
HSPB1 Is Essential for Inducing Resistance to Proteotoxic Stress in Beta-Cells

 , , , ,
, , , ,
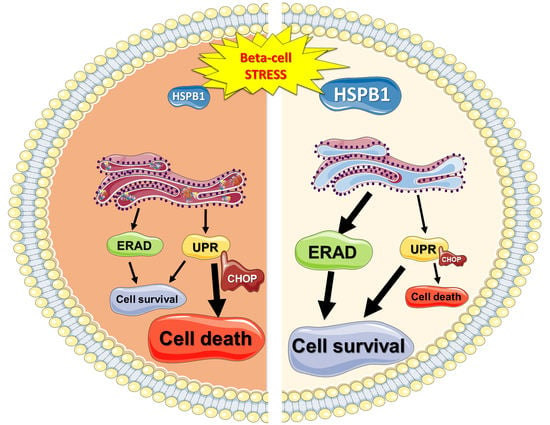
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Mouse Islets Isolation and Culture
2.2. Cell Culture of Min6 Cells
2.3. HSPB1 Silencing and Overexpression in Mouse Islets
2.3.1. HSPB1 Silencing
2.3.2. Transient HSPB1 Overexpression
2.4. Cell Treatments
2.5. Cell Viability Evaluated by Microscopy
2.6. Glucose-Induced Insulin Release Assay
2.7. Western Blots
2.8. Study of the Rate of Protein Degradation by the Ubiquitin-Proteasome System
2.9. Statistical Analysis
3. Results
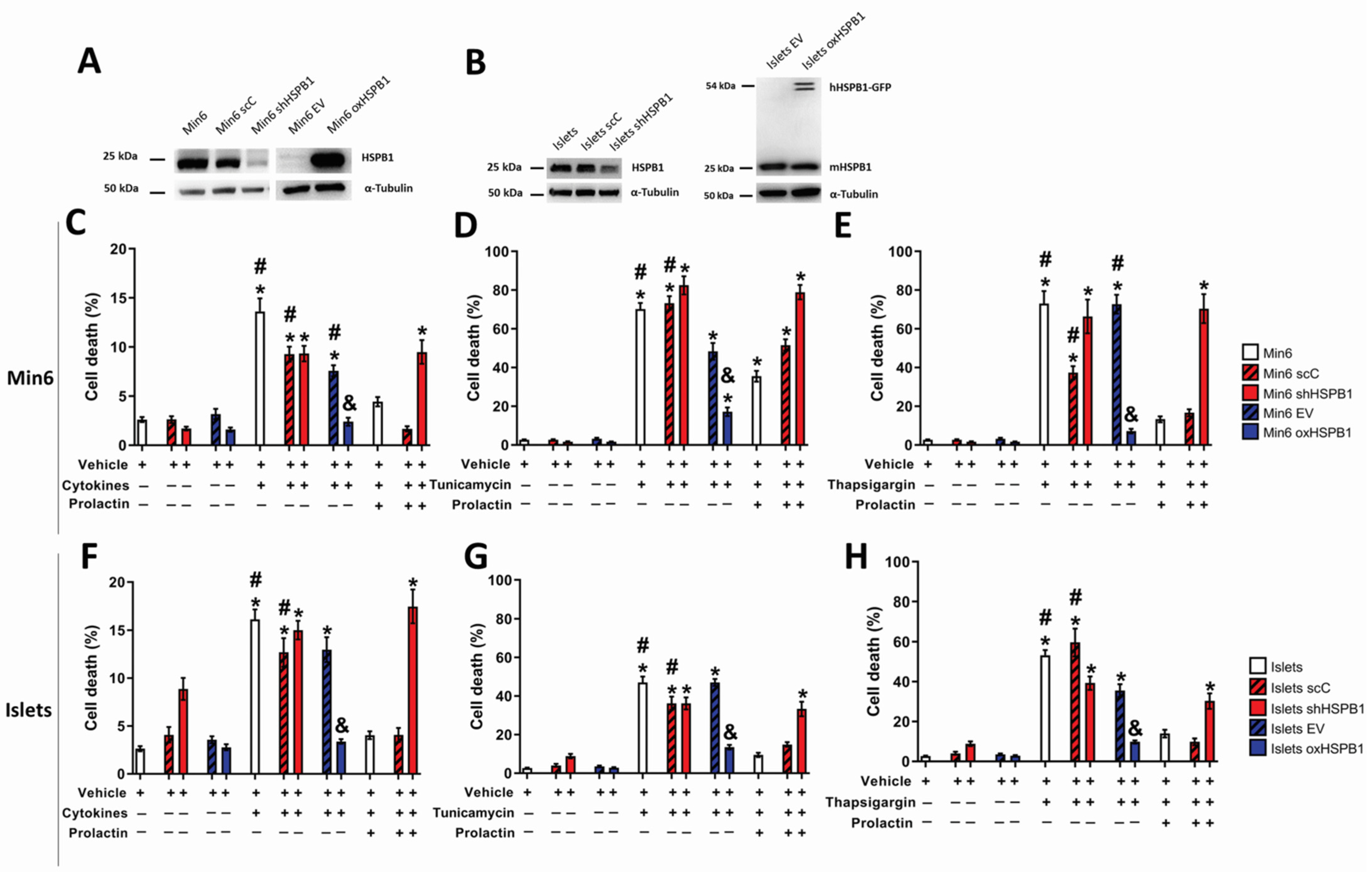
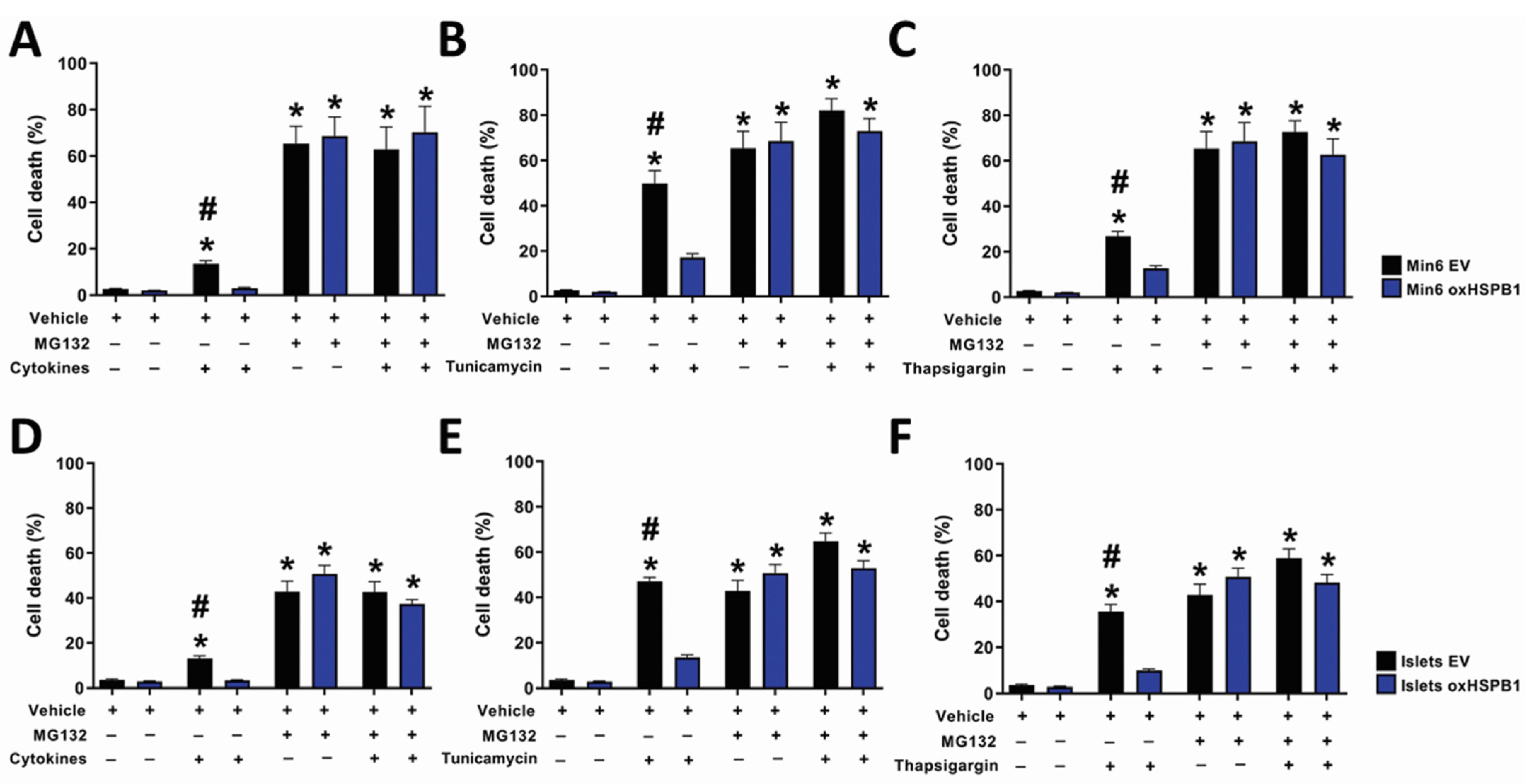
3.1. HSPB1 Is Required for Beta-Cell Cytoprotection against ER Stress-Induced Apoptosis
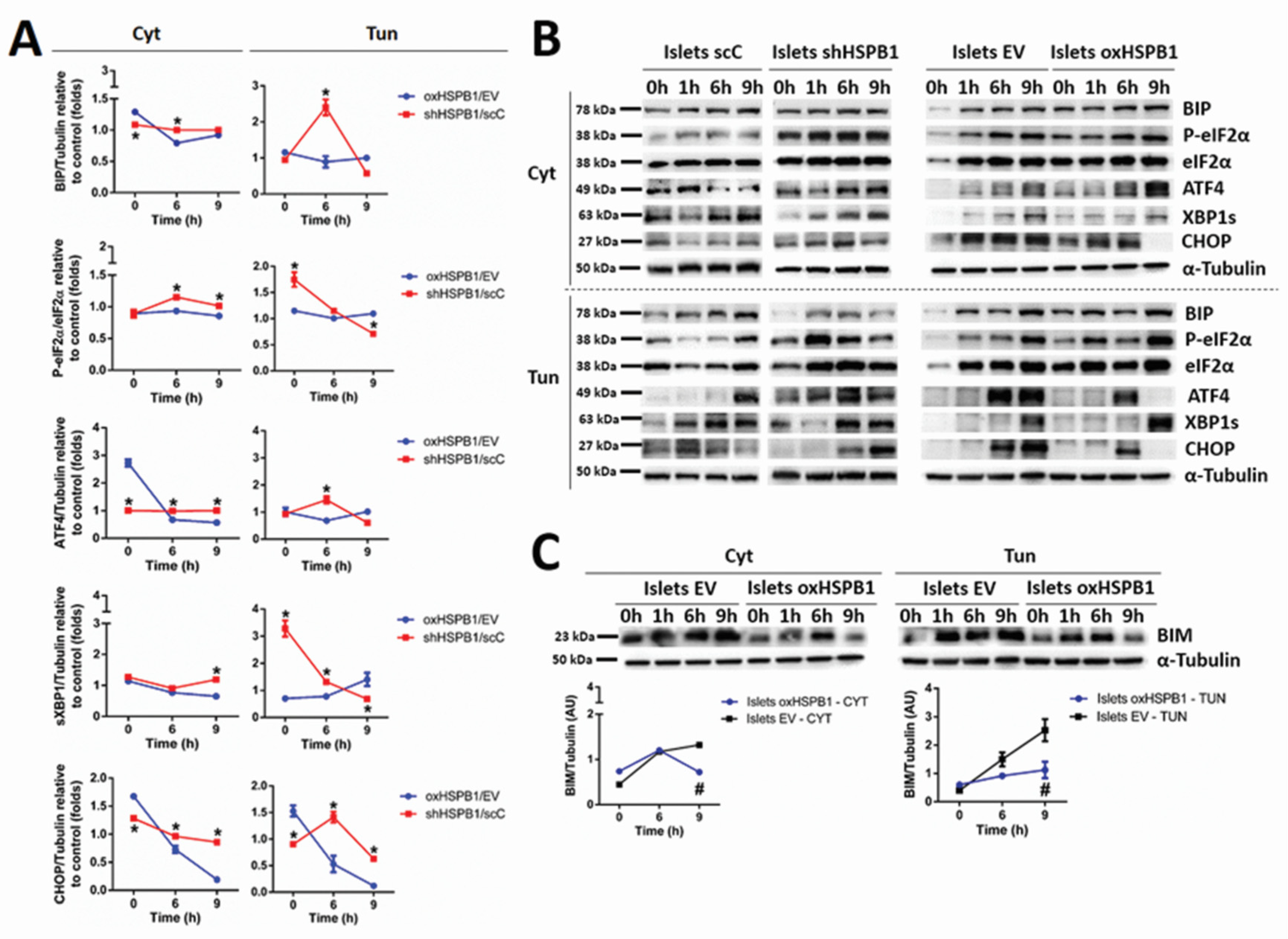
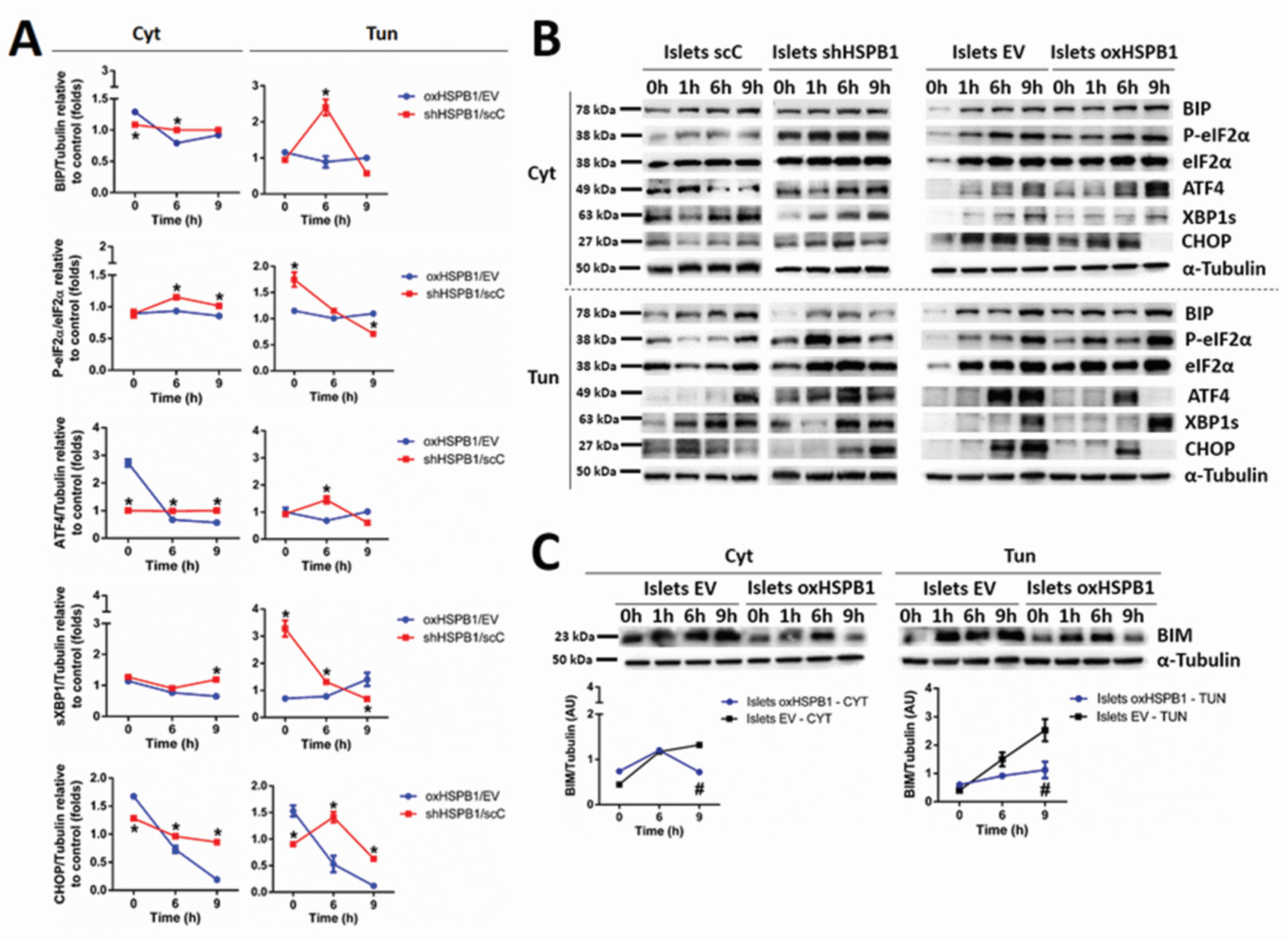
3.2. HSPB1 Modulates UPR in Mouse Islets and Min6 Cells
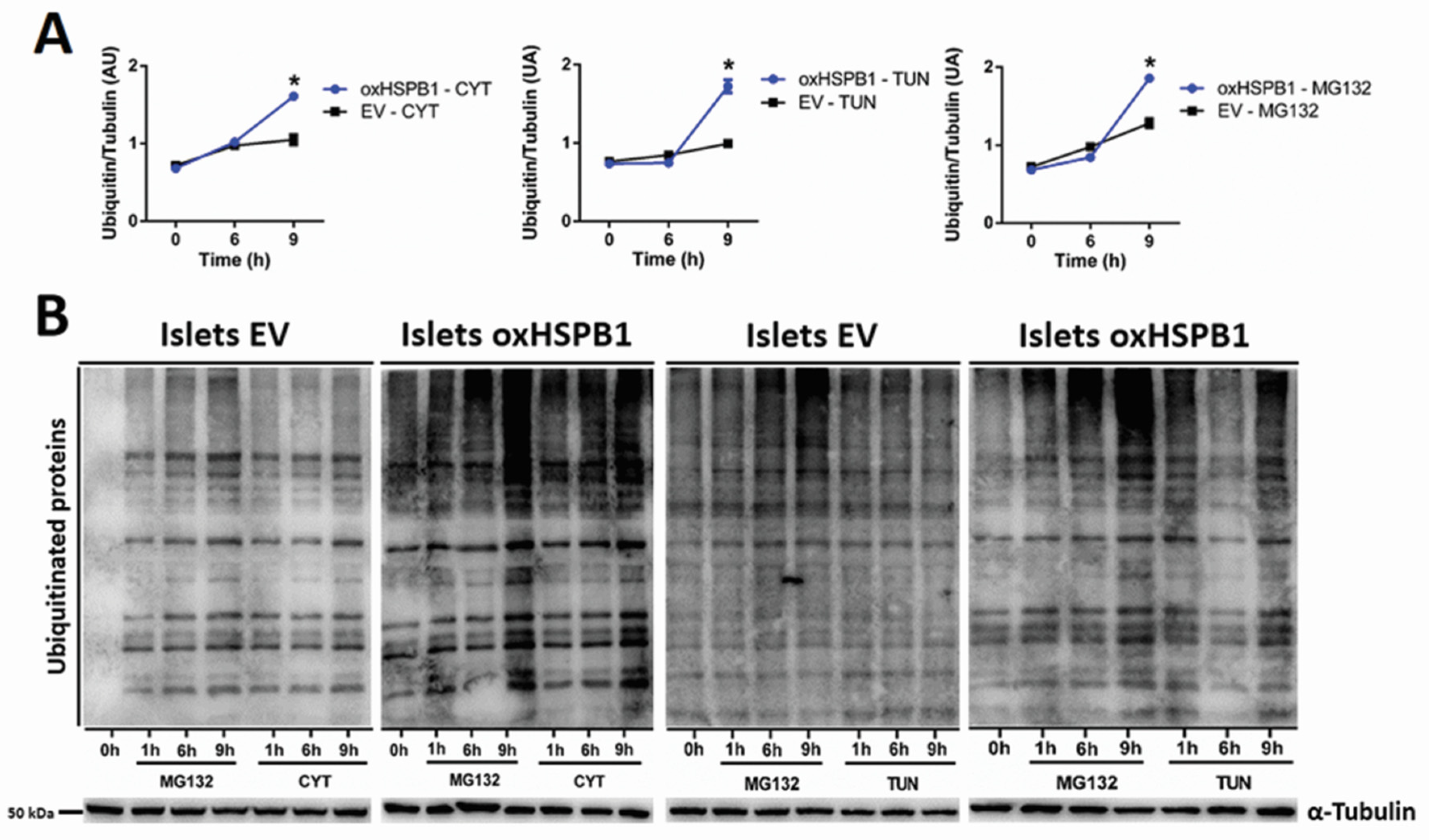
3.3. HSPB1-Mediated Beta-Cell Survival Requires a Functional Ubiquitin-Proteasome Protein Degradation System
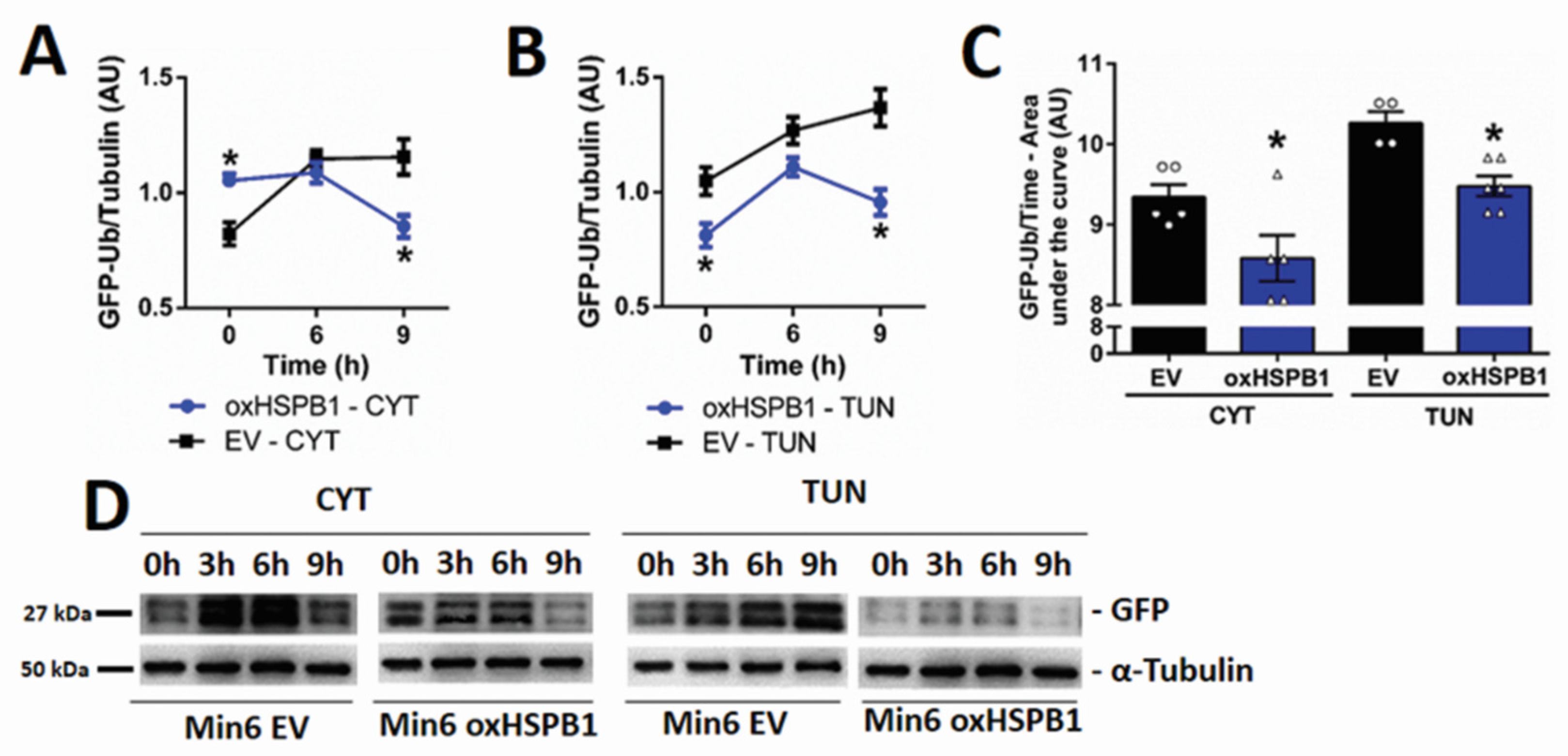
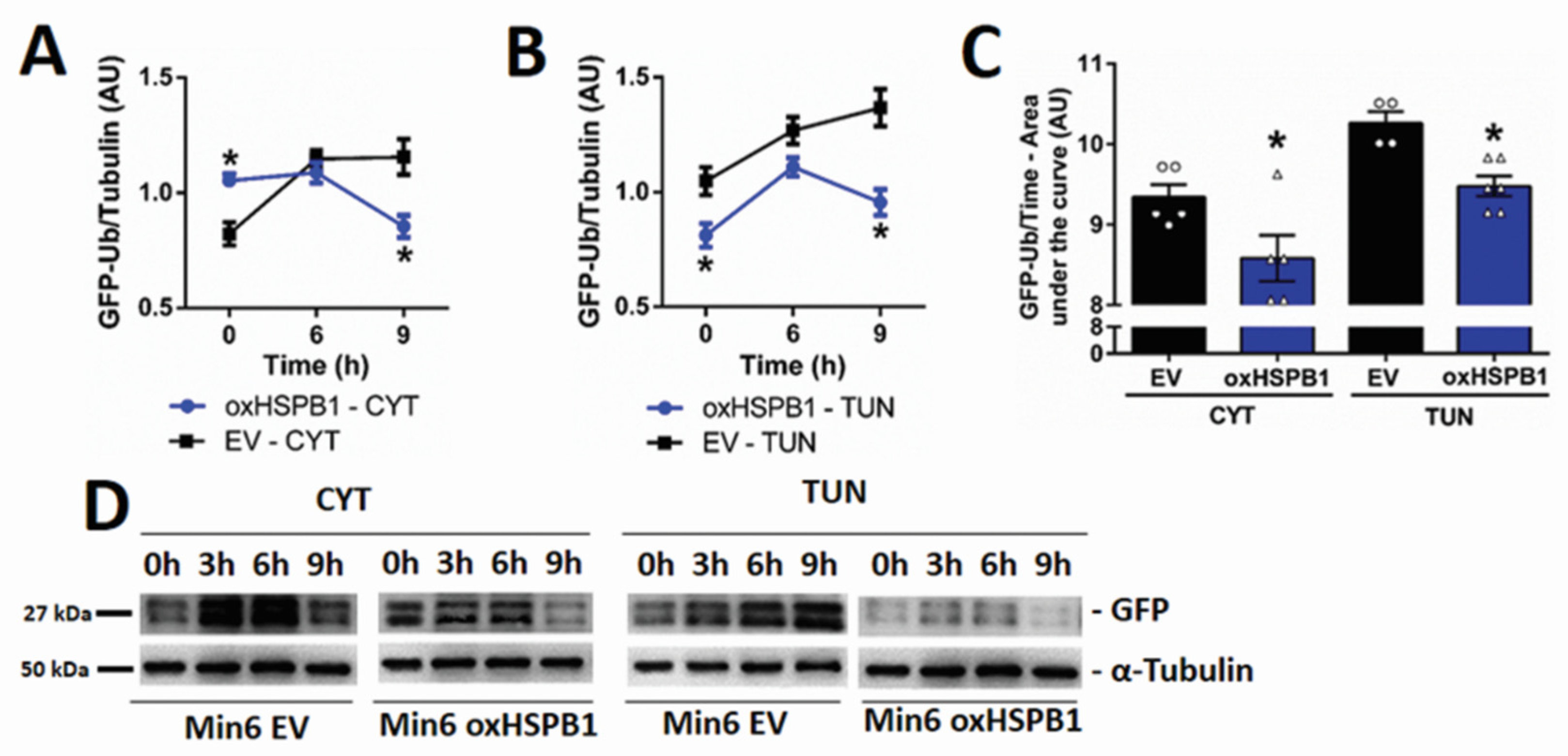
3.4. HSPB1 Accelerates the Rate of Proteasomal Protein Degradation
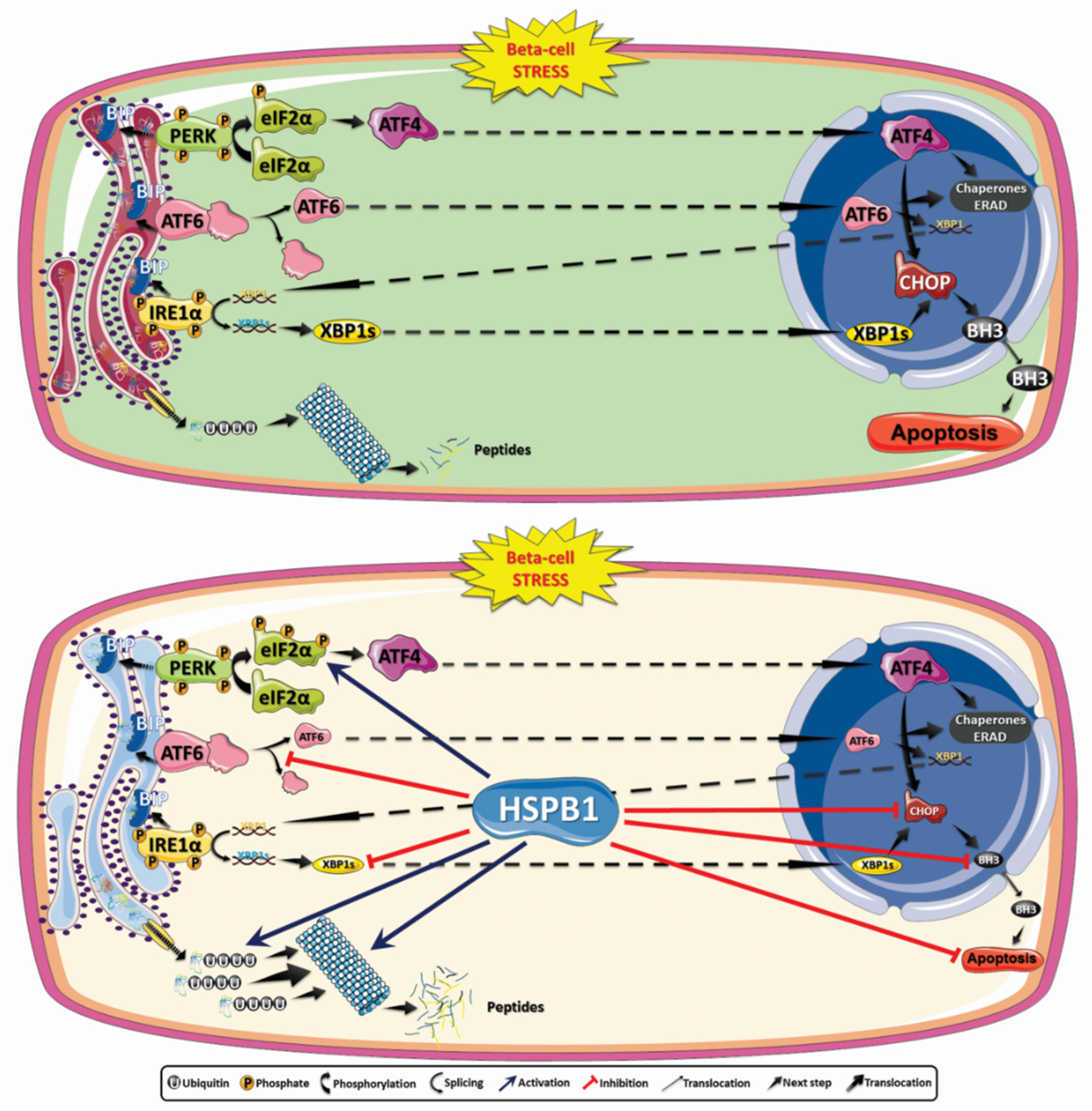
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ashoor, M.F.; Bintouq, A.K.; Rutter, M.K.; Malik, R.A. Pancreatic islet cell transplantation as a treatment for brittle type 1 diabetes: A case report and review of the literature. J. Taibah Univ. Med. Sci. 2016, 11, 395–400. [Google Scholar] [CrossRef]
- Shapiro, A.M.J.; Pokrywczynska, M.; Ricordi, C. Clinical pancreatic islet transplantation. Nat. Rev. Endocrinol. 2016, 13, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Gamble, A.; Pepper, A.R.; Bruni, A.; Shapiro, A.M.J. The journey of islet cell transplantation and future development. Islets 2018, 10, 80–94. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, A.; Brayman, K. Update on Islet Cell Transplantation for Type 1 Diabetes. Semin. Intervent. Radiol. 2012, 29, 090–098. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, B.D.B.; Platt, J.L.; Rabe, F.L.; Dunn, D.L.; Bach, F.H.; Sutherland, D.E.R. Differential Roles of Mac-1+ Cells, and CD4+ and CD8+ T Lymphocytes in Primary Nonfunction and Classic Rejection of Islet Allografts. J. Exp. Med. 1990, 172, 291–302. [Google Scholar] [CrossRef] [Green Version]
- Freemark, M.; Avril, I.; Fleenor, D.O.N.; Driscoll, P.; Petro, A.N.N.; Opara, E.; Kendall, W.; Oden, J.O.N.; Bridges, S.; Binart, N.; et al. Targeted deletion of the PRL receptor: Effects on islet development, insulin production, and glucose tolerance. Endocrinology 2002, 143, 1378–1385. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Mita, A.; Ricordi, C.; Messinger, S.; Miki, A.; Sakuma, Y.; Timoneri, F.; Barker, S.; Fornoni, A.; Molano, R.D.; et al. Prolactin supplementation to culture medium improves beta-cell survival. Transplantation 2010, 89, 1328–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Guthalu, N.K.; Filipowska, J.; Hampton, R.F.; Leblanc, S.; Garcia-Ocana, A.; Vasavada, R.C. Lactogens reduce endoplasmic reticulum stress-induced rodent and human β-cell death and diabetes incidence in Akita mice. Diabetes 2020, 69, 1–46. [Google Scholar] [CrossRef] [PubMed]
- Wailemann, R.A.; Terra, L.F.; Oliveira, T.C.; Dos Santos, A.F.; Gomes, V.M.; Labriola, L. Heat shock protein B1 is required for the prolactin-induced cytoprotective effects on pancreatic islets. Mol. Cell. Endocrinol. 2018, 477, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Terra, L.F.; Wailemann, R.A.M.; dos Santos, A.F.; Gomes, V.M.; Silva, R.P.; Laporte, A.; Meotti, F.C.; Terra, W.R.; Palmisano, G.; Lortz, S.; et al. Heat shock protein B1 is a key mediator of prolactin-induced beta-cell cytoprotection against oxidative stress. Free Radic. Biol. Med. 2019, 134, 394–405. [Google Scholar] [CrossRef] [PubMed]
- Bonam, S.R.; Ruff, M.; Muller, S. HSPA8/HSC70 in Immune Disorders: A Molecular Rheostat that Adjusts Chaperone-Mediated Autophagy Substrates. Cells 2019, 8, 849. [Google Scholar] [CrossRef] [Green Version]
- Kampinga, H.H.; Garrido, C. HSPBs: Small proteins with big implications in human disease. Int. J. Biochem. Cell Biol. 2012, 44, 1706–1710. [Google Scholar] [CrossRef] [PubMed]
- Garrido, C.; Paul, C.; Seigneuric, R.; Kampinga, H.H. The small heat shock proteins family: The long forgotten chaperones. Int. J. Biochem. Cell Biol. 2012, 44, 1588–1592. [Google Scholar] [CrossRef]
- Mule, S.N.; Gomes, V.D.M.; Wailemann, R.A.M.; Macedo-da-Silva, J.; Rosa-Fernandes, L.; Larsen, M.R.; Labriola, L.; Palmisano, G. HSPB1 influences mitochondrial respiration in ER-stressed beta cells. Biochim. Biophys. Acta—Proteins Proteomics 2021, 1869, 140680. [Google Scholar] [CrossRef] [PubMed]
- Cnop, M.; Welsh, N.; Jonas, J.; Jo, A.; Lenzen, S. Mechanisms of Pancreatic Beta Cell Death in Type 1 and Type 2 Diabetes. Diabetes 2005, 54, 97–107. [Google Scholar] [CrossRef] [Green Version]
- Eizirik, D.L.; Cardozo, A.K.; Cnop, M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr. Rev. 2008, 29, 42–61. [Google Scholar] [CrossRef] [Green Version]
- Cardozo, A.K.; Ortis, F.; Storling, J.; Feng, Y.; Rasschaert, J.; Tonnesen, M.; Eylen, V.; Mandrup-poulsen, T. Endoplasmic Reticulum Stress in Pancreatic β-Cells. Diabetes 2005, 54, 452–461. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.; Ricordi, C.; Mita, A.; Miki, A.; Sakuma, Y.; Molano, R.D.; Fornoni, A.; Pileggi, A.; Inverardi, L.; Ichii, H. β-Cell Specific Cytoprotection by Prolactin on Human Islets. Transplant. Proc. 2008, 40, 382–383. [Google Scholar] [CrossRef] [Green Version]
- Özcan, U.; Yilmaz, E.; Özcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Görgün, C.Z.; Hotamisligil, G.S. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef] [Green Version]
- Brozzi, F.; Eizirik, D.L. ER stress and the decline and fall of pancreatic beta cells in type 1 diabetes. Ups. J. Med. Sci. 2016, 121, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling—From basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Publ. Gr. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Schwesinger, C. The ubiquitin–proteasome system in kidney physiology and disease. Nat. Rev. Nephrol. 2019, 15, 393–411. [Google Scholar] [CrossRef]
- Tsai, Y.C.; Weissman, A.M. The unfolded protein response, degradation from the endoplasmic reticulum, and cancer. Genes Cancer 2010, 1, 764–778. [Google Scholar] [CrossRef]
- Cnop, M.; Foufelle, F.; Velloso, L.A. Endoplasmic reticulum stress, obesity and diabetes. Trends Mol. Med. 2012, 18, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, S.G.; Gromada, J.; Urano, F. Endoplasmic reticulum stress and pancreatic beta cell death Protein homeostasis in the β cell. Trends Endocrinol. Metab. 2012, 22, 266–274. [Google Scholar] [CrossRef] [Green Version]
- Delbridge, A.R.D.; Grabow, S.; Strasser, A.; Vaux, D.L. Thirty years of BCL-2: Translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer 2016, 16, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Einsele-Scholz, S.; Malmsheimer, S.; Bertram, K.; Stehle, D.; Johänning, J.; Manz, M.; Daniel, P.T.; Gillissen, B.F.; Schulze-Osthoff, K.; Essmann, F. Bok is a genuine multi-BH-domain protein that triggers apoptosis in the absence of Bax and Bak. J. Cell Sci. 2016, 129, 3054. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, H.; Asano, T.; Tsukuda, K.; Katagiri, H.; Inukai, K.; Anai, M.; Kikuchi, M.; Yazaki, Y.; Miyazaki, J.I.; Oka, Y. Pancreatic beta cell line MIN6 exhibits characteristics of glucose metabolism and glucose-stimulated insulin secretion similar to those of normal islets. Diabetologia 1993, 36, 1139–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiscornia, G.; Singer, O.; Verma, I.M. Production and purification of lentiviral vectors. Nat. Protoc. 2006, 1, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Tiscornia, G.; Singer, O.; Verma, I.M. Design and cloning of lentiviral vectors expressing small interfering RNAs. Nat. Protoc. 2006, 1, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Voss, O.H.; Batra, S.; Kolattukudy, S.J.; Gonzalez-Mejia, M.E.; Smith, J.B.; Doseff, A.I. Binding of caspase-3 prodomain to heat shock protein 27 regulates monocyte apoptosis by inhibiting caspase-3 proteolytic activation. J. Biol. Chem. 2007, 282, 25088–25099. [Google Scholar] [CrossRef] [Green Version]
- Labriola, L.; Montor, W.R.; Krogh, K.; Lojudice, F.H.; Genzini, T.; Goldberg, A.C.; Eliaschewitz, F.G.; Sogayar, M.C. Beneficial effects of prolactin and laminin on human pancreatic islet-cell cultures. Mol. Cell. Endocrinol. 2007, 263, 120–133. [Google Scholar] [CrossRef] [PubMed]
- Lindsten, K.; Menéndez-Benito, V.; Masucci, M.G.; Dantuma, N.P. A transgenic mouse model of the ubiquitin/proteasome system. Nat. Biotechnol. 2003, 21, 897–902. [Google Scholar] [CrossRef] [PubMed]
- Dantuma, N.P.; Lindsten, K.; Glas, R.; Jellne, M.; Masucci, M.G. Short-lived green fluorescent proteins for quantifying ubiquitin/proteasome-dependent proteolysis in living cells. Nat. Biotechnol. 2000, 18, 538–543. [Google Scholar] [CrossRef]
- Nikesitch, N.; Lee, J.M.; Ling, S.; Roberts, T.L. Endoplasmic reticulum stress in the development of multiple myeloma and drug resistance. Clin. Transl. Immunol. 2018, 7, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Sommer, T.; Wolf, D.H. Endoplasmic reticulum degradation: Reverse protein flow of no return. FASEB J. 1997, 11, 1227–1233. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, J.L.; McCracken, A.A. ER protein quality control and proteasome-mediated protein degradation. Semin. Cell Dev. Biol. 1999, 10, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Karagöz, G.E.; Acosta-Alvear, D.; Walter, P. The Unfolded Protein Response: Detecting and Responding to Fluctuations in the Protein-Folding Capacity of the Endoplasmic Reticulum. Cold Spring Harb. Perspect. Biol. 2019, 11, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, K.; Haslbeck, M.; Buchner, J. The Heat Shock Response: Life on the Verge of Death. Mol. Cell 2010, 40, 253–266. [Google Scholar] [CrossRef]
- Terra, L.F.; Garay-Malpartida, M.H.; Wailemann, R.A.M.; Sogayar, M.C.; Labriola, L. Recombinant human prolactin promotes human beta cell survival via inhibition of extrinsic and intrinsic apoptosis pathways. Diabetologia 2011, 54, 1388–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labriola, L.; Ferreira, G.B.; Montor, W.R.; Demasi, M.A.A.; Pimenta, D.C.; Lojudice, F.H.; Genzini, T.; Goldberg, A.C.; Eliaschewitz, F.G.; Sogayar, M.C. Prolactin-induced changes in protein expression in human pancreatic islets. Mol. Cell. Endocrinol. 2007, 264, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Amen, O.M.; Sarker, S.D.; Ghildyal, R.; Arya, A. Endoplasmic reticulum stress activates unfolded protein response signaling and mediates inflammation, obesity, and cardiac dysfunction: Therapeutic and molecular approach. Front. Pharmacol. 2019, 10, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Kong, F.J.; Wu, J.H.; Sun, S.Y.; Zhou, J.Q. The endoplasmic reticulum stress/autophagy pathway is involved in cholesterol-induced pancreatic β-cell injury. Sci. Rep. 2017, 7, 44746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, A.P.; Klocke, B.J.; Ballestas, M.E.; Roth, K.A. CHOP potentially co-operates with FOXO3a in neuronal cells to regulate PUMA and BIM expression in response to ER stress. PLoS ONE 2012, 7, e39586. [Google Scholar] [CrossRef] [PubMed]
- Allagnat, F.; Christulia, F.; Ortis, F.; Pirot, P.; Lortz, S.; Lenzen, S.; Eizirik, D.L.; Cardozo, A.K. Sustained production of spliced X-box binding protein 1 (XBP1) induces pancreatic beta cell dysfunction and apoptosis. Diabetologia 2010, 53, 1120–1130. [Google Scholar] [CrossRef] [Green Version]
- Sharma, R.B.; Darko, C.; Alonso, L.C. Intersection of the ATF6 and XBP1 ER stress pathways in mouse islet cells. J. Biol. Chem. 2020, 295, 14164–14177. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liang, Y.; Lin, Y.; Liu, Y.; You, Y.; Yin, W. IRE1α-TRAF2-ASK1 pathway is involved in CSTMP-induced apoptosis and ER stress in human non-small cell lung cancer A549 cells. Biomed. Pharmacother. 2016, 82, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Lei, K.; Davis, R.J. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 2432–2437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litwak, S.A.; Pang, L.; Galic, S.; Igoillo-Esteve, M.; Stanley, W.J.; Turatsinze, J.V.; Loh, K.; Thomas, H.E.; Sharma, A.; Trepo, E.; et al. JNK activation of BIM promotes hepatic oxidative stress, steatosis, and insulin resistance in obesity. Diabetes 2017, 66, 2973–2986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurzov, E.N.; Ortis, F.; Cunha, D.A.; Gosset, G.; Li, M.; Cardozo, A.K.; Eizirik, D.L. Signaling by IL-1β+IFN-γ and ER stress converge on DP5/Hrk activation: A novel mechanism for pancreatic β-cell apoptosis. Cell Death Differ. 2009, 16, 1539–1550. [Google Scholar] [CrossRef] [PubMed]
- Cnop, M.; Toivonen, S.; Igoillo-Esteve, M.; Salpea, P. Endoplasmic reticulum stress and eIF2α phosphorylation: The Achilles heel of pancreatic β cells. Mol. Metab. 2017, 6, 1024–1039. [Google Scholar] [CrossRef]
- Chan, J.Y.; Luzuriaga, J.; Maxwell, E.L.; West, P.K.; Bensellam, M.; Laybutt, D.R. The balance between adaptive and apoptotic unfolded protein responses regulates b-cell death under ER stress conditions through. Mol. Cell. Endocrinol. 2015, 413, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Back, S.H.; Kaufman, R.J. Endoplasmic Reticulum Stress and Type 2 Diabetes. Annu Rev Biochem 2012, 81, 767–793. [Google Scholar] [CrossRef] [Green Version]
- Eizirik, D.L.; Cnop, M. ER stress in pancreatic β cells: The thin red line between adaptation and failure. Sci. Signal. 2010, 3, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Weiss, M.A.; Arunagiri, A.; Yong, J.; Rege, N.; Sun, J.; Haataja, L.; Kaufman, R.J.; Arvan, P. Biosynthesis, structure, and folding of the insulin precursor protein. Diabetes Obes. Metab. 2018, 20, 28–50. [Google Scholar] [CrossRef]
- Shen, Y.; Ballar, P.; Apostolou, A.; Doong, H.; Fang, S. ER stress differentially regulates the stabilities of ERAD ubiquitin ligases and their substrates. Biochem. Biophys. Res. Commun. 2007, 352, 919–924. [Google Scholar] [CrossRef]
- Vembar, S.S.; Brodsky, J.L. One step at a time: Endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 2008, 9, 944–957. [Google Scholar] [CrossRef] [PubMed]
- Stolz, A.; Wolf, D.H. Endoplasmic reticulum associated protein degradation: A chaperone assisted journey to hell. Biochim. Biophys. Acta Mol. Cell Res. 2010, 1803, 694–705. [Google Scholar] [CrossRef] [Green Version]
- Hedhli, N.; Wang, L.; Wang, Q.; Rashed, E.; Tian, Y.; Sui, X.; Madura, K.; Depre, C. Proteasome activation during cardiac hypertrophy by the chaperone H11 Kinase/Hsp. Cardiovasc. Res. 2008, 77, 497–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arrigo, A.P.; Gibert, B. Protein interactomes of three stress inducible small heat shock proteins: HspB1, HspB5 and HspB8. Int. J. Hyperth. 2013, 29, 409–422. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.; Mnich, K.; Oommen, D.; Chakravarthy, R.; Almeida-Souza, L.; Krols, M.; Saveljeva, S.; Doyle, K.; Gupta, S.; Timmerman, V.; et al. HSPB1 facilitates ERK-mediated phosphorylation and degradation of BIM to attenuate endoplasmic reticulum stress-induced apoptosis. Cell Death Dis. 2017, 8, e3026. [Google Scholar] [CrossRef]
- Nogueira, T.C.; Paula, F.M.; Villate, O.; Colli, M.L.; Moura, R.F.; Cunha, D.A.; Marselli, L.; Marchetti, P.; Cnop, M.; Julier, C.; et al. GLIS3, a Susceptibility Gene for Type 1 and Type 2 Diabetes, Modulates Pancreatic Beta Cell Apoptosis via Regulation of a Splice Variant of the BH3-Only Protein Bim. PLoS Genet. 2013, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Santin, I.; Moore, F.; Colli, M.L.; Gurzov, E.N.; Marselli, L.; Marchetti, P.; Eizirik, D.L. PTPN2, a candidate gene for type 1 diabetes, modulates pancreatic β-cell apoptosis via regulation of the BH3-only protein bim. Diabetes 2011, 60, 3279–3288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parcellier, A.; Schmitt, E.; Gurbuxani, S.; Seigneurin-Berny, D.; Pance, A.; Chantôme, A.; Plenchette, S.; Khochbin, S.; Solary, E.; Garrido, C. HSP27 is a ubiquitin-binding protein involved in I-kappaBalpha proteasomal degradation. Mol. Cell. Biol. 2003, 23, 5790–5802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacy, P.E. The Pancreatic Beta Cell. N. Engl. J. Med. 1967, 276, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Al-Awar, A.; Kupai, K.; Veszelka, M.; Szucs, G.; Attieh, Z.; Murlasits, Z.; Török, S.; Pósa, A.; Varga, C. Experimental Diabetes Mellitus in Different Animal Models. J. Diabetes Res. 2016, 2016, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomes, V.M.; Wailemann, R.A.M.; Arini, G.S.; Oliveira, T.C.; Almeida, D.R.Q.; dos Santos, A.F.; Terra, L.F.; Lortz, S.; Labriola, L. HSPB1 Is Essential for Inducing Resistance to Proteotoxic Stress in Beta-Cells. Cells 2021, 10, 2178. https://doi.org/10.3390/cells10092178
Gomes VM, Wailemann RAM, Arini GS, Oliveira TC, Almeida DRQ, dos Santos AF, Terra LF, Lortz S, Labriola L. HSPB1 Is Essential for Inducing Resistance to Proteotoxic Stress in Beta-Cells. Cells. 2021; 10(9):2178. https://doi.org/10.3390/cells10092178
Chicago/Turabian StyleGomes, Vinícius M., Rosangela A. M. Wailemann, Gabriel S. Arini, Talita C. Oliveira, Daria R. Q. Almeida, Ancély F. dos Santos, Letícia F. Terra, Stephan Lortz, and Leticia Labriola. 2021. "HSPB1 Is Essential for Inducing Resistance to Proteotoxic Stress in Beta-Cells" Cells 10, no. 9: 2178. https://doi.org/10.3390/cells10092178
APA StyleGomes, V. M., Wailemann, R. A. M., Arini, G. S., Oliveira, T. C., Almeida, D. R. Q., dos Santos, A. F., Terra, L. F., Lortz, S., & Labriola, L. (2021). HSPB1 Is Essential for Inducing Resistance to Proteotoxic Stress in Beta-Cells. Cells, 10(9), 2178. https://doi.org/10.3390/cells10092178