
The Phenotypic Responses of Vascular Smooth Muscle Cells Exposed to Mechanical Cues



Abstract
1. Introduction
Mechanical Forces and Smooth Muscle Cells
2. How Do We Measure Smooth Muscle Phenotypic Modulation?
3. Vascular Mechanical Microenvironment
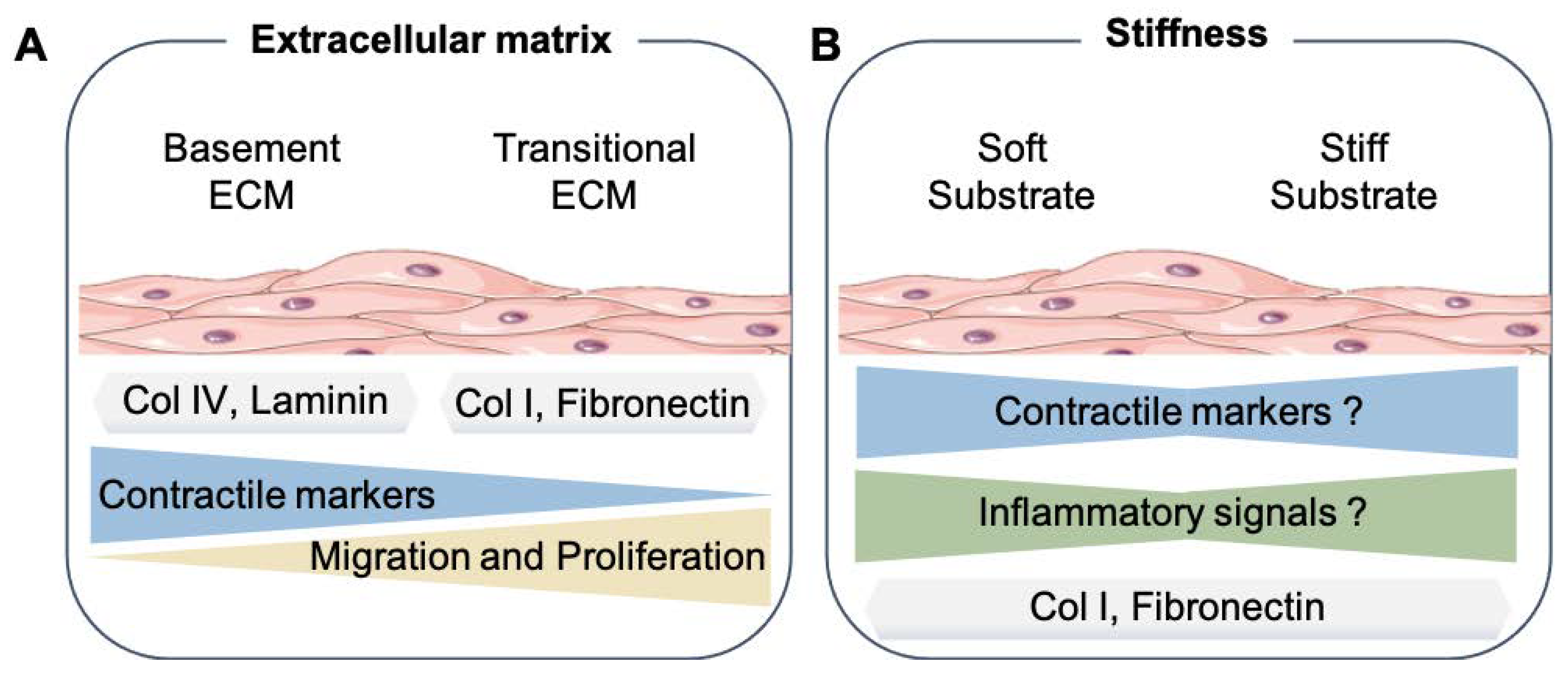
3.1. Smooth Muscle Phenotype and Extracellular Matrix Proteins
3.2. Influence of Stiffness on Smooth Muscle Phenotype
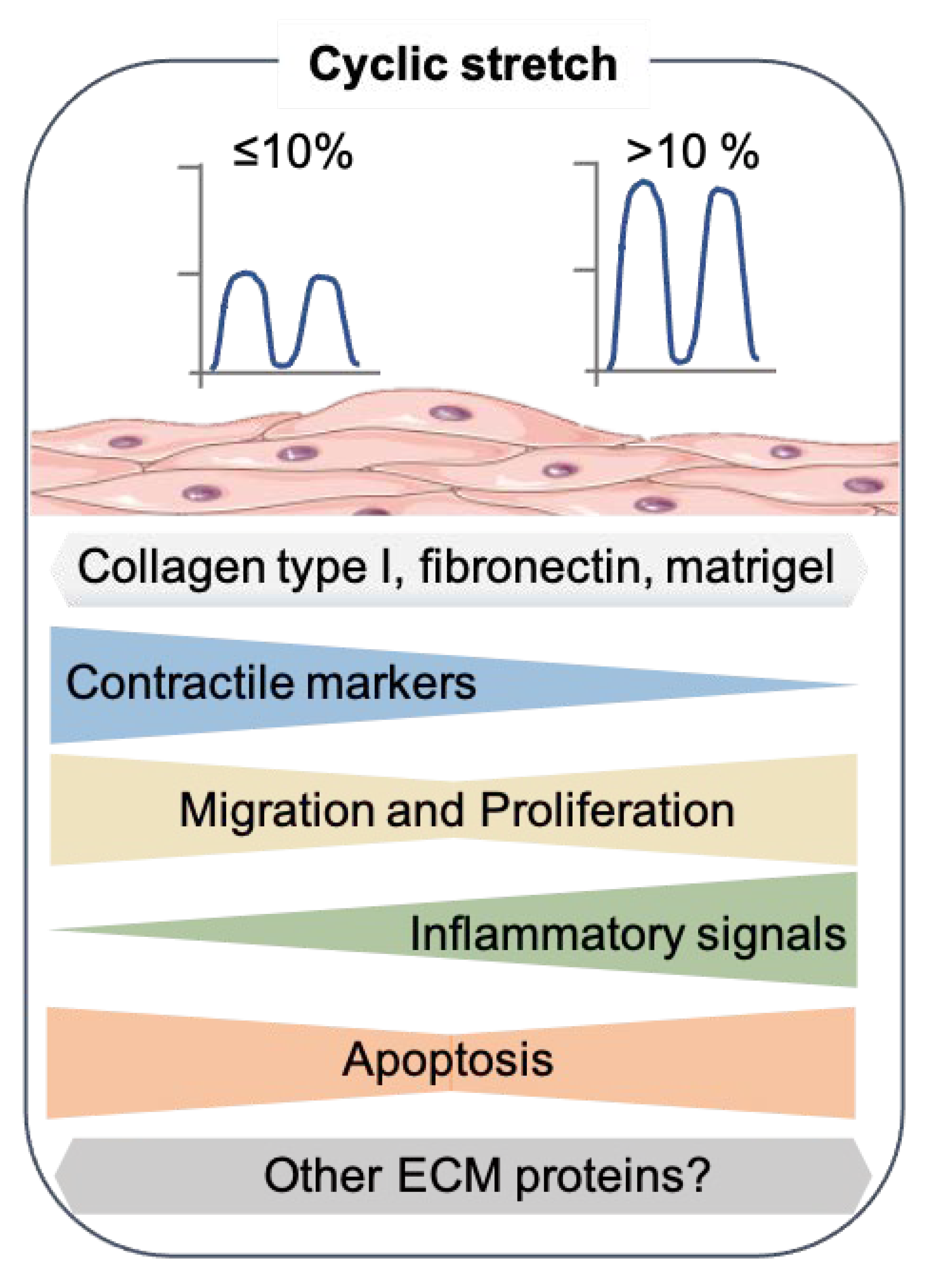
4. Cyclic Mechanical Stretch
4.1. In Vitro Modeling of the Cyclic Stretch
4.2. Effect of the Cyclic Stretch on SMC Marker Gene Expression
4.3. Other Aspects of SMC Phenotypic Modulation
4.3.1. Effect of Cyclic Stretch on SMC Migration
4.3.2. Effect of Cyclic Stretch on SMC Proliferation
4.3.3. Effect of Cyclic Stretch on SMC Apoptosis
5. Fluid Shear Stress and SMCs
5.1. In Vitro Modeling of Fluid Shear Stress
5.2. Shear Stress and Phenotypic Modulation of SMCs
6. Smooth Muscle Cell Mechanotransduction
Mechanotransduction Signaling Pathways in SMCs
7. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Murray, C.J.; Lopez, A.D. Measuring the global burden of disease. N. Engl. J. Med. 2013, 369, 448–457. [Google Scholar] [CrossRef]
- Song, P.; Fang, Z.; Wang, H.; Cai, Y.; Rahimi, K.; Zhu, Y.; Fowkes, F.G.R.; Fowkes, F.J.I.; Rudan, I. Global and regional prevalence, burden, and risk factors for carotid atherosclerosis: A systematic review, meta-analysis, and modelling study. Lancet Glob. Health 2020, 8, e721–e729. [Google Scholar] [CrossRef]
- Owens, G.K. Regulation of differentiation of vascular smooth muscle cells. Physiol. Rev. 1995, 75, 487–517. [Google Scholar] [CrossRef] [PubMed]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef]
- Hahn, C.; Schwartz, M.A. Mechanotransduction in vascular physiology and atherogenesis. Nat. Rev. Mol. Cell Biol. 2009, 10, 53–62. [Google Scholar] [CrossRef]
- Baeyens, N.; Bandyopadhyay, C.; Coon, B.G.; Yun, S.; Schwartz, M.A. Endothelial fluid shear stress sensing in vascular health and disease. J. Clin. Investig. 2016, 126, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, Y.S.; Chien, S. Shear stress-initiated signaling and its regulation of endothelial function. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2191–2198. [Google Scholar] [CrossRef]
- Roux, E.; Bougaran, P.; Dufourcq, P.; Couffinhal, T. Fluid Shear Stress Sensing by the Endothelial Layer. Front. Physiol. 2020, 11, 861. [Google Scholar] [CrossRef] [PubMed]
- Gordon, E.; Schimmel, L.; Frye, M. The Importance of Mechanical Forces for in vitro Endothelial Cell Biology. Front. Physiol. 2020, 11, 684. [Google Scholar] [CrossRef] [PubMed]
- Dessalles, C.A.; Leclech, C.; Castagnino, A.; Barakat, A.I. Integration of substrate- and flow-derived stresses in endothelial cell mechanobiology. Commun. Biol. 2021, 4, 764. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, K.S.; Gotlieb, A.I. The role of shear stress in the pathogenesis of atherosclerosis. Lab. Invest. 2005, 85, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Anwar, M.A.; Shalhoub, J.; Lim, C.S.; Gohel, M.S.; Davies, A.H. The effect of pressure-induced mechanical stretch on vascular wall differential gene expression. J. Vasc. Res. 2012, 49, 463–478. [Google Scholar] [CrossRef]
- Jufri, N.F.; Mohamedali, A.; Avolio, A.; Baker, M.S. Mechanical stretch: Physiological and pathological implications for human vascular endothelial cells. Vascular. Cell 2015, 7, 8. [Google Scholar] [CrossRef]
- Isnard, R.N.; Pannier, B.M.; Laurent, S.; London, G.M.; Diebold, B.; Safar, M.E. Pulsatile diameter and elastic modulus of the aortic arch in essential hypertension: A noninvasive study. J. Am. Coll. Cardiol. 1989, 13, 399–405. [Google Scholar] [CrossRef]
- O’Rourke, M. Mechanical principles in arterial disease. Hypertension 1995, 26, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Mantella, L.E.; Quan, A.; Verma, S. Variability in vascular smooth muscle cell stretch-induced responses in 2D culture. Vasc. Cell 2015, 7, 7. [Google Scholar] [CrossRef]
- Liu, S.; Tao, R.; Wang, M.; Tian, J.; Genin, G.M.; Lu, T.J.; Xu, F. Regulation of Cell Behavior by Hydrostatic Pressure. Appl. Mech. Rev. 2019, 71, 0408031. [Google Scholar] [CrossRef]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef]
- Liu, M.; Gomez, D. Smooth Muscle Cell Phenotypic Diversity. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1715–1723. [Google Scholar] [CrossRef]
- Kawai-Kowase, K.; Owens, G.K. Multiple repressor pathways contribute to phenotypic switching of vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 2007, 292, C59–C69. [Google Scholar] [CrossRef]
- Zhang, M.J.; Zhou, Y.; Chen, L.; Wang, Y.Q.; Wang, X.; Pi, Y.; Gao, C.Y.; Li, J.C.; Zhang, L.L. An overview of potential molecular mechanisms involved in VSMC phenotypic modulation. Histochem. Cell Biol. 2016, 145, 119–130. [Google Scholar] [CrossRef]
- Xu, J.; Shi, G.P. Vascular wall extracellular matrix proteins and vascular diseases. Biochim. Biophys. Acta—Mol. Basis Dis. 2014, 1842, 2106–2119. [Google Scholar] [CrossRef] [PubMed]
- Del Monte-Nieto, G.; Fischer, J.W.; Gorski, D.J.; Harvey, R.P.; Kovacic, J.C. Basic Biology of Extracellular Matrix in the Cardiovascular System, Part 1/4: JACC Focus Seminar. J. Am. Coll. Cardiol. 2020, 75, 2169–2188. [Google Scholar] [CrossRef]
- Holm Nielsen, S.; Jonasson, L.; Kalogeropoulos, K.; Karsdal, M.A.; Reese-Petersen, A.L.; Auf dem Keller, U.; Genovese, F.; Nilsson, J.; Goncalves, I. Exploring the role of extracellular matrix proteins to develop biomarkers of plaque vulnerability and outcome. J. Intern. Med. 2020, 287, 493–513. [Google Scholar] [CrossRef] [PubMed]
- Barallobre-Barreiro, J.; Loeys, B.; Mayr, M.; Rienks, M.; Verstraeten, A.; Kovacic, J.C. Extracellular Matrix in Vascular Disease, Part 2/4: JACC Focus Seminar. J. Am. Coll. Cardiol. 2020, 75, 2189–2203. [Google Scholar] [CrossRef] [PubMed]
- Orr, A.W.; Lee, M.Y.; Lemmon, J.A.; Yurdagul, A.; Gomez, M.F.; Schoppee Bortz, P.D.; Wamhoff, B.R. Molecular mechanisms of collagen isotype-specific modulation of smooth muscle cell phenotype. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 225–231. [Google Scholar] [CrossRef]
- Hedin, U.; Bottger, B.A.; Forsberg, E.; Johansson, S.; Thyberg, J. Diverse effects of fibronectin and laminin on phenotypic properties of cultured arterial smooth muscle cells. J. Cell Biol. 1988, 107, 307–319. [Google Scholar] [CrossRef]
- Yamamoto, M.; Yamamoto, K.; Noumura, T. Type I collagen promotes modulation of cultured rabbit arterial smooth muscle cells from a contractile to a synthetic phenotype. Exp. Cell Res. 1993, 204, 121–129. [Google Scholar] [CrossRef]
- Timraz, S.B.H.; Rezgui, R.; Boularaoui, S.M.; Teo, J.C.M. Stiffness of Extracellular Matrix Components Modulates the Phenotype of Human Smooth Muscle Cells in Vitro and Allows for the Control of Properties of Engineered Tissues. Procedia Engineer. 2015, 110, 29–36. [Google Scholar] [CrossRef]
- Oh, Y.S. Arterial Stiffness and Hypertension. Clin. Hypertens 2018, 24, 17. [Google Scholar] [CrossRef]
- Pelham, R.J., Jr.; Wang, Y. Cell locomotion and focal adhesions are regulated by substrate flexibility. Proc. Natl. Acad. Sci. USA 1997, 94, 13661–13665. [Google Scholar] [CrossRef]
- Polacheck, W.J.; Chen, C.S. Measuring cell-generated forces: A guide to the available tools. Nat. Methods 2016, 13, 415–423. [Google Scholar] [CrossRef]
- Kothapalli, D.; Liu, S.L.; Bae, Y.H.; Monslow, J.; Xu, T.; Hawthorne, E.A.; Byfield, F.J.; Castagnino, P.; Rao, S.; Rader, D.J.; et al. Cardiovascular protection by ApoE and ApoE-HDL linked to suppression of ECM gene expression and arterial stiffening. Cell Rep. 2012, 2, 1259–1271. [Google Scholar] [CrossRef]
- Xie, S.A.; Zhang, T.; Wang, J.; Zhao, F.; Zhang, Y.P.; Yao, W.J.; Hur, S.S.; Yeh, Y.T.; Pang, W.; Zheng, L.S.; et al. Matrix stiffness determines the phenotype of vascular smooth muscle cell in vitro and in vivo: Role of DNA methyltransferase 1. Biomaterials 2018, 155, 203–216. [Google Scholar] [CrossRef]
- Shao, Y.; Li, G.; Huang, S.; Li, Z.; Qiao, B.; Chen, D.; Li, Y.; Liu, H.; Du, J.; Li, P. Effects of Extracellular Matrix Softening on Vascular Smooth Muscle Cell Dysfunction. Cardiovasc. Toxicol. 2020, 20, 548–556. [Google Scholar] [CrossRef]
- Mao, X.; Mao, L.; Zhang, H.; Tan, Y.; Wang, H. Substrate Stiffness Affected the Inflammatory Response of SMCs. J. Biosci. Med. 2021, 9, 44–54. [Google Scholar] [CrossRef]
- Rickel, A.P.; Sanyour, H.J.; Leyda, N.A.; Hong, Z.K. Extracellular Matrix Proteins and Substrate Stiffness Synergistically Regulate Vascular Smooth Muscle Cell Migration and Cortical Cytoskeleton Organization. Acs. Appl. Bio. Mater. 2020, 3, 2360–2369. [Google Scholar] [CrossRef]
- McDaniel, D.P.; Shaw, G.A.; Elliott, J.T.; Bhadriraju, K.; Meuse, C.; Chung, K.H.; Plant, A.L. The stiffness of collagen fibrils influences vascular smooth muscle cell phenotype. Biophys. J. 2007, 92, 1759–1769. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.K.; Xu, T.; Assoian, R.K.; Rader, D.J. Mining the Stiffness-Sensitive Transcriptome in Human Vascular Smooth Muscle Cells Identifies Long Noncoding RNA Stiffness Regulators. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H.; Yang, G.; Li, Z.; Shen, W. Fibroblast responses to cyclic mechanical stretching depend on cell orientation to the stretching direction. J. Biomech. 2004, 37, 573–576. [Google Scholar] [CrossRef] [PubMed]
- Ursekar, C.P.; Teo, S.K.; Hirata, H.; Harada, I.; Chiam, K.H.; Sawada, Y. Design and construction of an equibiaxial cell stretching system that is improved for biochemical analysis. PLoS ONE 2014, 9, e90665. [Google Scholar] [CrossRef]
- Wada, S.; Kanzaki, H.; Narimiya, T.; Nakamura, Y. Novel device for application of continuous mechanical tensile strain to mammalian cells. Biol. Open 2017, 6, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Bono, N.; Pezzoli, D.; Levesque, L.; Loy, C.; Candiani, G.; Fiore, G.B.; Mantovani, D. Unraveling the role of mechanical stimulation on smooth muscle cells: A comparative study between 2D and 3D models. Biotechnol. Bioeng. 2016, 113, 2254–2263. [Google Scholar] [CrossRef]
- Huang, K.; Yan, Z.Q.; Zhao, D.; Chen, S.G.; Gao, L.Z.; Zhang, P.; Shen, B.R.; Han, H.C.; Qi, Y.X.; Jiang, Z.L. SIRT1 and FOXO Mediate Contractile Differentiation of Vascular Smooth Muscle Cells under Cyclic Stretch. Cell. Physiol. Biochem. 2015, 37, 1817–1829. [Google Scholar] [CrossRef]
- Yao, Q.P.; Zhang, P.; Qi, Y.X.; Chen, S.G.; Shen, B.R.; Han, Y.; Yan, Z.Q.; Jiang, Z.L. The role of SIRT6 in the differentiation of vascular smooth muscle cells in response to cyclic strain. Int. J. Biochem. Cell Biol. 2014, 49, 98–104. [Google Scholar] [CrossRef]
- Mao, X.; Said, R.; Louis, H.; Max, J.P.; Bourhim, M.; Challande, P.; Wahl, D.; Li, Z.; Regnault, V.; Lacolley, P. Cyclic stretch-induced thrombin generation by rat vascular smooth muscle cells is mediated by the integrin αvβ3 pathway. Cardiovasc. Res. 2012, 96, 513–523. [Google Scholar] [CrossRef]
- Rodríguez, A.I.; Csányi, G.; Ranayhossaini, D.J.; Feck, D.M.; Blose, K.J.; Assatourian, L.; Vorp, D.A.; Pagano, P.J. MEF2B-Nox1 signaling is critical for stretch-induced phenotypic modulation of vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 430–438. [Google Scholar] [CrossRef]
- Pfisterer, L.; Feldner, A.; Hecker, M.; Korff, T. Hypertension impairs myocardin function: A novel mechanism facilitating arterial remodelling. Cardiovasc. Res. 2012, 96, 120–129. [Google Scholar] [CrossRef]
- Wang, Y.; Cao, W.; Cui, J.; Yu, Y.; Zhao, Y.; Shi, J.; Wu, J.; Xia, Z.; Yu, B.; Liu, J. Arterial Wall Stress Induces Phenotypic Switching of Arterial Smooth Muscle Cells in Vascular Remodeling by Activating the YAP/TAZ Signaling Pathway. Cell Physiol. Biochem. 2018, 51, 842–853. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.J.; Zhao, H.C.; Zhang, P.; Huo, B.; Shen, B.R.; Yan, Z.Q.; Qi, Y.X.; Jiang, Z.L. Involvement of BK channel in differentiation of vascular smooth muscle cells induced by mechanical stretch. Int. J. Biochem. Cell Biol. 2015, 59, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Song, J.T.; Qu, H.Y.; Bi, C.L.; Huang, X.Z.; Liu, X.X.; Zhang, M. Mechanical stretch suppresses microRNA-145 expression by activating extracellular signal-regulated kinase 1/2 and upregulating angiotensin- converting enzyme to alter vascular smooth muscle cell phenotype. PLoS ONE 2014, 9, e96338. [Google Scholar] [CrossRef] [PubMed]
- Mantella, L.E.; Singh, K.K.; Sandhu, P.; Kantores, C.; Ramadan, A.; Khyzha, N.; Quan, A.; Al-Omran, M.; Fish, J.E.; Jankov, R.P.; et al. Fingerprint of long non-coding RNA regulated by cyclic mechanical stretch in human aortic smooth muscle cells: Implications for hypertension. Mol. Cell. Biochem. 2017, 435, 163–173. [Google Scholar] [CrossRef]
- Qin, H.; Ishiwata, T.; Wang, R.; Kudo, M.; Yokoyama, M.; Naito, Z.; Asano, G. Effects of extracellular matrix on phenotype modulation and MAPK transduction of rat aortic smooth muscle cells in vitro. Exp. Mol. Pathol. 2000, 69, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.Z.; Wang, B.W.; Shyu, K.G. Effects of cyclic stretch on the molecular regulation of myocardin in rat aortic vascular smooth muscle cells. J. Biomed. Sci. 2013, 20, 50. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Wong, M.; Smith, Q.; Baker, A.B. A novel system for studying mechanical strain waveform-dependent responses in vascular smooth muscle cells. Lab. Chip 2013, 13, 4573–4582. [Google Scholar] [CrossRef]
- Zampetaki, A.; Zhang, Z.; Hu, Y.; Xu, Q. Biomechanical stress induces IL-6 expression in smooth muscle cells via Ras/Rac1-p38 MAPK-NF-kappaB signaling pathways. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2946–H2954. [Google Scholar] [CrossRef]
- Zhao, J.; Nishimura, Y.; Kimura, A.; Ozawa, K.; Kondo, T.; Tanaka, T.; Yoshizumi, M. Chemokines protect vascular smooth muscle cells from cell death induced by cyclic mechanical stretch. Sci. Rep. 2017, 7, 16128. [Google Scholar] [CrossRef]
- Song, J.; Qu, H.; Hu, B.; Bi, C.; Li, M.; Wang, L.; Huang, X.; Zhang, M. Physiological cyclic stretch up-regulates angiotensin-converting enzyme 2 expression to reduce proliferation and migration of vascular smooth muscle cells. Biosci. Rep. 2020, 40, BSR20192012. [Google Scholar] [CrossRef]
- Qi, Y.X.; Qu, M.J.; Yan, Z.Q.; Zhao, D.; Jiang, X.H.; Shen, B.R.; Jiang, Z.L. Cyclic strain modulates migration and proliferation of vascular smooth muscle cells via Rho-GDIα, Rac1, and p38 pathway. J. Cell. Biochem. 2010, 109, 906–914. [Google Scholar] [CrossRef]
- Li, C.; Wernig, F.; Leitges, M.; Hu, Y.; Xu, Q. Mechanical stress-activated PKCdelta regulates smooth muscle cell migration. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2003, 17, 2106–2108. [Google Scholar] [CrossRef]
- Chapman, G.B.; Durante, W.; Hellums, J.D.; Schafer, A.I. Physiological cyclic stretch causes cell cycle arrest in cultured vascular smooth muscle cells. Am. J. Physiol. —Heart Circ. Physiol. 2000, 278, H748–H754. [Google Scholar] [CrossRef] [PubMed]
- Schad, J.F.; Meltzer, K.R.; Hicks, M.R.; Beutler, D.S.; Cao, T.V.; Standley, P.R. Cyclic strain upregulates VEGF and attenuates proliferation of vascular smooth muscle cells. Vascular. Cell 2011, 3, 21. [Google Scholar] [CrossRef]
- Ping, S.; Li, Y.; Liu, S.; Zhang, Z.; Wang, J.; Zhou, Y.; Liu, K.; Huang, J.; Chen, D.; Wang, J.; et al. Simultaneous increases in proliferation and apoptosis of vascular smooth muscle cells accelerate diabetic mouse venous atherosclerosis. PLoS ONE 2015, 10, e0141375. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Zhang, D.; Li, Q.; Liu, Y.; Jing, S.; Cui, J.; Xu, W.; Li, S.; Liu, J.; Yu, B. Biomechanical Stretch Induces Inflammation, Proliferation, and Migration by Activating NFAT5 in Arterial Smooth Muscle Cells. Inflammation 2017, 40, 2129–2136. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liu, X.; Li, M.; Zhang, Y.; Chen, W.; Zhang, M.; Zhang, C.; Zhang, M. Mechanical Stretch Induces Smooth Muscle Cell Dysfunction by Regulating ACE2 via P38/ATF3 and Post-transcriptional Regulation by miR-421. Front. Physiol. 2020, 11, 540591. [Google Scholar] [CrossRef]
- Song, J.t.; Hu, B.; Qu, H.y.; Bi, C.l.; Huang, X.z.; Zhang, M. Mechanical Stretch Modulates MicroRNA 21 Expression, Participating in Proliferation and Apoptosis in Cultured Human Aortic Smooth Muscle Cells. PLoS ONE 2012, 7, e47657. [Google Scholar] [CrossRef]
- Zheng, T.-F.; Liu, X.-L.; Li, X.; Wang, Q.-Q.; Zhao, Y.-C.; Li, X.; Li, M.-M.; Zhang, Y.; Zhang, M.; Zhang, W.-C.; et al. Dickkopf-1 promotes Vascular Smooth Muscle Cell proliferation and migration through upregulating UHRF1 during Cyclic Stretch application. Int. J. Biol. Sci. 2021, 17, 1234–1249. [Google Scholar] [CrossRef]
- Basatemur, G.L.; Jorgensen, H.F.; Clarke, M.C.H.; Bennett, M.R.; Mallat, Z. Vascular smooth muscle cells in atherosclerosis. Nat. Rev. Cardiol. 2019, 16, 727–744. [Google Scholar] [CrossRef]
- Yu, H.; Clarke, M.C.; Figg, N.; Littlewood, T.D.; Bennett, M.R. Smooth muscle cell apoptosis promotes vessel remodeling and repair via activation of cell migration, proliferation, and collagen synthesis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2402–2409. [Google Scholar] [CrossRef] [PubMed]
- Mayr, M.; Xu, Q. Smooth muscle cell apoptosis in arteriosclerosis. Exp. Gerontol. 2001, 36, 969–987. [Google Scholar] [CrossRef]
- Wang, J.; Liu, K.; Wang, H.; Li, Z.; Li, Y.; Ping, S.; Bardeesi, A.S.A.; Guo, Y.; Zhou, Y.; Pei, T.; et al. Role of nifedipine and hydrochlorothiazide in MAPK activation and vascular smooth muscle cell proliferation and apoptosis. Herz 2017, 42, 573–584. [Google Scholar] [CrossRef]
- Wang, L.; Deng, L.; Lin, N.; Shi, Y.; Chen, J.; Zhou, Y.; Chen, D.; Liu, S.; Li, C. Berberine inhibits proliferation and apoptosis of vascular smooth muscle cells induced by mechanical stretch via the PDI/ERS and MAPK pathways. Life Sci. 2020, 259, 118253. [Google Scholar] [CrossRef] [PubMed]
- Ping, S.; Liu, S.; Zhou, Y.; Li, Z.; Li, Y.; Liu, K.; Bardeesi, A.S.; Wang, L.; Chen, J.; Deng, L.; et al. Protein disulfide isomerase-mediated apoptosis and proliferation of vascular smooth muscle cells induced by mechanical stress and advanced glycosylation end products result in diabetic mouse vein graft atherosclerosis. Cell Death Dis. 2017, 8, e2818. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Ozawa, K.; Kyotani, Y.; Nagayama, K.; Ito, S.; Komatsubara, A.T.; Tsuji, Y.; Yoshizumi, M. Azelnidipine inhibits cultured rat aortic smooth muscle cell death induced by cyclic mechanical stretch. PLoS ONE 2014, 9, e102813. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Nakahira, K.; Kimura, A.; Kyotani, Y.; Yoshizumi, M. Upregulation of iNOS Protects Cyclic Mechanical Stretch-Induced Cell Death in Rat Aorta Smooth Muscle Cells. Int. J. Mol. Sci. 2020, 21, 8660. [Google Scholar] [CrossRef]
- Cheng, W.P.; Wang, B.W.; Chen, S.C.; Chang, H.; Shyu, K.G. Mechanical stretch induces the apoptosis regulator PUMA in vascular smooth muscle cells. Cardiovasc. Res. 2012, 93, 181–189. [Google Scholar] [CrossRef]
- Jia, L.X.; Zhang, W.M.; Zhang, H.J.; Li, T.T.; Wang, Y.L.; Qin, Y.W.; Gu, H.; Du, J. Mechanical stretch-induced endoplasmic reticulum stress, apoptosis and inflammation contribute to thoracic aortic aneurysm and dissection. J. Pathol. 2015, 236, 373–383. [Google Scholar] [CrossRef]
- Wang, D.M.; Tarbell, J.M. Modeling interstitial flow in an artery wall allows estimation of wall shear stress on smooth muscle cells. J. Biomech. Eng. 1995, 117, 358–363. [Google Scholar] [CrossRef]
- Rizzo, V. Enhanced interstitial flow as a contributing factor in neointima formation: (shear) stressing vascular wall cell types other than the endothelium. Am. J. Physiol.-Heart C 2009, 297, H1196–H1197. [Google Scholar] [CrossRef][Green Version]
- Shi, Z.D.; Abraham, G.; Tarbell, J.M. Shear stress modulation of smooth muscle cell marker genes in 2-D and 3-D depends on mechanotransduction by heparan sulfate proteoglycans and ERK1/2. PLoS ONE 2010, 5, e12196. [Google Scholar] [CrossRef]
- Hsu, S.; Chu, J.S.; Chen, F.F.; Wang, A.; Li, S. Effects of Fluid Shear Stress on a Distinct Population of Vascular Smooth Muscle Cells. Cell Mol. Bioeng. 2011, 4, 627–636. [Google Scholar] [CrossRef]
- Perisic Matic, L.; Rykaczewska, U.; Razuvaev, A.; Sabater-Lleal, M.; Lengquist, M.; Miller, C.L.; Ericsson, I.; Rohl, S.; Kronqvist, M.; Aldi, S.; et al. Phenotypic Modulation of Smooth Muscle Cells in Atherosclerosis Is Associated With Downregulation of LMOD1, SYNPO2, PDLIM7, PLN, and SYNM. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1947–1961. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhao, M.; Liu, A.; Lv, M.; Zhang, J.; Li, Y.; Yang, X.; Wu, Z. Shear Stress Induces Phenotypic Modulation of Vascular Smooth Muscle Cells via AMPK/mTOR/ULK1-Mediated Autophagy. Cell Mol. Neurobiol. 2018, 38, 541–548. [Google Scholar] [CrossRef]
- Ekstrand, J.; Razuvaev, A.; Folkersen, L.; Roy, J.; Hedin, U. Tissue factor pathway inhibitor-2 is induced by fluid shear stress in vascular smooth muscle cells and affects cell proliferation and survival. J. Vasc. Surg. 2010, 52, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Fan, Y.; Deng, X. Vascular smooth muscle cell glycocalyx modulates shear-induced proliferation, migration, and NO production responses. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H76–H83. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, T.N.; Shepherd, B.R.; Asada, H.; Teso, D.; Muto, A.; Fancher, T.; Pimiento, J.M.; Maloney, S.P.; Dardik, A. Laminar shear stress stimulates vascular smooth muscle cell apoptosis via the Akt pathway. J. Cell Physiol. 2008, 216, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Albarran-Juarez, J.; Iring, A.; Wang, S.; Joseph, S.; Grimm, M.; Strilic, B.; Wettschureck, N.; Althoff, T.F.; Offermanns, S. Piezo1 and Gq/G11 promote endothelial inflammation depending on flow pattern and integrin activation. J. Exp. Med. 2018, 215, 2655–2672. [Google Scholar] [CrossRef] [PubMed]
- Haga, M.; Yamashita, A.; Paszkowiak, J.; Sumpio, B.E.; Dardik, A. Oscillatory shear stress increases smooth muscle cell proliferation and Akt phosphorylation. J. Vasc. Surg. 2003, 37, 1277–1284. [Google Scholar] [CrossRef]
- Martino, F.; Perestrelo, A.R.; Vinarsky, V.; Pagliari, S.; Forte, G. Cellular Mechanotransduction: From Tension to Function. Front. Physiol. 2018, 9, 824. [Google Scholar] [CrossRef]
- Lehoux, S.; Castier, Y.; Tedgui, A. Molecular mechanisms of the vascular responses to haemodynamic forces. J. Intern. Med. 2006, 259, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, H.; Yoshimoto, T.; Sugiyama, T.; Hirata, Y. Activation of cell adhesion kinase beta by mechanical stretch in vascular smooth muscle cells. Endocrinology 2003, 144, 2304–2310. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Kollar, B.; Nahar, T.; Suresh Babu, S.; Wojtowicz, A.; Sticht, C.; Gretz, N.; Wagner, A.H.; Korff, T.; Hecker, M. Loss of the mechanotransducer zyxin promotes a synthetic phenotype of vascular smooth muscle cells. J. Am. Heart Assoc. 2015, 4, e001712. [Google Scholar] [CrossRef] [PubMed]
- Kona, S.; Chellamuthu, P.; Xu, H.; Hills, S.R.; Nguyen, K.T. Effects of cyclic strain and growth factors on vascular smooth muscle cell responses. Open Biomed. Eng. J. 2009, 3, 28–38. [Google Scholar] [CrossRef][Green Version]
- Morrow, D.; Sweeney, C.; Birney, Y.A.; Cummins, P.M.; Walls, D.; Redmond, E.M.; Cahill, P.A. Cyclic strain inhibits Notch receptor signaling in vascular smooth muscle cells in vitro. Circ. Res. 2005, 96, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Akazawa, H.; Qin, Y.; Sano, M.; Takano, H.; Minamino, T.; Makita, N.; Iwanaga, K.; Zhu, W.; Kudoh, S.; et al. Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat. Cell Biol. 2004, 6, 499–506. [Google Scholar] [CrossRef]
- Mohanty, M.J.; Li, X. Stretch-induced Ca(2+) release via an IP(3)-insensitive Ca(2+) channel. Am. J. Physiol. Cell Physiol. 2002, 283, C456–C462. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, S.H.; Tribe, R.M.; Songu-Mize, E. Cyclic stretch decreases TRPC4 protein and capacitative calcium entry in rat vascular smooth muscle cells. Life Sci. 2008, 83, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.Y.; Chiu, J.J. Mechanical Regulation of Epigenetic Modifications in Vascular Biology and Pathobiology. In Vascular Mechanobiology in Physiology and Disease. Cardiac and Vascular Biology; Duncker, D.J., Hecker, M., Eds.; Springer: Cham, Switzerland, 2021; Volume 8, pp. 241–276. [Google Scholar] [CrossRef]
- Chen, J.; Zhou, Y.; Liu, S.; Li, C. Biomechanical signal communication in vascular smooth muscle cells. J. Cell Commun. Signal. 2020, 14, 357–376. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.Q.; Yao, Q.P.; Zhang, M.L.; Qi, Y.X.; Guo, Z.Y.; Shen, B.R.; Jiang, Z.L. Histone deacetylases modulate vascular smooth muscle cell migration induced by cyclic mechanical strain. J. Biomech. 2009, 42, 945–948. [Google Scholar] [CrossRef]
- Halka, A.T.; Turner, N.J.; Carter, A.; Ghosh, J.; Murphy, M.O.; Kirton, J.P.; Kielty, C.M.; Walker, M.G. The effects of stretch on vascular smooth muscle cell phenotype in vitro. Cardiovasc. Pathol. 2008, 17, 98–102. [Google Scholar] [CrossRef]
- Numaguchi, K.; Eguchi, S.; Yamakawa, T.; Motley, E.D.; Inagami, T. Mechanotransduction of rat aortic vascular smooth muscle cells requires RhoA and intact actin filaments. Circ. Res. 1999, 85, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Figtree, G.A.; Broadfoot, K.; Casadei, B.; Califf, R.; Crea, F.; Drummond, G.R.; Freedman, J.E.; Guzik, T.J.; Harrison, D.; Hausenloy, D.J.; et al. A call to action for new global approaches to cardiovascular disease drug solutions. Eur. Heart J. 2021, 42, 1464–1475. [Google Scholar] [CrossRef] [PubMed]
- Stegemann, J.P.; Hong, H.; Nerem, R.M. Mechanical, biochemical, and extracellular matrix effects on vascular smooth muscle cell phenotype. J. Appl. Physiol. 2005, 98, 2321–2327. [Google Scholar] [CrossRef]
- Jia, L.; Wang, L.; Wei, F.; Li, C.; Wang, Z.; Yu, H.; Chen, H.; Wang, B.; Jiang, A. Effects of Caveolin-1-ERK1/2 pathway on endothelial cells and smooth muscle cells under shear stress. Exp. Biol. Med. 2020, 245, 21–33. [Google Scholar] [CrossRef]
- van Haaften, E.E.; Wissing, T.B.; Kurniawan, N.A.; Smits, A.; Bouten, C.V.C. Human In Vitro Model Mimicking Material-Driven Vascular Regeneration Reveals How Cyclic Stretch and Shear Stress Differentially Modulate Inflammation and Matrix Deposition. Adv. Biosyst. 2020, 4, e1900249. [Google Scholar] [CrossRef]
- Dobnikar, L.; Taylor, A.L.; Chappell, J.; Oldach, P.; Harman, J.L.; Oerton, E.; Dzierzak, E.; Bennett, M.R.; Spivakov, M.; Jorgensen, H.F. Disease-relevant transcriptional signatures identified in individual smooth muscle cells from healthy mouse vessels. Nat. Commun. 2018, 9, 4567. [Google Scholar] [CrossRef]
- Pan, H.; Xue, C.; Auerbach, B.J.; Fan, J.; Bashore, A.C.; Cui, J.; Yang, D.Y.; Trignano, S.B.; Liu, W.; Shi, J.; et al. Single-Cell Genomics Reveals a Novel Cell State During Smooth Muscle Cell Phenotypic Switching and Potential Therapeutic Targets for Atherosclerosis in Mouse and Human. Circulation 2020, 142, 2060–2075. [Google Scholar] [CrossRef] [PubMed]
- Wirka, R.C.; Wagh, D.; Paik, D.T.; Pjanic, M.; Nguyen, T.; Miller, C.L.; Kundu, R.; Nagao, M.; Coller, J.; Koyano, T.K.; et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat. Med. 2019, 25, 1280–1289. [Google Scholar] [CrossRef]
- Wagh, K.; Ishikawa, M.; Garcia, D.A.; Stavreva, D.A.; Upadhyaya, A.; Hager, G.L. Mechanical Regulation of Transcription: Recent Advances. Trends Cell Biol. 2021, 31, 457–472. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Study | Stretch Intensity, Duration and Frequency | Matrix Coating | SMC-Source | SM Marker Expression |
|---|---|---|---|---|
| [43] | 7% for 2 and 5 days 1Hz | Collagen I | Human umbilical artery | CNN1 (=) ACTA2 (=) |
| [44] | 10% for 24 h 1.25 Hz | Collagen I | Sprague–Dawley rat thoracic aorta | CNN1 ↑ ACTA2 ↑ TAGLN ↑ |
| [45] | 10% for 24 h 1.25 Hz | Gelatin | Sprague–Dawley rat thoracic aorta | Cnn1 ↑ Acta2 ↑ Tagln ↑ |
| [46] | 10% for 24 h 1 Hz | Collagen I | Wistar rat thoracic aorta | Acta2 ↑ Myh11 ↑ |
| [47] | 10% for 24 h 1 Hz | Collagen I | Sprague–Dawley rat thoracic aorta | Cnn1 ↓ Smtn ↓ Opn ↑ |
| [48] | 13% for 24 h 0.5 Hz | Fibronectin | Human umbilical artery | CNN1 ↓ MYOCD ↓ MYH11 ↓ |
| [49] | 13% for 24 h 0.5 Hz | Matrigel | Human umbilical artery | CNN1 ↓ ACTA2 ↓ MYH11 ↓ |
| [50] | 15% for 24 h 1.25 Hz | Collagen I | Sprague–Dawley rat thoracic aorta | Cnn1 ↓ Acta2 ↓ Tagln ↓ |
| [51] | 16% for 12 h 1 Hz | Collagen I | Human aorta | CNN1 ↓ ACTA2 ↓ TAGLN ↓ MYOCD ↓ KLF4 ↑ |
| [52] | 20% for 24 h 1 Hz | Collagen I | Human aorta | CNN1 ↓ ACTA2 ↓ |
| Study | Stretch Intensity, Duration and Frequeny | Matrix Coating | Technique Used | SMC- Source | Migration Effect |
|---|---|---|---|---|---|
| [58] | 10% for 12 h 1 Hz | Collagen I | Scratch assay | Human aortic | Decreased |
| [47] | 10% for 24 h 1 Hz | Collagen I | Scratch assay | Sprague–Dawley rat thoracic aorta | Increased |
| [59] | 15% for 24 h 1.25 Hz | Collagen I | Transwell | Sprague–Dawley rat thoracic aorta | Increased |
| [60] | 20% for 3 h, 1 Hz | Collagen I | Scratch assay | 129/SV Mouse aortic | Increased |
| Study | Stretch Intensity, Duration and Frequeny | Matrix Coating | Technique Used | SMC Source | Proliferation Effect |
|---|---|---|---|---|---|
| [58] | 10% for 12 h 1 Hz | Collagen I | BrdU incorporation | Human aorta | Decreased |
| [62] | 10% for 48 h 1 Hz | Collagen I | Fluorescence spectroscopy | A7R5 rat thoracic aorta | Decreased |
| [61] | 10% for 4 days, 1 Hz | Collagen I | Cell counts | Sprague–Dawley rat thoracic aorta | Decreased |
| [63] | 10% for 1 h 1 Hz | Gelatin | Ki67 staining | C57BL/6J mouse aorta | Increased |
| [49] | 13% for 24 h 0.5 Hz | Matrigel | EdU incorporation | Human umbilical artery | Increased |
| [59] | 15% for 24 h 1.25 Hz | Collagen I | BrdU incorporation | Sprague–Dawley rat thoracic aorta | Increased |
| [66] | 16% for 12 h 1 Hz | Collagen I | BrdU incorporation | Human aorta | Increased |
| [52] | 20% for 24 h 1 Hz | Collagen I | Colorimetric assay | Human aorta | Increased |
| Study | Stretch Intensity, Duration, Frequency | Matrix Substrate | Technique Used | SMC Source | Apoptotic Effect |
|---|---|---|---|---|---|
| [72] | 10% for 1–24 h 1 Hz | Gelatin | TUNEL | C57BL/6J Mouse aortic | Increased |
| [71] | 10% for 1 h or 15 h 1 Hz | Gelatin | TUNEL | C57BL/6J Mouse aortic | Increased |
| [73] | 10%for 1 h 1 Hz | Gelatin | TUNEL | C57BL/6J mouse aorta | Increased |
| [75] | 15% for 4 h 1 Hz | Collagen I | LDHrelease | Sprague–Dawley rat thoracic aorta | Increased |
| [76] | 15% for 4 h 1 Hz | Collagen I | apoptosis marker genes | Sprague–Dawley rat thoracic aorta | Increased |
| [66] | 16% for 12 h 1 Hz | Collagen I | Cell sorting | Human aortic | Increased |
| [78] | 18% for 36 h | Collagen I | Cell sorting | C57B/L6 Mouse aortic | Increased |
| [65] | 18% for 12 h 1 Hz | Collagen I | Flow cytometry | Human aortic | Increased |
| [77] | 20% for 18 h 1 Hz | Collagen I | Cell sorting TUNEL | Human coronary | Increased |
| Study | Shear Stress Type, Intensity, and Duration | Material and Matrix Substrate | SMC Source | Technique Used | Effects on SM Phenotype |
|---|---|---|---|---|---|
| [80] | Laminar: 8 dynes/cm2 for 15 h | Plastic/ fibronectin | Sprague–Dawley rat thoracic aorta | Rotating disk | Acta2 ↓ Tagln ↓ Myh11 ↓ Smtn ↓ Cnn1 ↓ |
| [81] | Laminar: 12 dynes/cm2 for 24 h | Glass/ fibronectin | Human aorta | Parallel plate flow chamber | ↑ proliferation ↓ inflammation |
| [82] | Laminar: 14 dynes/cm2 for 24 h | Not stated | Rat aortic | Parallel plate flow chamber | Myh11 ↓ Smtn ↓ Acta2 ↓ |
| [83] | Laminar: 15 dynes/cm2 for 6, 12 and 24 h | Plastic/ coating not stated | Rat Brain arteries | Parallel plate flow chamber | ↑ proliferation ↑ migration Acta2 ↓ Tagln ↓ |
| [84] | Laminar: 14 dynes/cm2 for 24 h | Plastic/ coating not stated | Sprague–Dawley Rat aortic | Parallel plate flow chamber | ↓ proliferation |
| [85] | Laminar: 12 dynes/cm2 for 24 h | Glass/ coating not stated | Sprague–Dawley rat thoracic aorta | Parallel plate flow chamber | ↓ proliferation ↓ migration |
| [86] | Laminar: 11 dynes/cm2 for 24 h | Glass/ Collagen I | Bovine aortic | Parallel plate flow chamber | ↓ proliferation |
| [88] | Oscillatory: 14 dynes/cm2 for 3 and 5 days | Plastic/ Collagen I | Bovine aortic | Orbital shaker | ↑ proliferation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jensen, L.F.; Bentzon, J.F.; Albarrán-Juárez, J. The Phenotypic Responses of Vascular Smooth Muscle Cells Exposed to Mechanical Cues. Cells 2021, 10, 2209. https://doi.org/10.3390/cells10092209
Jensen LF, Bentzon JF, Albarrán-Juárez J. The Phenotypic Responses of Vascular Smooth Muscle Cells Exposed to Mechanical Cues. Cells. 2021; 10(9):2209. https://doi.org/10.3390/cells10092209
Chicago/Turabian StyleJensen, Lise Filt, Jacob Fog Bentzon, and Julian Albarrán-Juárez. 2021. "The Phenotypic Responses of Vascular Smooth Muscle Cells Exposed to Mechanical Cues" Cells 10, no. 9: 2209. https://doi.org/10.3390/cells10092209
APA StyleJensen, L. F., Bentzon, J. F., & Albarrán-Juárez, J. (2021). The Phenotypic Responses of Vascular Smooth Muscle Cells Exposed to Mechanical Cues. Cells, 10(9), 2209. https://doi.org/10.3390/cells10092209