Ribeiro, H.; Carvalho, T.; Almeida, L.; Marques, R.; Misic, A.M.; Bartow-McKenney, C.; Larson, D.M.; Pavan, W.J.; et al. Glial-cell-derived neuroregulators control type 3 innate lymphoid cells and gut defence. Nature 2016, 535, 440–443. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzalez Acera, M.; Bubeck, M.; Mascia, F.; Diemand, L.; Sturm, G.; Kühl, A.A.; Atreya, R.; Lie, D.C.; Neurath, M.F.; Schumann, M.; et al. Dynamic, Transient, and Robust Increase in the Innervation of the Inflamed Mucosa in Inflammatory Bowel Diseases. Cells 2021, 10, 2253. https://doi.org/10.3390/cells10092253
Gonzalez Acera M, Bubeck M, Mascia F, Diemand L, Sturm G, Kühl AA, Atreya R, Lie DC, Neurath MF, Schumann M, et al. Dynamic, Transient, and Robust Increase in the Innervation of the Inflamed Mucosa in Inflammatory Bowel Diseases. Cells. 2021; 10(9):2253. https://doi.org/10.3390/cells10092253
Chicago/Turabian StyleGonzalez Acera, Miguel, Marvin Bubeck, Fabrizio Mascia, Leonard Diemand, Gregor Sturm, Anja A. Kühl, Raja Atreya, Dieter Chichung Lie, Markus F. Neurath, Michael Schumann, and et al. 2021. "Dynamic, Transient, and Robust Increase in the Innervation of the Inflamed Mucosa in Inflammatory Bowel Diseases" Cells 10, no. 9: 2253. https://doi.org/10.3390/cells10092253
APA StyleGonzalez Acera, M., Bubeck, M., Mascia, F., Diemand, L., Sturm, G., Kühl, A. A., Atreya, R., Lie, D. C., Neurath, M. F., Schumann, M., Klose, C. S. N., Trajanoski, Z., Becker, C., & Patankar, J. V. (2021). Dynamic, Transient, and Robust Increase in the Innervation of the Inflamed Mucosa in Inflammatory Bowel Diseases. Cells, 10(9), 2253. https://doi.org/10.3390/cells10092253