
Maternal High-Fat Diet Promotes Abdominal Aortic Aneurysm Expansion in Adult Offspring by Epigenetic Regulation of IRF8-Mediated Osteoclast-like Macrophage Differentiation

 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Experimental Animals
2.2. Mouse Aneurysm Induction Model
2.3. Haemodynamic Analysis
2.4. Vessel Measurement and Histological Analysis
2.5. Immunohistochemical Analysis
2.6. Quantitative Real-Time Polymerase Chain Reaction (qPCR)
2.7. Ex Vivo MMP Activity
2.8. In Vitro Differentiation of BMDMs into Osteoclast-like Macrophages
2.9. Western Blot Analysis
2.10. ChIP Assay
2.11. Statistical Analysis
3. Results
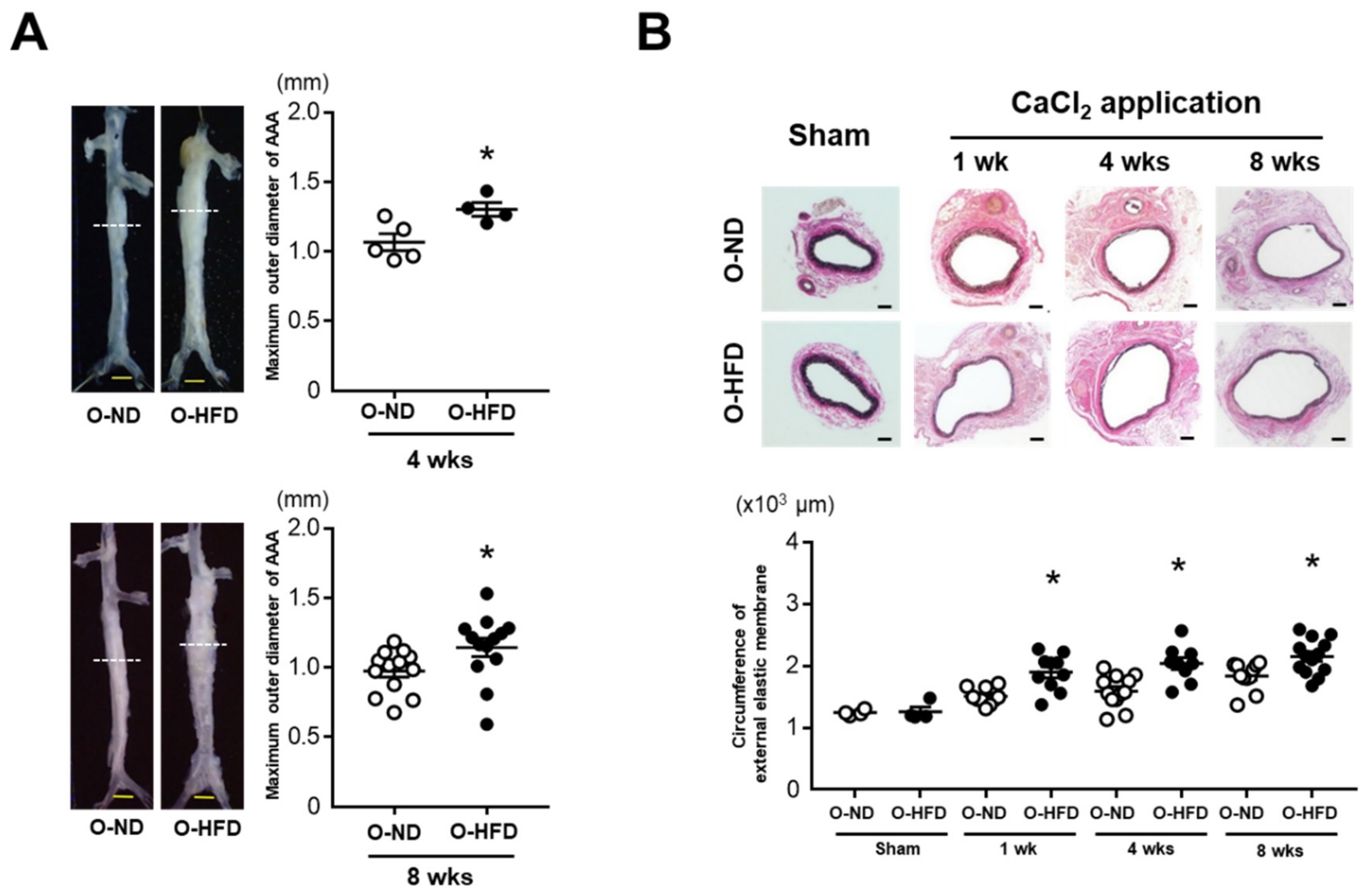
3.1. Maternal HFD Exaggerates AAA Development in Offspring
3.2. Maternal HFD Does Not Affect Accumulation of Classically Activated Macrophage
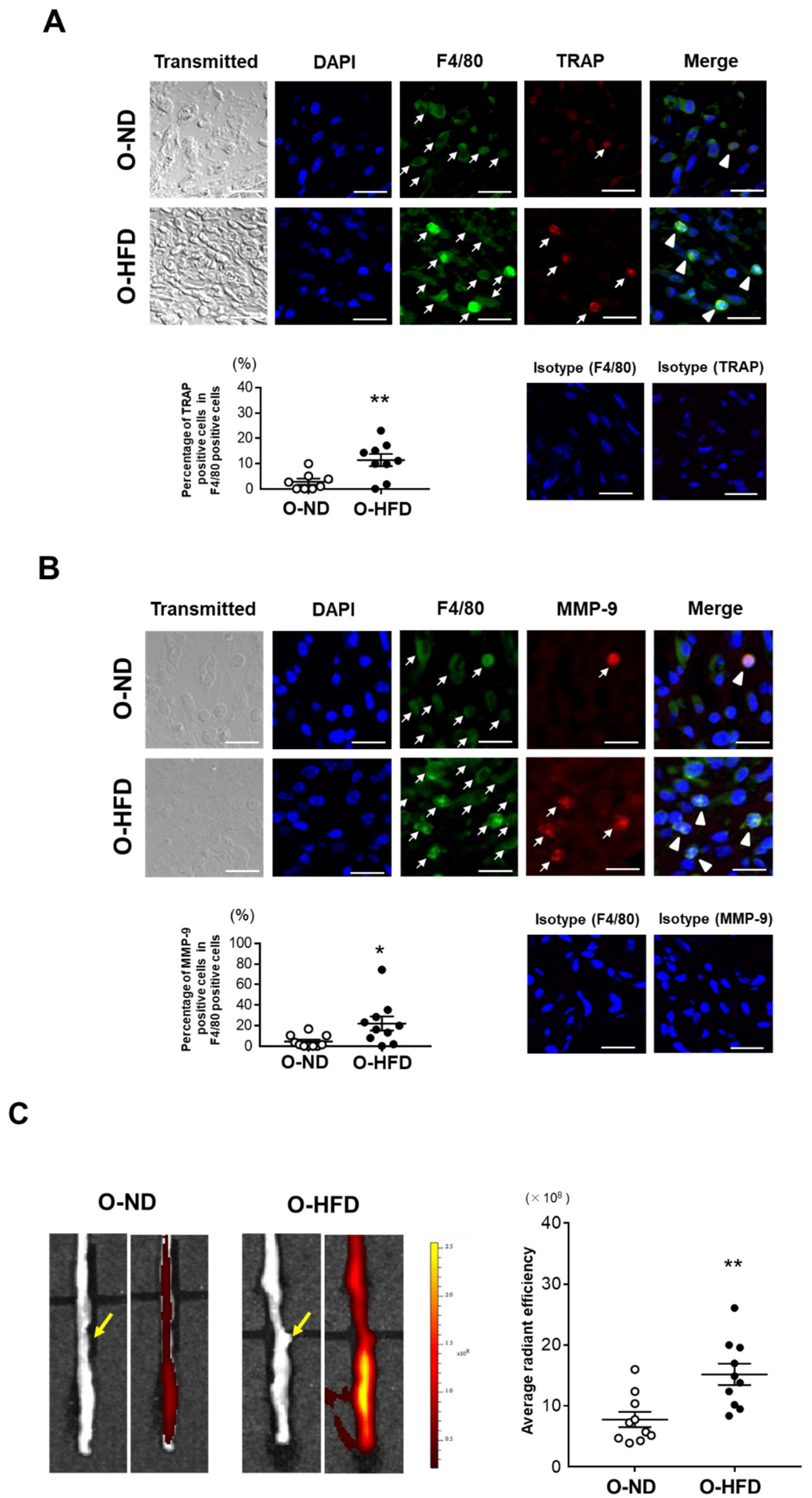
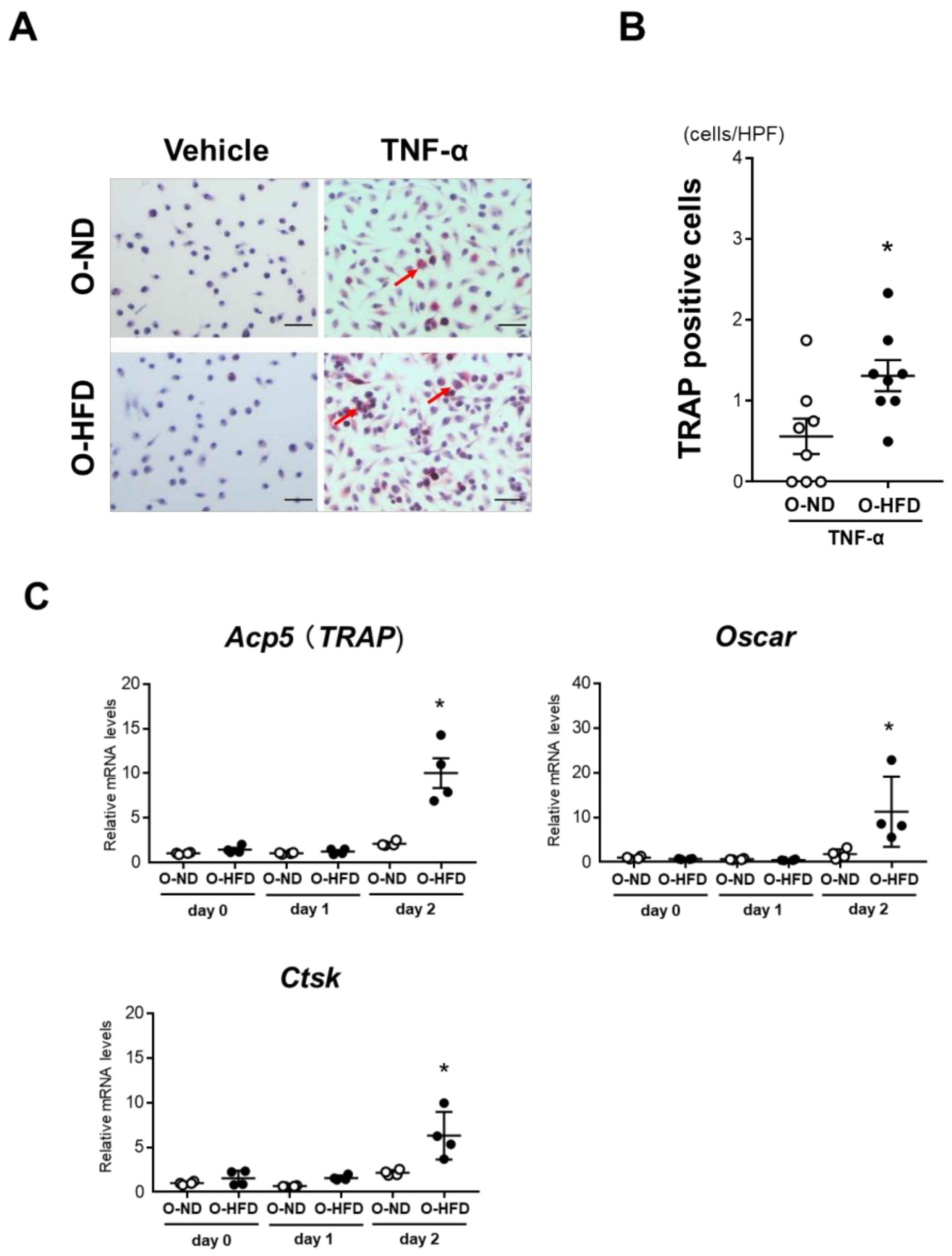
3.3. TRAP-Positive Macrophage Accumulation Is Enhanced in O-HFD
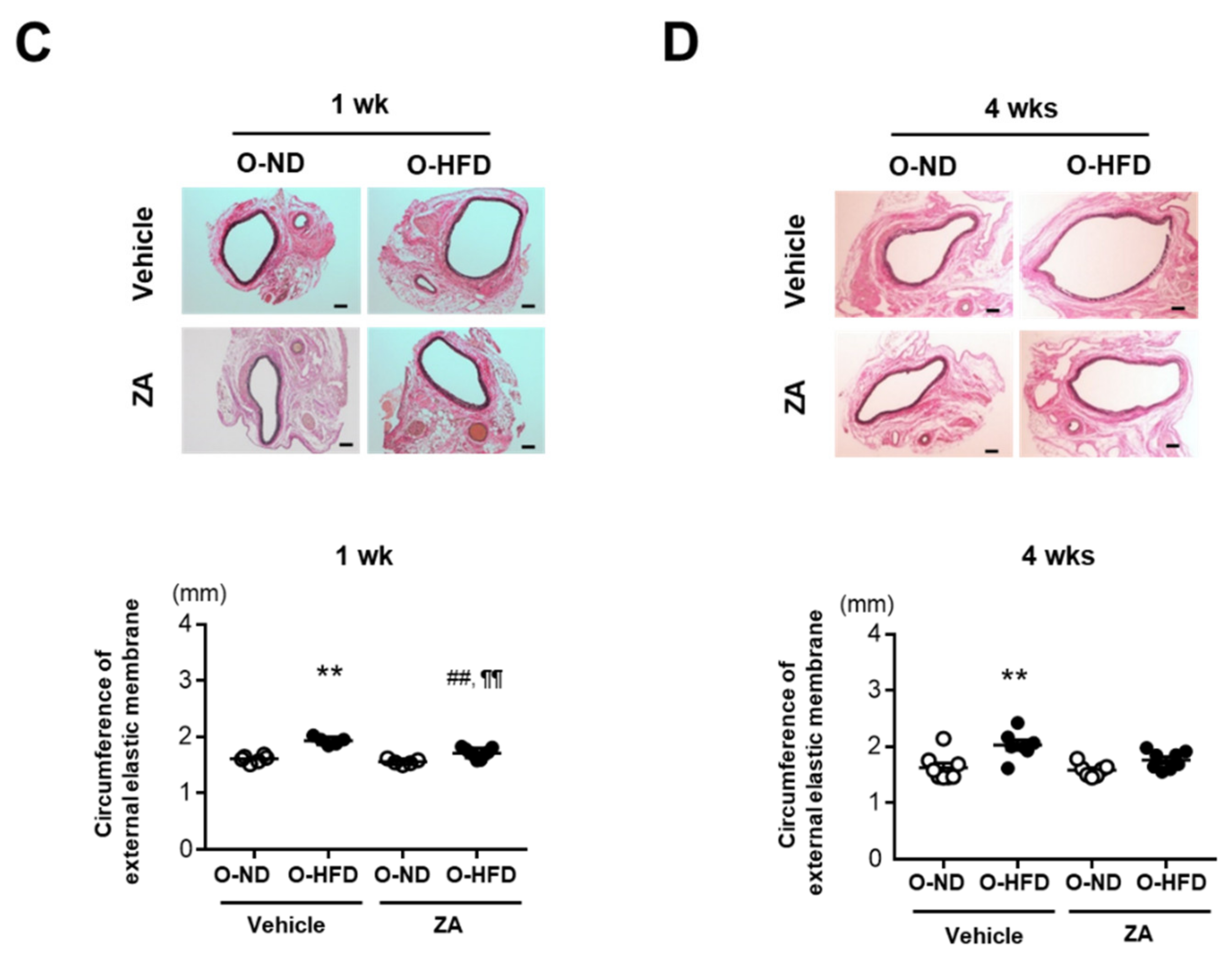
3.4. ZA Treatment Eliminates AAA Development in O-HFD
3.5. TNF-α-Induced Osteoclast-like Macrophage Differentiation Is Enhanced in BMDMs of O-HFD
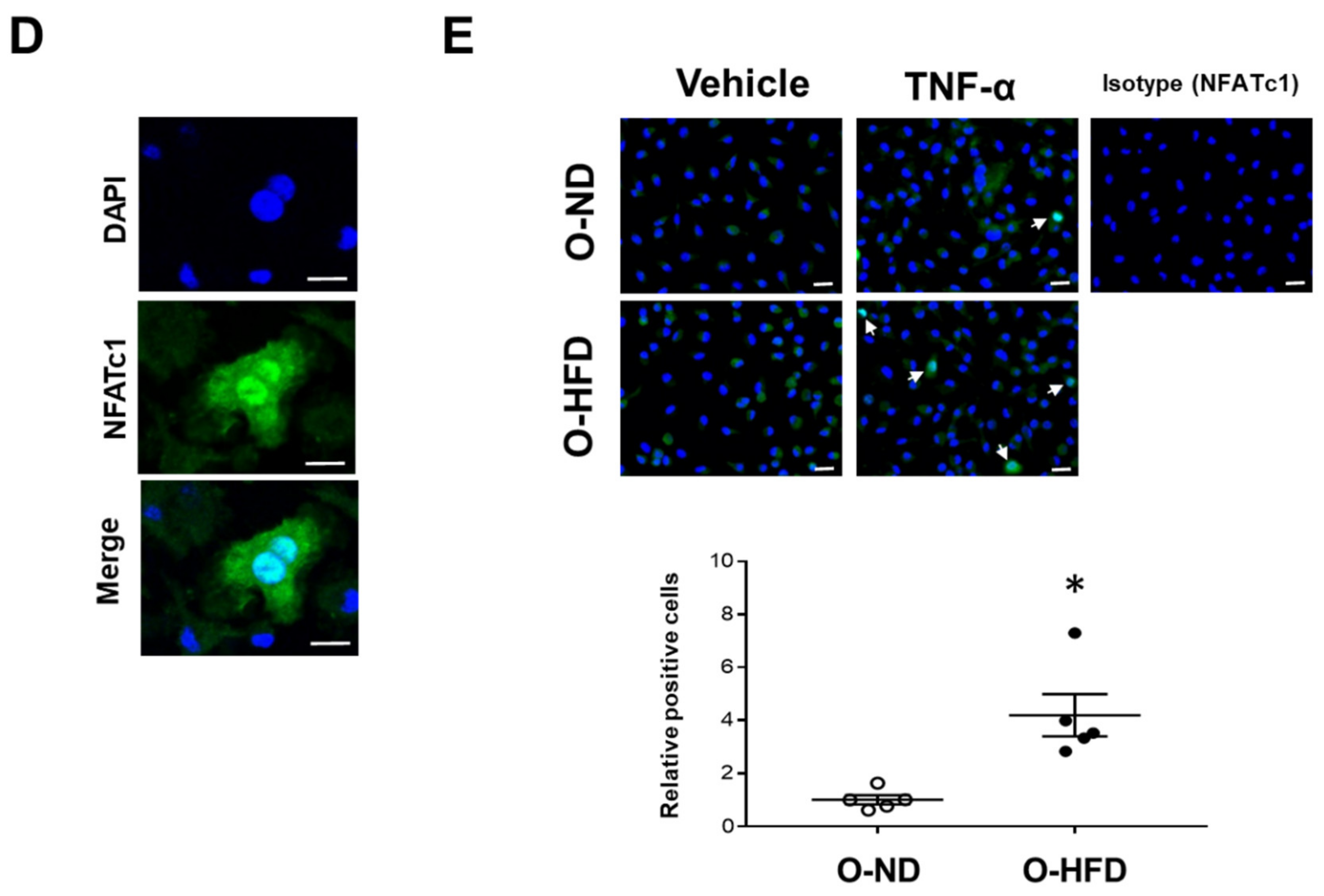
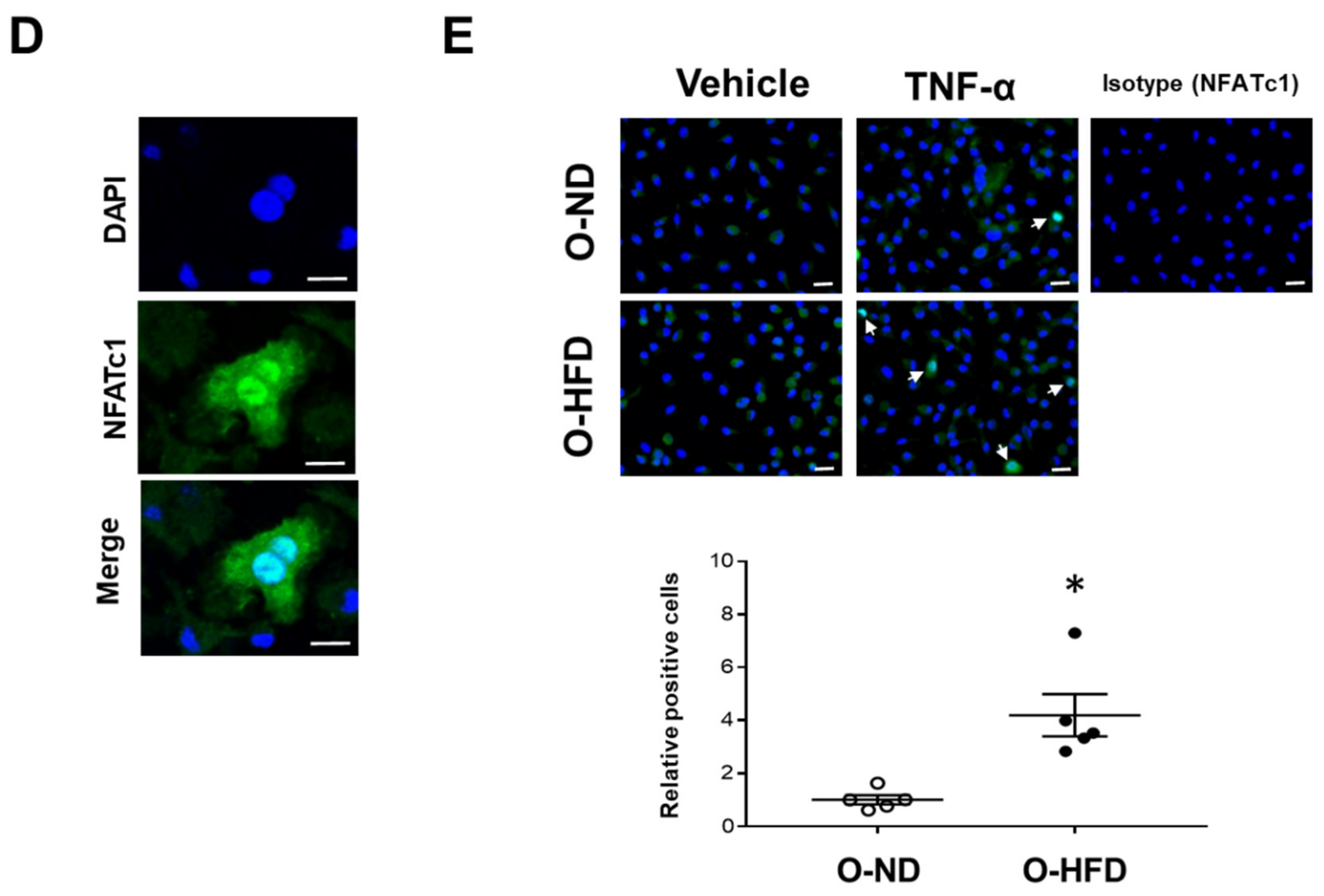
3.6. TNF-α-Induced NFATc1 Expression Is Increased in BMDMs of O-HFD
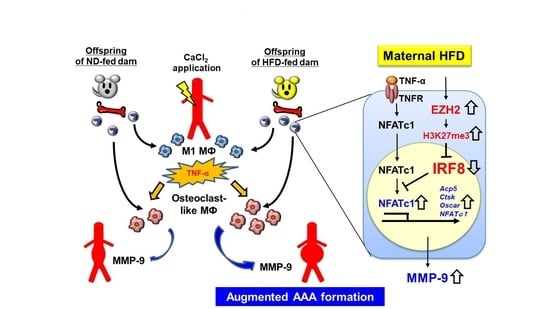
3.7. Expression of Transcriptional Repressor IRF-8 Is Decreased in BMDMs of O-HFD
3.8. EZH2 Inhibitor Restores IRF8 Expression and Ameliorates Differentiation of BMDMs into Osteoclast-like Macrophages in O-HFD
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Drake, A.J.; Reynolds, R.M. Impact of maternal obesity on offspring obesity and cardiometabolic disease risk. Reproduction 2010, 140, 387–398. [Google Scholar] [CrossRef]
- Reynolds, R.M.; Allan, K.M.; Raja, E.A.; Bhattacharya, S.; McNeill, G.; Hannaford, P.C.; Sarwar, N.; Lee, A.J.; Bhattacharya, S.; Norman, J.E. Maternal obesity during pregnancy and premature mortality from cardiovascular event in adult offspring: Follow-up of 1,323,275 person years. BMJ 2013, 347, f4539. [Google Scholar] [CrossRef] [Green Version]
- Godfrey, K.M.; Reynolds, R.M.; Prescott, S.L.; Nyirenda, M.; Jaddoe, V.W.; Eriksson, J.G.; Broekman, B.F. Influence of maternal obesity on the long-term health of offspring. Lancet Diabetes Endocrinol. 2017, 5, 53–64. [Google Scholar] [CrossRef] [Green Version]
- Napoli, C.; Infante, T.; Casamassimi, A. Maternal-foetal epigenetic interactions in the beginning of cardiovascular damage. Cardiovasc. Res. 2011, 92, 367–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleming, T.P.; Watkins, A.J.; Velazquez, M.A.; Mathers, J.C.; Prentice, A.M.; Stephenson, J.; Barker, M.; Saffery, R.; Yajnik, C.S.; Eckert, J.J.; et al. Origins of lifetime health around the time of conception: Causes and consequences. Lancet 2018, 391, 1842–1852. [Google Scholar] [CrossRef]
- Hjort, L.; Martino, D.; Grunnet, L.G.; Naeem, H.; Maksimovic, J.; Olsson, A.H.; Zhang, C.; Ling, C.; Olsen, S.F.; Saffery, R.; et al. Gestational diabetes and maternal obesity are associated with epigenome-wide methylation changes in children. JCI Insight 2018, 3, e122572. [Google Scholar] [CrossRef]
- Greco, C.M.; Condorelli, G. Epigenetic modifications and noncoding RNAs in cardiac hypertrophy and failure. Nat. Rev. Cardiol. 2015, 12, 488–497. [Google Scholar] [CrossRef]
- Rosa-Garrido, M.; Chapski, D.J.; Vondriska, T.M. Epigenomes in Cardiovascular Disease. Circ. Res. 2018, 122, 1586–1607. [Google Scholar] [CrossRef]
- Stratton, M.S.; Farina, F.M.; Elia, L. Epigenetics and vascular diseases. J. Mol. Cell. Cardiol. 2019, 133, 148–163. [Google Scholar] [CrossRef]
- Wakana, N.; Irie, D.; Kikai, M.; Terada, K.; Yamamoto, K.; Kawahito, H.; Kato, T.; Ogata, T.; Ueyama, T.; Matoba, S.; et al. Maternal high-fat diet exaggerates atherosclerosis in adult offspring by augmenting periaortic adipose tissue-specific proinflammatory response. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 558–569. [Google Scholar] [CrossRef] [Green Version]
- Wada, N.; Yamada, H.; Motoyama, S.; Saburi, M.; Sugimoto, T.; Kubota, H.; Miyawaki, D.; Wakana, N.; Kami, D.; Ogata, T.; et al. Maternal high-fat diet exaggerates diet-induced insulin resistance in adult offspring by enhancing inflammasome activation through noncanonical pathway of caspase-11. Mol. Metab. 2020, 37, 100988. [Google Scholar] [CrossRef] [PubMed]
- Kent, K.C. Abdominal aortic aneurysms. N. Engl. J. Med. 2014, 371, 2101–2108. [Google Scholar] [CrossRef] [PubMed]
- Golledge, J. Abdominal aortic aneurysm: Update on pathogenesis and medical treatments. Nat. Rev. Cardiol. 2019, 16, 225–242. [Google Scholar] [CrossRef]
- Lindeman, J.H.; Matsumura, J.S. Pharmacologic management of aneurysms. Circ. Res. 2019, 124, 631–646. [Google Scholar] [CrossRef] [PubMed]
- Raffort, J.; Lareyre, F.; Clément, M.; Hassen-Khodja, R.; Chinetti, G.; Mallat, Z. Monocytes and macrophages in abdominal aortic aneurysm. Nat. Rev. Cardiol. 2017, 14, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Quintana, R.A.; Taylor, W.R. Cellular Mechanisms of Aortic Aneurysm Formation. Circ. Res. 2019, 124, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Takei, Y.; Tanaka, T.; Kent, K.C.; Yamanouchi, D. Osteoclastogenic Differentiation of Macrophages in the Development of Abdominal Aortic Aneurysms. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1962–1971. [Google Scholar] [CrossRef] [Green Version]
- Sénémaud, J.; Caligiuri, G.; Etienne, H.; Delbosc, S.; Michel, J.B.; Coscas, R. Translational Relevance and Recent Advances of Animal Models of Abdominal Aortic Aneurysm. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Fukamizu, A.; Sugimura, K.; Takimoto, E.; Sugiyama, F.; Seo, M.S.; Takahashi, S.; Hatae, T.; Kajiwara, N.; Yagami, K.; Murakami, K. Chimeric renin-angiotensin system demonstrates sustained increase in blood pressure of transgenic mice carrying both human renin and human angiotensinogen genes. J. Biol. Chem. 1993, 268, 11617–11621. [Google Scholar] [CrossRef]
- Sheth, R.A.; Maricevich, M.; Mahmood, U. In vivo optical molecular imaging of matrix metalloproteinase activity in abdominal aortic aneurysms correlates with treatment effects on growth rate. Atherosclerosis 2010, 212, 181–187. [Google Scholar] [CrossRef] [Green Version]
- Khallou-Laschet, J.; Varthaman, A.; Fornasa, G.; Compain, C.; Gaston, A.T.; Clement, M.; Dussiot, M.; Levillain, O.; Graff-Dubois, S.; Nicoletti, A.; et al. Macrophage plasticity in experimental atherosclerosis. PLoS ONE 2010, 5, e8852. [Google Scholar] [CrossRef] [Green Version]
- Takayanagi, H.; Kim, S.; Koga, T.; Nishina, H.; Isshiki, M.; Yoshida, H.; Saiura, A.; Isobe, M.; Yokochi, T.; Inoue, J.; et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev. Cell 2002, 3, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Rao, K.; Xiong, H.; Gagnidze, K.; Li, F.; Horvath, C.; Plevy, S. Activation of the murine interleukin-12 p40 promoter by functional interactions between NFAT and ICSBP. J. Biol. Chem. 2003, 278, 39372–39382. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Takami, M.; Yamada, A.; Wang, X.; Koga, T.; Hu, X.; Tamura, T.; Ozato, K.; Choi, Y.; Ivashkiv, L.B.; et al. Interferon regulatory factor-8 regulates bone metabolism by suppressing osteoclastogenesis. Nat. Med. 2009, 15, 1066–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivashkiv, L.B.; Zhao, B.; Park-Min, K.H.; Takami, M. Feedback inhibition of osteoclastogenesis during inflammation by IL-10, M-CSF receptor shedding, and induction of IRF8. Ann. N. Y. Acad. Sci. 2011, 1237, 88–94. [Google Scholar] [CrossRef] [Green Version]
- Asagiri, M.; Sato, K.; Usami, T.; Ochi, S.; Nishina, H.; Yoshida, H.; Morita, I.; Wagner, E.F.; Mak, T.W.; Serfling, E.; et al. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J. Exp. Med. 2005, 202, 1261–1269. [Google Scholar] [CrossRef] [Green Version]
- Fang, C.; Qiao, Y.; Mun, S.H.; Lee, M.J.; Murata, K.; Bae, S.; Zhao, B.; Park-Min, K.H.; Ivashkiv, L.B. Cutting Edge: EZH2 Promotes Osteoclastogenesis by Epigenetic Silencing of the Negative Regulator IRF8. J. Immunol. 2016, 196, 4452–4456. [Google Scholar] [CrossRef]
- Police, S.B.; Thatcher, S.E.; Charnigo, R.; Daugherty, A.; Cassis, L.A. Obesity promotes inflammation in periaortic adipose tissue and angiotensin II-induced abdominal aortic aneurysm formation. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1458–1464. [Google Scholar] [CrossRef] [Green Version]
- Police, S.B.; Putnam, K.; Thatcher, S.; Batifoulier-Yiannikouris, F.; Daugherty, A.; Cassis, L.A. Weight loss in obese C57BL/6 mice limits adventitial expansion of established angiotensin II-induced abdominal aortic aneurysms. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1932–H1938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopal, K.; Kumar, K.; Nandini, R.; Jahan, P.; Kumar, M.J. High fat diet containing cholesterol induce aortic aneurysm through recruitment and proliferation of circulating agranulocytes in apoE knock out mice model. J. Thromb. Thrombolysis 2010, 30, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Howatt, D.A.; Balakrishnan, A.; Graham, M.J.; Mullick, A.E.; Daugherty, A. Hypercholesterolemia Induced by a PCSK9 Gain-of-Function Mutation Augments Angiotensin II-Induced Abdominal Aortic Aneurysms in C57BL/6 Mice-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1753–1757. [Google Scholar] [CrossRef] [Green Version]
- Longo, G.M.; Xiong, W.; Greiner, T.C.; Zhao, Y.; Fiotti, N.; Baxter, B.T. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J. Clin. Investig. 2002, 110, 625–632. [Google Scholar] [CrossRef]
- Nosoudi, N.; Nahar-Gohad, P.; Sinha, A.; Chowdhury, A.; Gerard, P.; Carsten, C.G.; Gray, B.H.; Vyavahare, N.R. Prevention of abdominal aortic aneurysm progression by targeted inhibition of matrix metalloproteinase activity with batimastat-loaded nanoparticles. Circ. Res. 2015, 117, e80–e89. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Kelly, M.; Takei, Y.; Yamanouchi, D. RANKL-mediated osteoclastogenic differentiation of macrophages in the abdominal aorta of angiotensin II-infused apolipoprotein E knockout mice. J. Vasc. Surg. 2018, 68, 48S–59S.e1. [Google Scholar] [CrossRef]
- Hughes, D.E.; Wright, K.R.; Uy, H.L.; Sasaki, A.; Yoneda, T.; Roodman, G.D.; Mundy, G.R.; Boyce, B.F. Bisphosphonates promote apoptosis in murine osteoclasts in vitro and in vivo. J. Bone Miner. Res. 1995, 10, 1478–1487. [Google Scholar] [CrossRef]
- Fleisch, H. Development of bisphosphonates. Breast Cancer Res. 2002, 4, 30–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokota, K.; Sato, K.; Miyazaki, T.; Aizaki, Y.; Tanaka, S.; Sekikawa, M.; Kozu, N.; Kadono, Y.; Oda, H.; Mimura, T. Characterization and Function of Tumor Necrosis Factor and Interleukin-6-Induced Osteoclasts in Rheumatoid Arthritis. Arthritis Rheumatol. 2021, 73, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.B.; Ruan, C.C.; Lin, J.R.; Xu, L.; Chen, X.H.; Du, Y.N.; Fu, M.X.; Kong, L.R.; Zhu, D.L.; Gao, P.J. Perivascular Adipose Tissue-Derived PDGF-D Contributes to Aortic Aneurysm Formation during Obesity. Diabetes 2018, 67, 1549–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina-Sánchez, P.; del Campo, L.; Esteban, V.; Rius, C.; Chèvre, R.; Fuster, J.J.; Ferrer, M.; Redondo, J.M.; Andrés, V. Defective p27 phosphorylation at serine 10 affects ZA vascular reactivity and increases abdominal aortic aneurysm development via Cox-2 activation. J. Mol. Cell. Cardiol. 2018, 116, 5–15. [Google Scholar] [CrossRef]
- Di Gregoli, K.; Mohamad Anuar, N.N.; Bianco, R.; White, S.J.; Newby, A.C.; George, S.J.; Johnson, J.L. MicroRNA-181b Controls Atherosclerosis and Aneurysms through Regulation of TIMP-3 and Elastin. Circ. Res. 2017, 120, 49–65. [Google Scholar] [CrossRef] [Green Version]
- Ono, T.; Nakashima, T. Recent advances in osteoclast biology. Histochem. Cell Biol. 2018, 149, 325–341. [Google Scholar] [CrossRef]
- Nishikawa, K.; Nakashima, T.; Hayashi, M.; Fukunaga, T.; Kato, S.; Kodama, T.; Takahashi, S.; Calame, K.; Takayanagi, H. Blimp1-mediated repression of negative regulators is required for osteoclast differentiation. Proc. Natl. Acad. Sci. USA 2010, 107, 3117–3122. [Google Scholar] [CrossRef] [Green Version]
- Yasui, T.; Hirose, J.; Tsutsumi, S.; Nakamura, K.; Aburatani, H.; Tanaka, S. Epigenetic regulation of osteoclast differentiation: Possible involvement of Jmjd3 in the histone demethylation of Nfatc1. J. Bone Miner. Res. 2011, 26, 2665–2671. [Google Scholar] [CrossRef]
- Sugatani, T.; Vacher, J.; Hruska, K.A. A microRNA expression signature of osteoclastogenesis. Blood 2011, 117, 3648–3657. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Wei, W.; Dechow, P.C.; Wan, Y. HDAC7 inhibits osteoclastogenesis by reversing RANKL-triggered β-catenin switch. Mol. Endocrinol. 2013, 27, 325–335. [Google Scholar] [CrossRef] [Green Version]
- Dou, C.; Li, N.; Ding, N.; Liu, C.; Yang, X.; Kang, F.; Cao, Z.; Quan, H.; Hou, T.; Xu, J.; et al. HDAC2 regulates FoxO1 during RANKL-induced osteoclastogenesis. Am. J. Physiol.-Cell Physiol. 2016, 310, C780–C787. [Google Scholar] [CrossRef]
- Lozano, C.; Duroux-Richard, I.; Firat, H.; Schordan, E.; Apparailly, F. MicroRNAs: Key Regulators to Understand Osteoclast Differentiation? Front. Immunol. 2019, 10, 375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamik, J.; Pulugulla, S.H.; Zhang, P.; Sun, Q.; Lontos, K.; Macar, D.A.; Auron, P.E.; Galson, D.L. EZH2 Supports Osteoclast Differentiation and Bone Resorption via Epigenetic and Cytoplasmic Targets. J. Bone Miner. Res. 2020, 35, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, K.; Iwamoto, Y.; Kobayashi, Y.; Katsuoka, F.; Kawaguchi, S.; Tsujita, T.; Nakamura, T.; Kato, S.; Yamamoto, M.; Takayanagi, H.; et al. DNA methyltransferase 3a regulates osteoclast differentiation by coupling to an S-adenosylmethionine-producing metabolic pathway. Nat. Med. 2015, 21, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Raffort, J.; Lareyre, F.; Clement, M.; Mallat, Z. Micro-RNAs in abdominal aortic aneurysms: Insights from animal models and relevance to human disease. Cardiovasc. Res. 2016, 110, 165–177. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.Z.; Wang, F.; Gao, P.; Pei, J.F.; Liu, Y.; Xu, T.T.; Tang, X.; Fu, W.Y.; Lu, J.; Yan, Y.F.; et al. Age-Associated Sirtuin 1 Reduction in Vascular Smooth Muscle Links Vascular Senescence and Inflammation to Abdominal Aortic Aneurysm. Circ. Res. 2016, 119, 1076–1088. [Google Scholar] [CrossRef] [PubMed]
- Nakao, T.; Horie, T.; Baba, O.; Nishiga, M.; Nishino, T.; Izuhara, M.; Kuwabara, Y.; Nishi, H.; Usami, S.; Nakazeki, F.; et al. Genetic Ablation of MicroRNA-33 Attenuates Inflammation and Abdominal Aortic Aneurysm Formation via Several Anti-Inflammatory Pathways. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2161–2170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Maegdefessel, L. Non-coding RNA Contribution to Thoracic and Abdominal Aortic Aneurysm Disease Development and Progression. Front. Physiol. 2017, 8, 429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Boon, R.A.; Maegdefessel, L.; Dimmeler, S.; Jo, H. Role of Noncoding RNAs in the Pathogenesis of Abdominal Aortic Aneurysm. Circ. Res. 2019, 124, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.M.; Gallagher, K.A. Epigenetic Mechanisms in Monocytes/Macrophages Regulate Inflammation in Cardiometabolic and Vascular Disease. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 623–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lino Cardenas, C.L.; Kessinger, C.W.; MacDonald, C.; Jassar, A.S.; Isselbacher, E.M.; Jaffer, F.A.; Lindsay, M.E. Inhibition of the methyltranferase EZH2 improves aortic performance in experimental thoracic aortic aneurysm. JCI Insight 2018, 3, e97493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.H.; Roberts, C.W. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef]
- Huang, X.; Yan, J.; Zhang, M.; Wang, Y.; Chen, Y.; Fu, X.; Wei, R.; Zheng, X.L.; Liu, Z.; Zhang, X.; et al. Targeting Epigenetic Crosstalk as a Therapeutic Strategy for EZH2-Aberrant Solid Tumors. Cell 2018, 175, 186–199.e19. [Google Scholar] [CrossRef] [Green Version]
- Croucher, P.I.; McDonald, M.M.; Martin, T.J. Bone metastasis: The importance of the neighbourhood. Nat. Rev. Cancer 2016, 16, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Armenian, S.H.; Xu, L.; Ky, B.; Sun, C.; Farol, L.T.; Pal, S.K.; Douglas, P.S.; Bhatia, S.; Chao, C. Cardiovascular Disease among Survivors of Adult-Onset Cancer: A Community-Based Retrospective Cohort Study. J. Clin. Oncol. 2016, 34, 1122–1130. [Google Scholar] [CrossRef]
- Sturgeon, K.M.; Deng, L.; Bluethmann, S.M.; Zhou, S.; Trifiletti, D.M.; Jiang, C.; Kelly, S.P.; Zaorsky, N.G. A population-based study of cardiovascular disease mortality risk in US cancer patients. Eur. Heart J. 2019, 40, 3889–3897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Assis, S.; Warri, A.; Cruz, M.I.; Laja, O.; Tian, Y.; Zhang, B.; Wang, Y.; Huang, T.H.; Hilakivi-Clarke, L. High-fat or ethinyl-oestradiol intake during pregnancy increases mammary cancer risk in several generations of offspring. Version 2. Nat. Commun. 2012, 3, 1053. [Google Scholar] [CrossRef] [Green Version]
- Benesh, E.C.; Humphrey, P.A.; Wang, Q.; Moley, K.H. Maternal high-fat diet induces hyperproliferation and alters Pten/Akt signaling in prostates of offspring. Sci. Rep. 2013, 3, 3466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.R.; Zhao, H.; Lazarenko, O.P.; Blackburn, M.L.; Shankar, K. Maternal regulation of SATB2 in osteo-progeniters impairs skeletal development in offspring. FASEB J. 2020, 34, 2511–2523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Saben, J.L.; He, G.; Moley, K.H.; Long, F. Diet-Induced Metabolic Dysregulation in Female Mice Causes Osteopenia in Adult Offspring. J. Endocr. Soc. 2020, 4, bvaa028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, C.L.; van Poznak, C.; Lacchetti, C.; Kirshner, J.; Eastell, R.; Gagel, R.; Smith, S.; Edwards, B.J.; Frank, E.; Lyman, G.H.; et al. Management of Osteoporosis in Survivors of Adult Cancers with Nonmetastatic Disease: ASCO Clinical Practice Guideline. J. Clin. Oncol. 2019, 37, 2916–2946. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saburi, M.; Yamada, H.; Wada, N.; Motoyama, S.; Sugimoto, T.; Kubota, H.; Miyawaki, D.; Wakana, N.; Kami, D.; Ogata, T.; et al. Maternal High-Fat Diet Promotes Abdominal Aortic Aneurysm Expansion in Adult Offspring by Epigenetic Regulation of IRF8-Mediated Osteoclast-like Macrophage Differentiation. Cells 2021, 10, 2224. https://doi.org/10.3390/cells10092224
Saburi M, Yamada H, Wada N, Motoyama S, Sugimoto T, Kubota H, Miyawaki D, Wakana N, Kami D, Ogata T, et al. Maternal High-Fat Diet Promotes Abdominal Aortic Aneurysm Expansion in Adult Offspring by Epigenetic Regulation of IRF8-Mediated Osteoclast-like Macrophage Differentiation. Cells. 2021; 10(9):2224. https://doi.org/10.3390/cells10092224
Chicago/Turabian StyleSaburi, Makoto, Hiroyuki Yamada, Naotoshi Wada, Shinichiro Motoyama, Takeshi Sugimoto, Hiroshi Kubota, Daisuke Miyawaki, Noriyuki Wakana, Daisuke Kami, Takehiro Ogata, and et al. 2021. "Maternal High-Fat Diet Promotes Abdominal Aortic Aneurysm Expansion in Adult Offspring by Epigenetic Regulation of IRF8-Mediated Osteoclast-like Macrophage Differentiation" Cells 10, no. 9: 2224. https://doi.org/10.3390/cells10092224
APA StyleSaburi, M., Yamada, H., Wada, N., Motoyama, S., Sugimoto, T., Kubota, H., Miyawaki, D., Wakana, N., Kami, D., Ogata, T., & Matoba, S. (2021). Maternal High-Fat Diet Promotes Abdominal Aortic Aneurysm Expansion in Adult Offspring by Epigenetic Regulation of IRF8-Mediated Osteoclast-like Macrophage Differentiation. Cells, 10(9), 2224. https://doi.org/10.3390/cells10092224