
DNA Methylation of TGFβ Target Genes: Epigenetic Control of TGFβ Functional Duality in Liver Cancer

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Human HCC Samples
2.2. Culture of HCC Cells
2.3. Gene Expression Profiling
2.4. DNA Extraction
2.5. Promoter Methylation Analysis
3. Results
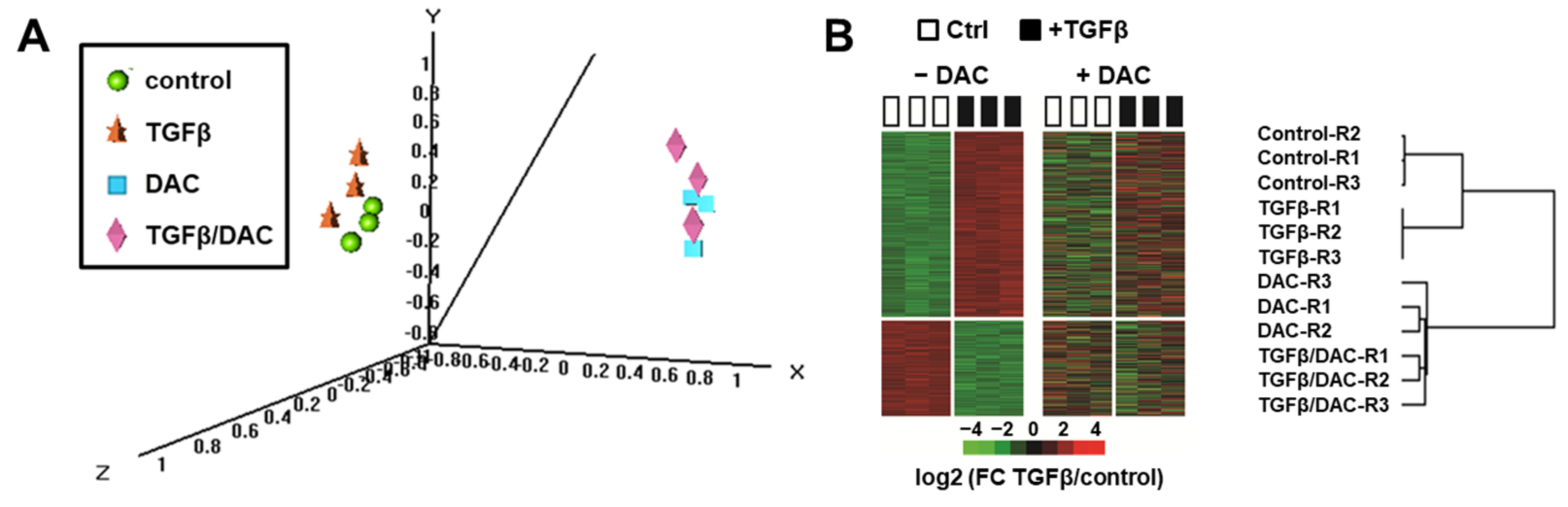
3.1. Decitabine Impairs the Transcriptional Response of SNU449 HCC Cells to TGFβ
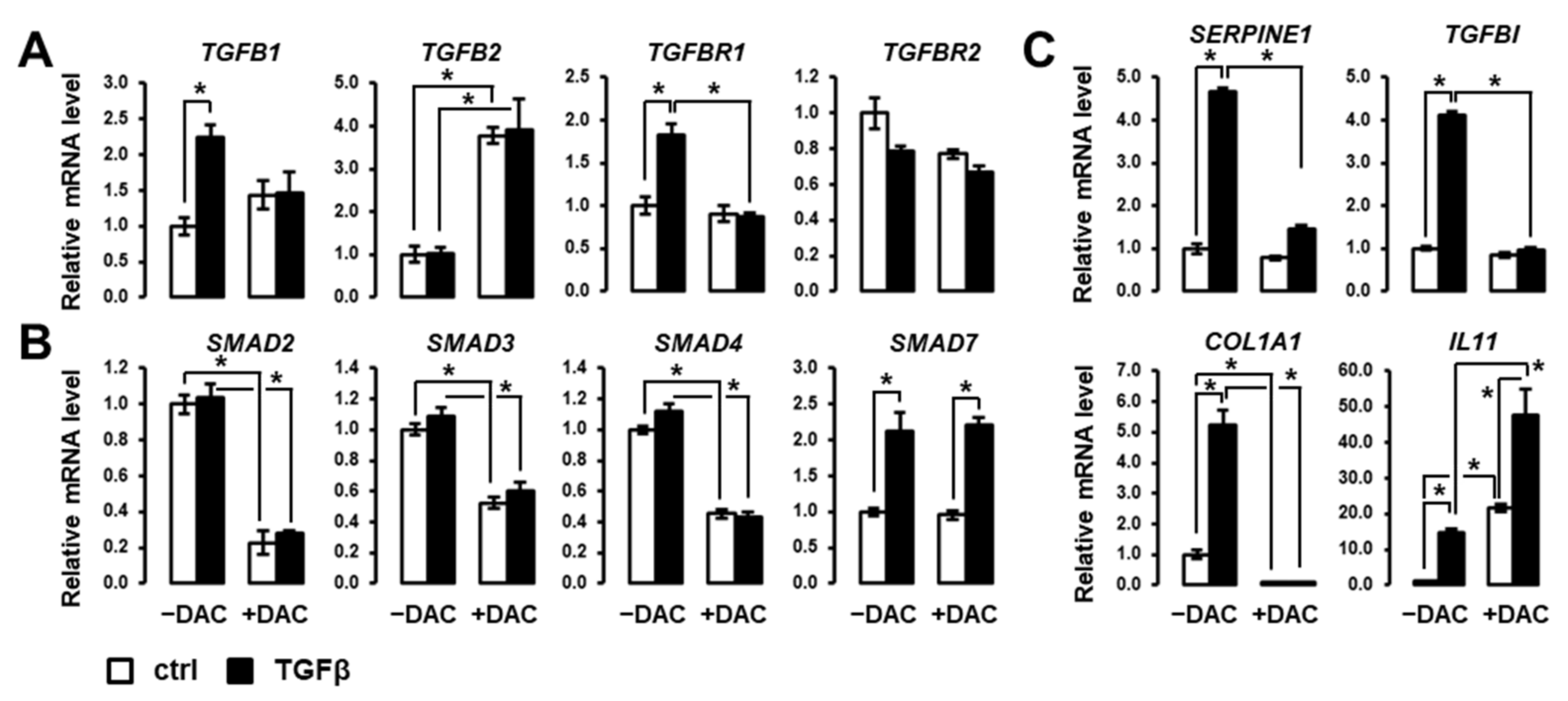
3.2. Decitabine Inhibits the Expression of Members of Canonical SMAD/TGFβ Signaling Pathway
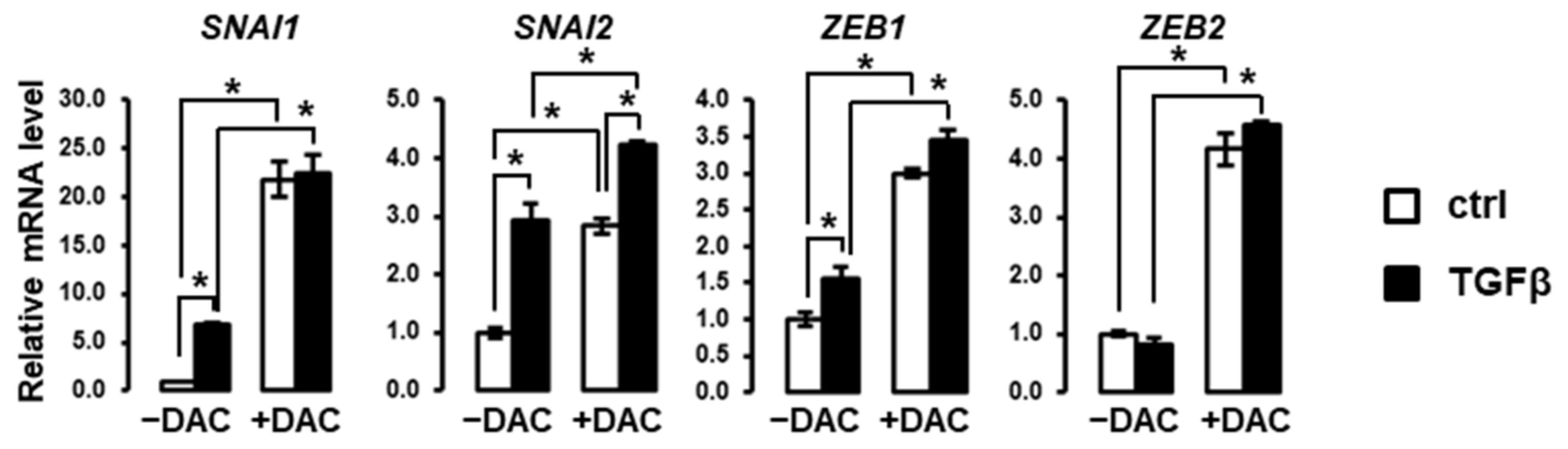
3.3. Decitabine Induces the Expression of EMT-Related Transcription Factors in SNU449 Cells
3.4. Promoter Methylation of TGFβ-Associated Genes in SNU449 HCC Cells
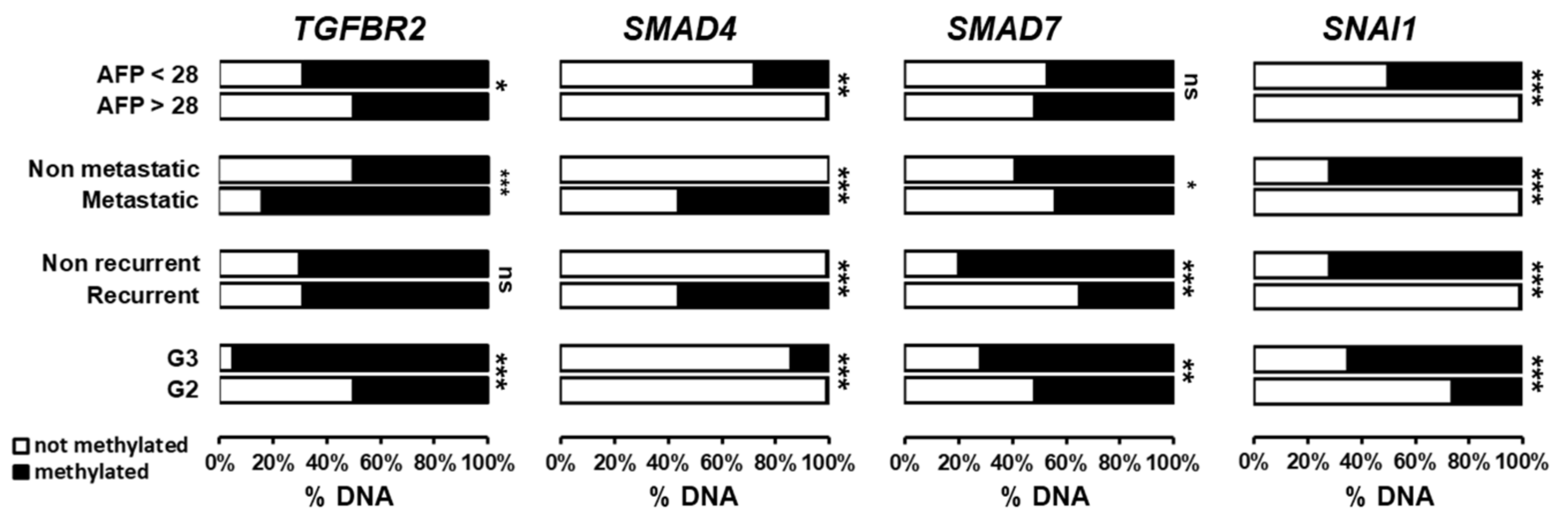
3.5. Clinical Relevance of Promoter Methylation of TGFβ-Associated Genes in Human HCC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Huang, D.Q.; El-Serag, H.B.; Loomba, R. Global epidemiology of NAFLD-related HCC: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 223–238. [Google Scholar] [CrossRef]
- El-Zayadi, A.R.; Badran, H.M.; Barakat, E.M.; Attia Mel, D.; Shawky, S.; Mohamed, M.K.; Selim, O.; Saeid, A. Hepatocellular carcinoma in Egypt: A single center study over a decade. World J. Gastroenterol. 2005, 11, 5193–5198. [Google Scholar] [CrossRef] [PubMed]
- Lehman, E.M.; Wilson, M.L. Epidemiology of hepatitis viruses among hepatocellular carcinoma cases and healthy people in Egypt: A systematic review and meta-analysis. Int. J. Cancer 2009, 124, 690–697. [Google Scholar] [CrossRef]
- Haber, P.K.; Puigvehi, M.; Castet, F.; Lourdusamy, V.; Montal, R.; Tabrizian, P.; Buckstein, M.; Kim, E.; Villanueva, A.; Schwartz, M.; et al. Evidence-based management of HCC: Systematic review and meta-analysis of randomized controlled trials (2002-2020). Gastroenterology 2021, 161, 879–898. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef]
- Llovet, J.M.; Hernandez-Gea, V. Hepatocellular carcinoma: Reasons for phase III failure and novel perspectives on trial design. Clin. Cancer Res. 2014, 20, 2072–2079. [Google Scholar] [CrossRef] [PubMed]
- Rebouissou, S.; Nault, J.C. Advances in molecular classification and precision oncology in hepatocellular carcinoma. J. Hepatol. 2020, 72, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Bevant, K.; Coulouarn, C. Landscape of genomic alterations in hepatocellular carcinoma: Current knowledge and perspectives for targeted therapies. Hepatobiliary Surg. Nutr. 2017, 6, 404–407. [Google Scholar] [CrossRef]
- Allain, C.; Angenard, G.; Clement, B.; Coulouarn, C. Integrative Genomic Analysis Identifies the Core Transcriptional Hallmarks of Human Hepatocellular Carcinoma. Cancer Res. 2016, 76, 6374–6381. [Google Scholar] [CrossRef] [PubMed]
- Coulouarn, C.; Factor, V.M.; Thorgeirsson, S.S. Transforming growth factor-beta gene expression signature in mouse hepatocytes predicts clinical outcome in human cancer. Hepatology 2008, 47, 2059–2067. [Google Scholar] [CrossRef]
- David, C.J.; Massague, J. Contextual determinants of TGFbeta action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Dropmann, A.; Dediulia, T.; Breitkopf-Heinlein, K.; Korhonen, H.; Janicot, M.; Weber, S.N.; Thomas, M.; Piiper, A.; Bertran, E.; Fabregat, I.; et al. TGF-beta1 and TGF-beta2 abundance in liver diseases of mice and men. Oncotarget 2016, 7, 19499–19518. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, I.; Moreno-Caceres, J.; Sanchez, A.; Dooley, S.; Dewidar, B.; Giannelli, G.; Ten Dijke, P.; IT-LIVER Consortium. TGF-beta signalling and liver disease. FEBS J. 2016, 283, 2219–2232. [Google Scholar] [CrossRef] [PubMed]
- Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, A.N. TGF-beta in Hepatic Stellate Cell Activation and Liver Fibrogenesis-Updated 2019. Cells 2019, 8, 1419. [Google Scholar] [CrossRef] [PubMed]
- Giannelli, G.; Mikulits, W.; Dooley, S.; Fabregat, I.; Moustakas, A.; ten Dijke, P.; Portincasa, P.; Winter, P.; Janssen, R.; Leporatti, S.; et al. The rationale for targeting TGF-beta in chronic liver diseases. Eur. J. Clin. Invest. 2016, 46, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Ikushima, H.; Miyazono, K. TGFbeta signalling: A complex web in cancer progression. Nat. Rev. Cancer 2010, 10, 415–424. [Google Scholar] [CrossRef]
- Massague, J. TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Fernandez-Barrena, M.G.; Arechederra, M.; Colyn, L.; Berasain, C.; Avila, M.A. Epigenetics in hepatocellular carcinoma development and therapy: The tip of the iceberg. JHEP Rep. 2020, 2, 100167. [Google Scholar] [CrossRef]
- Nishiyama, A.; Nakanishi, M. Navigating the DNA methylation landscape of cancer. Trends Genet. 2021. [Google Scholar] [CrossRef] [PubMed]
- Merdrignac, A.; Angenard, G.; Allain, C.; Petitjean, K.; Bergeat, D.; Bellaud, P.; Fautrel, A.; Turlin, B.; Clement, B.; Dooley, S.; et al. A novel transforming growth factor beta-induced long noncoding RNA promotes an inflammatory microenvironment in human intrahepatic cholangiocarcinoma. Hepatol. Commun. 2018, 2, 254–269. [Google Scholar] [CrossRef]
- Johnstone, C.N.; Chand, A.; Putoczki, T.L.; Ernst, M. Emerging roles for IL-11 signaling in cancer development and progression: Focus on breast cancer. Cytokine Growth Factor Rev. 2015, 26, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Jenkins, B.J. Recent insights into targeting the IL-6 cytokine family in inflammatory diseases and cancer. Nat. Rev. Immunol. 2018, 18, 773–789. [Google Scholar] [CrossRef] [PubMed]
- Eden, A.; Gaudet, F.; Waghmare, A.; Jaenisch, R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science 2003, 300, 455. [Google Scholar] [CrossRef] [PubMed]
- Zucman-Rossi, J.; Villanueva, A.; Nault, J.C.; Llovet, J.M. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology 2015, 149, 1226–1239. [Google Scholar] [CrossRef]
- Ding, X.; He, M.; Chan, A.W.H.; Song, Q.X.; Sze, S.C.; Chen, H.; Man, M.K.H.; Man, K.; Chan, S.L.; Lai, P.B.S.; et al. Genomic and Epigenomic Features of Primary and Recurrent Hepatocellular Carcinomas. Gastroenterology 2019, 157, 1630–1645. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Ancey, P.B.; Cros, M.P.; Durand, G.; Le Calvez-Kelm, F.; Hernandez-Vargas, H.; Herceg, Z. Dynamic imbalance between cancer cell subpopulations induced by transforming growth factor beta (TGF-beta) is associated with a DNA methylome switch. BMC Genom. 2014, 15, 435. [Google Scholar] [CrossRef] [PubMed]
- Galle, E.; Thienpont, B.; Cappuyns, S.; Venken, T.; Busschaert, P.; Van Haele, M.; Van Cutsem, E.; Roskams, T.; van Pelt, J.; Verslype, C.; et al. DNA methylation-driven EMT is a common mechanism of resistance to various therapeutic agents in cancer. Clin. Epigenet. 2020, 12, 27. [Google Scholar] [CrossRef]
- Heldin, C.H.; Miyazono, K.; ten Dijke, P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature 1997, 390, 465–471. [Google Scholar] [CrossRef]
- Papoutsoglou, P.; Louis, C.; Coulouarn, C. Transforming Growth Factor-Beta (TGFbeta) Signaling Pathway in Cholangiocarcinoma. Cells 2019, 8, 960. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Navarro-Serer, B.; Jeong, Y.J.; Chianchiano, P.; Xia, L.; Luchini, C.; Veronese, N.; Dowiak, C.; Ng, T.; Trujillo, M.A.; et al. Pattern of Invasion in Human Pancreatic Cancer Organoids Is Associated with Loss of SMAD4 and Clinical Outcome. Cancer Res. 2020, 80, 2804–2817. [Google Scholar] [CrossRef] [PubMed]
- Oyanagi, H.; Shimada, Y.; Nagahashi, M.; Ichikawa, H.; Tajima, Y.; Abe, K.; Nakano, M.; Kameyama, H.; Takii, Y.; Kawasaki, T.; et al. SMAD4 alteration associates with invasive-front pathological markers and poor prognosis in colorectal cancer. Histopathology 2019, 74, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Mishra, L.; Deng, C.X. The role of TGF-beta/SMAD4 signaling in cancer. Int. J. Biol. Sci. 2018, 14, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Aitchison, A.A.; Veerakumarasivam, A.; Vias, M.; Kumar, R.; Hamdy, F.C.; Neal, D.E.; Mills, I.G. Promoter methylation correlates with reduced Smad4 expression in advanced prostate cancer. Prostate 2008, 68, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Hahn, S.A.; Schutte, M.; Hoque, A.T.; Moskaluk, C.A.; da Costa, L.T.; Rozenblum, E.; Weinstein, C.L.; Fischer, A.; Yeo, C.J.; Hruban, R.H.; et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996, 271, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sheng, J.; Dai, D.; Liu, T.; Qi, F. Smad4 acts as tumor suppressor by antagonizing lymphangiogenesis in colorectal cancer. Pathol. Res. Pract. 2015, 211, 286–292. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Seki, T.; Okazaki, K. TGF-beta signal shifting between tumor suppression and fibro-carcinogenesis in human chronic liver diseases. J. Gastroenterol. 2014, 49, 971–981. [Google Scholar] [CrossRef]
- Valcourt, U.; Kowanetz, M.; Niimi, H.; Heldin, C.H.; Moustakas, A. TGF-beta and the Smad signaling pathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Mol. Biol. Cell 2005, 16, 1987–2002. [Google Scholar] [CrossRef]
- Yuan, T.; Chen, Z.; Yan, F.; Qian, M.; Luo, H.; Ye, S.; Cao, J.; Ying, M.; Dai, X.; Gai, R.; et al. Deubiquitinating enzyme USP10 promotes hepatocellular carcinoma metastasis through deubiquitinating and stabilizing Smad4 protein. Mol. Oncol. 2020, 14, 197–210. [Google Scholar] [CrossRef]
- Vincent, T.; Neve, E.P.; Johnson, J.R.; Kukalev, A.; Rojo, F.; Albanell, J.; Pietras, K.; Virtanen, I.; Philipson, L.; Leopold, P.L.; et al. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-beta mediated epithelial-mesenchymal transition. Nat. Cell Biol. 2009, 11, 943–950. [Google Scholar] [CrossRef]
- Moon, H.; Ju, H.L.; Chung, S.I.; Cho, K.J.; Eun, J.W.; Nam, S.W.; Han, K.H.; Calvisi, D.F.; Ro, S.W. Transforming Growth Factor-beta Promotes Liver Tumorigenesis in Mice via Up-regulation of Snail. Gastroenterology 2017, 153, 1378–1391. [Google Scholar] [CrossRef]
- Wang, H.; Song, X.; Liao, H.; Wang, P.; Zhang, Y.; Che, L.; Zhang, J.; Zhou, Y.; Cigliano, A.; Ament, C.; et al. Overexpression of Mothers Against Decapentaplegic Homolog 7 Activates the Yes-Associated Protein/NOTCH Cascade and Promotes Liver Carcinogenesis in Mice and Humans. Hepatology 2021, 74, 248–263. [Google Scholar] [CrossRef]
- Park, Y.N.; Chae, K.J.; Oh, B.K.; Choi, J.; Choi, K.S.; Park, C. Expression of Smad7 in hepatocellular carcinoma and dysplastic nodules: Resistance mechanism to transforming growth factor-beta. Hepatogastroenterology 2004, 51, 396–400. [Google Scholar] [PubMed]
- Xia, H.; Ooi, L.L.; Hui, K.M. MicroRNA-216a/217-induced epithelial-mesenchymal transition targets PTEN and SMAD7 to promote drug resistance and recurrence of liver cancer. Hepatology 2013, 58, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Dillard, D.G.; Muller, S.; Cohen, C.; Bloch, D.; Del Gaudio, J.M.; Gal, A.A. High tumor grade in salivary gland mucoepidermoid carcinomas and loss of expression of transforming growth factor beta receptor type II. Arch. Otolaryngol. Head Neck Surg. 2001, 127, 683–686. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gobbi, H.; Arteaga, C.L.; Jensen, R.A.; Simpson, J.F.; Dupont, W.D.; Olson, S.J.; Schuyler, P.A.; Plummer, W.D., Jr.; Page, D.L. Loss of expression of transforming growth factor beta type II receptor correlates with high tumour grade in human breast in-situ and invasive carcinomas. Histopathology 2000, 36, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Wang, J.; Yumoto, K.; Jung, Y.; Cackowski, F.C.; Decker, A.M.; Li, Y.; Franceschi, R.T.; Pienta, K.J.; Taichman, R.S. DNMT1 Regulates Epithelial-Mesenchymal Transition and Cancer Stem Cells, Which Promotes Prostate Cancer Metastasis. Neoplasia 2016, 18, 553–566. [Google Scholar] [CrossRef]
- Wang, X.; Liang, Z.; Xu, X.; Li, J.; Zhu, Y.; Meng, S.; Li, S.; Wang, S.; Xie, B.; Ji, A.; et al. miR-148a-3p represses proliferation and EMT by establishing regulatory circuits between ERBB3/AKT2/c-myc and DNMT1 in bladder cancer. Cell Death Dis. 2016, 7, e2503. [Google Scholar] [CrossRef] [PubMed]
- Ateeq, B.; Unterberger, A.; Szyf, M.; Rabbani, S.A. Pharmacological inhibition of DNA methylation induces proinvasive and prometastatic genes in vitro and in vivo. Neoplasia 2008, 10, 266–278. [Google Scholar] [CrossRef]
- Jiang, H.; Cao, H.J.; Ma, N.; Bao, W.D.; Wang, J.J.; Chen, T.W.; Zhang, E.B.; Yuan, Y.M.; Ni, Q.Z.; Zhang, F.K.; et al. Chromatin remodeling factor ARID2 suppresses hepatocellular carcinoma metastasis via DNMT1-Snail axis. Proc. Natl. Acad. Sci. USA 2020, 117, 4770–4780. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, J.U.; Factor, V.M.; Thorgeirsson, S.S. Epigenetic regulation of cancer stem cells in liver cancer: Current concepts and clinical implications. J. Hepatol. 2010, 53, 568–577. [Google Scholar] [CrossRef]
- Marquardt, J.U.; Raggi, C.; Andersen, J.B.; Seo, D.; Avital, I.; Geller, D.; Lee, Y.H.; Kitade, M.; Holczbauer, A.; Gillen, M.C.; et al. Human hepatic cancer stem cells are characterized by common stemness traits and diverse oncogenic pathways. Hepatology 2011, 54, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zuo, X.; Pu, L.; Zhang, Y.; Han, G.; Zhang, L.; Wu, Z.; You, W.; Qin, J.; Dai, X.; et al. Hypomethylation-mediated activation of cancer/testis antigen KK-LC-1 facilitates hepatocellular carcinoma progression through activating the Notch1/Hes1 signalling. Cell Prolif. 2019, 52, e12581. [Google Scholar] [CrossRef]
- Henrique, R.; Jeronimo, C. DNA hypomethylation in plasma as a cancer biomarker: When less is more? Expert Rev. Mol. Diagn. 2014, 14, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Ding, B.; Lou, W.; Lin, S. Promoter Hypomethylation and miR-145-5p Downregulation- Mediated HDAC11 Overexpression Promotes Sorafenib Resistance and Metastasis of Hepatocellular Carcinoma Cells. Front. Cell Dev. Biol. 2020, 8, 724. [Google Scholar] [CrossRef] [PubMed]
- Azad, N.; Zahnow, C.A.; Rudin, C.M.; Baylin, S.B. The future of epigenetic therapy in solid tumours--lessons from the past. Nat. Rev. Clin. Oncol. 2013, 10, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Berdasco, M.; Esteller, M. Clinical epigenetics: Seizing opportunities for translation. Nat. Rev. Genet. 2019, 20, 109–127. [Google Scholar] [CrossRef] [PubMed]
- Bitzer, M.; Horger, M.; Giannini, E.G.; Ganten, T.M.; Worns, M.A.; Siveke, J.T.; Dollinger, M.M.; Gerken, G.; Scheulen, M.E.; Wege, H.; et al. Resminostat plus sorafenib as second-line therapy of advanced hepatocellular carcinoma—The SHELTER study. J. Hepatol. 2016, 65, 280–288. [Google Scholar] [CrossRef]
- Fan, H.; Lu, X.; Wang, X.; Liu, Y.; Guo, B.; Zhang, Y.; Zhang, W.; Nie, J.; Feng, K.; Chen, M.; et al. Low-dose decitabine-based chemoimmunotherapy for patients with refractory advanced solid tumors: A phase I/II report. J. Immunol. Res. 2014, 2014, 371087. [Google Scholar] [CrossRef]
- Yeo, W.; Chan, S.L.; Mo, F.K.; Chu, C.M.; Hui, J.W.; Tong, J.H.; Chan, A.W.; Koh, J.; Hui, E.P.; Loong, H.; et al. Phase I/II study of temsirolimus for patients with unresectable Hepatocellular Carcinoma (HCC)- a correlative study to explore potential biomarkers for response. BMC Cancer 2015, 15, 395. [Google Scholar] [CrossRef]
- Gailhouste, L.; Liew, L.C.; Yasukawa, K.; Hatada, I.; Tanaka, Y.; Nakagama, H.; Ochiya, T. Differentiation Therapy by Epigenetic Reconditioning Exerts Antitumor Effects on Liver Cancer Cells. Mol. Ther. 2018, 26, 1840–1854. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.K.; Li, Y.; Pandit, H.; Li, S.; Pulliam, Z.; Zheng, Q.; Yu, Y.; Martin, R.C.G. Epigenetic modulation enhances immunotherapy for hepatocellular carcinoma. Cell Immunol. 2019, 336, 66–74. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bévant, K.; Desoteux, M.; Abdel Wahab, A.H.A.; Abdel Wahab, S.A.; Metwally, A.M.; Coulouarn, C. DNA Methylation of TGFβ Target Genes: Epigenetic Control of TGFβ Functional Duality in Liver Cancer. Cells 2021, 10, 2207. https://doi.org/10.3390/cells10092207
Bévant K, Desoteux M, Abdel Wahab AHA, Abdel Wahab SA, Metwally AM, Coulouarn C. DNA Methylation of TGFβ Target Genes: Epigenetic Control of TGFβ Functional Duality in Liver Cancer. Cells. 2021; 10(9):2207. https://doi.org/10.3390/cells10092207
Chicago/Turabian StyleBévant, Kevin, Matthis Desoteux, Abdel Hady A. Abdel Wahab, Sabrin A. Abdel Wahab, Ayman Mohamed Metwally, and Cédric Coulouarn. 2021. "DNA Methylation of TGFβ Target Genes: Epigenetic Control of TGFβ Functional Duality in Liver Cancer" Cells 10, no. 9: 2207. https://doi.org/10.3390/cells10092207
APA StyleBévant, K., Desoteux, M., Abdel Wahab, A. H. A., Abdel Wahab, S. A., Metwally, A. M., & Coulouarn, C. (2021). DNA Methylation of TGFβ Target Genes: Epigenetic Control of TGFβ Functional Duality in Liver Cancer. Cells, 10(9), 2207. https://doi.org/10.3390/cells10092207