
Proteomic Analysis of Hydromethylthionine in the Line 66 Model of Frontotemporal Dementia Demonstrates Actions on Tau-Dependent and Tau-Independent Networks


 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
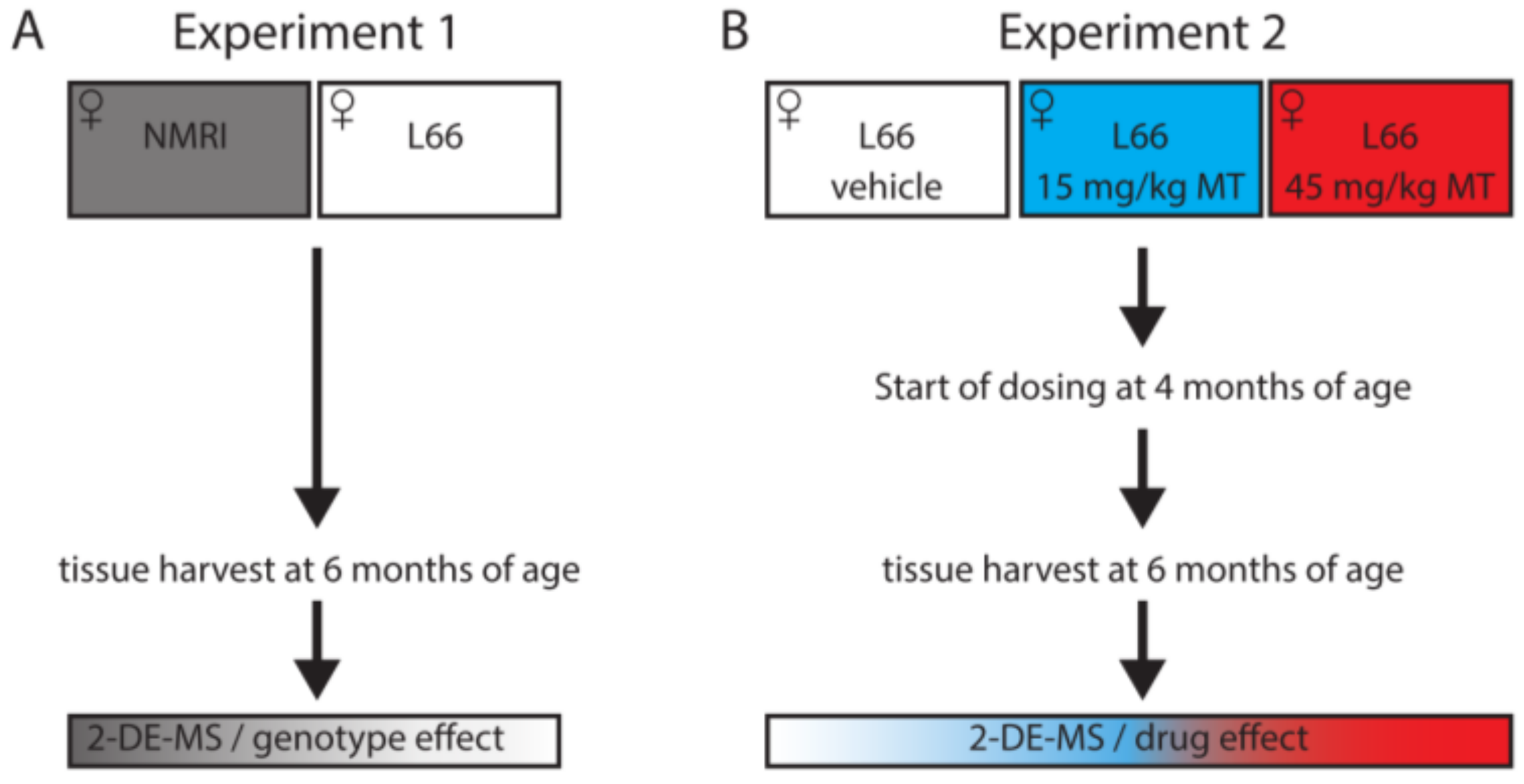
2.2. Drugs and Treatment Cohorts
2.3. Sample Collection
2.4. Immunohistochemistry and Neuronal Cell Counting
2.5. Urea Protein Extraction
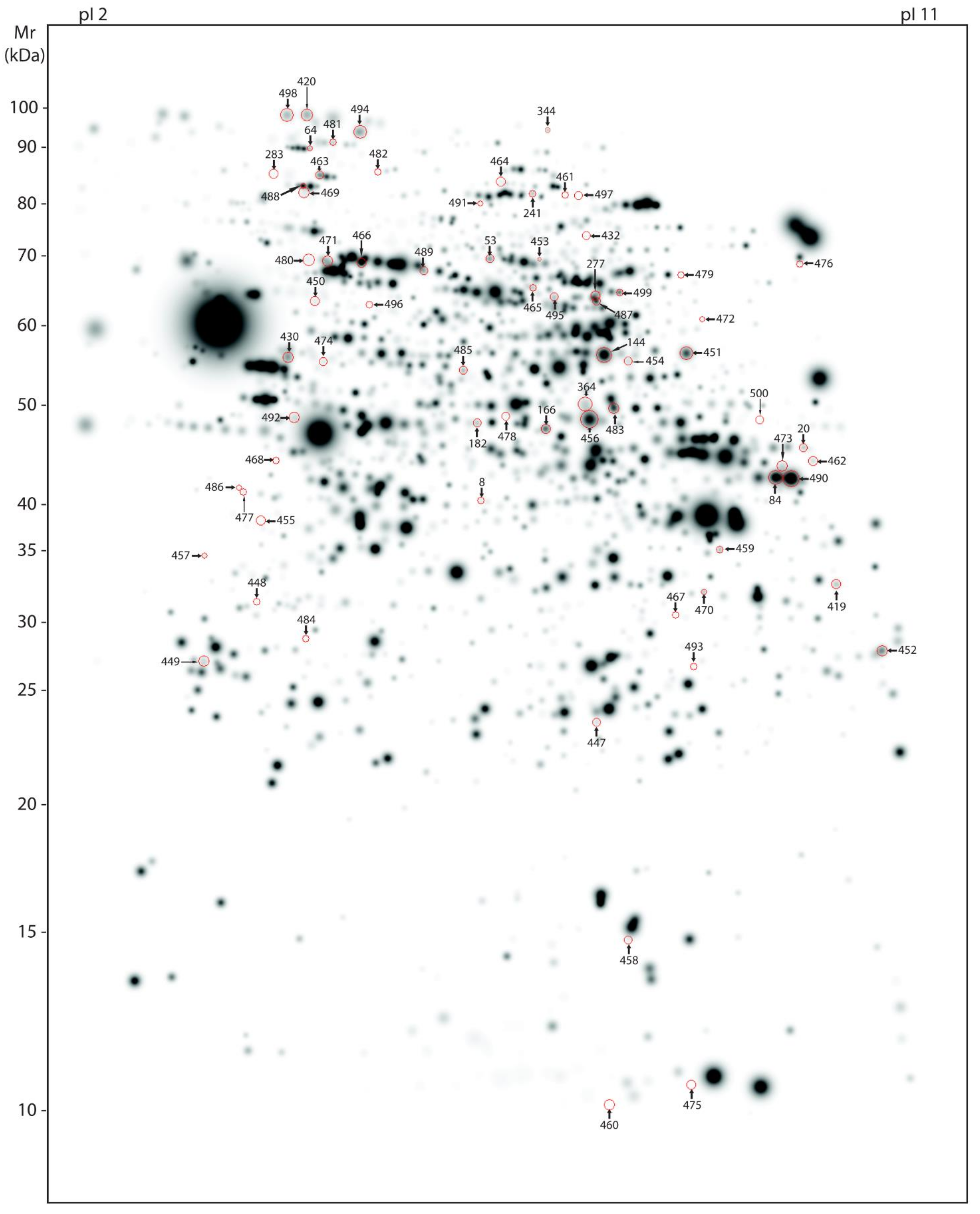
2.6. 2-DE and Comparative Gel Analysis
2.7. Trypsin Digestion and MS-MS Analyses for Protein Identification
2.8. Generation of Expression Heatmaps, Protein Classification and Pathway Analyses
2.9. SDS-Page and Immunoblotting
2.10. Data Analyses
3. Results
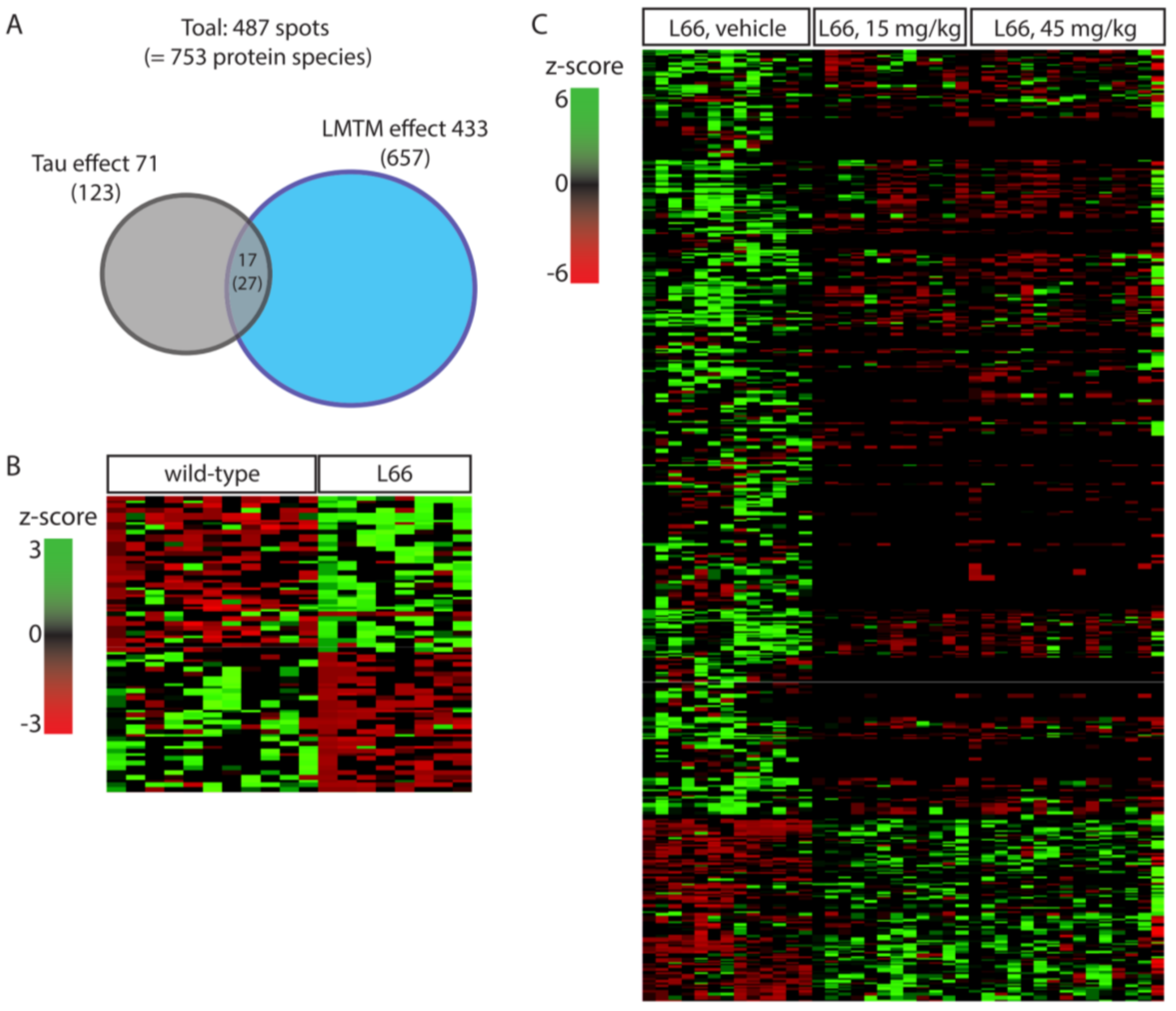
3.1. Impact of Tau-Overexpression on the Neuroproteome in L66 Mice
3.2. LMTM-Dependent Protein Expression in L66 Mice
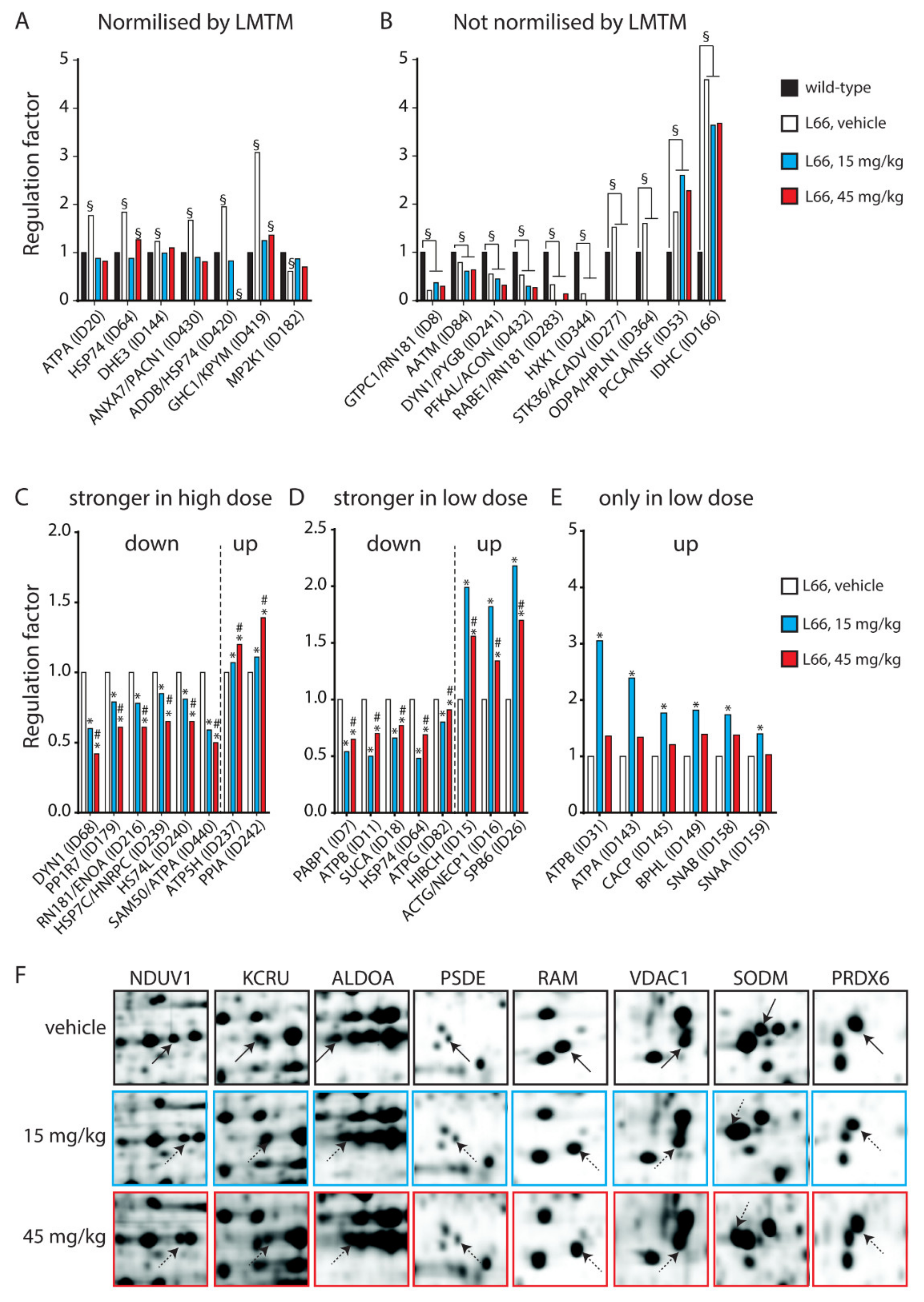
3.3. Tau-Dependent Pathway Rescue by LMTM
3.4. Tau-Independent Pathway Rescue by LMTM
4. Discussion
4.1. L66 Mice Show Common Pathways Dysregulated in FTD Patients
4.2. Tau Accumulation in L66 Mice Affects Multiple Brain Proteome Networks
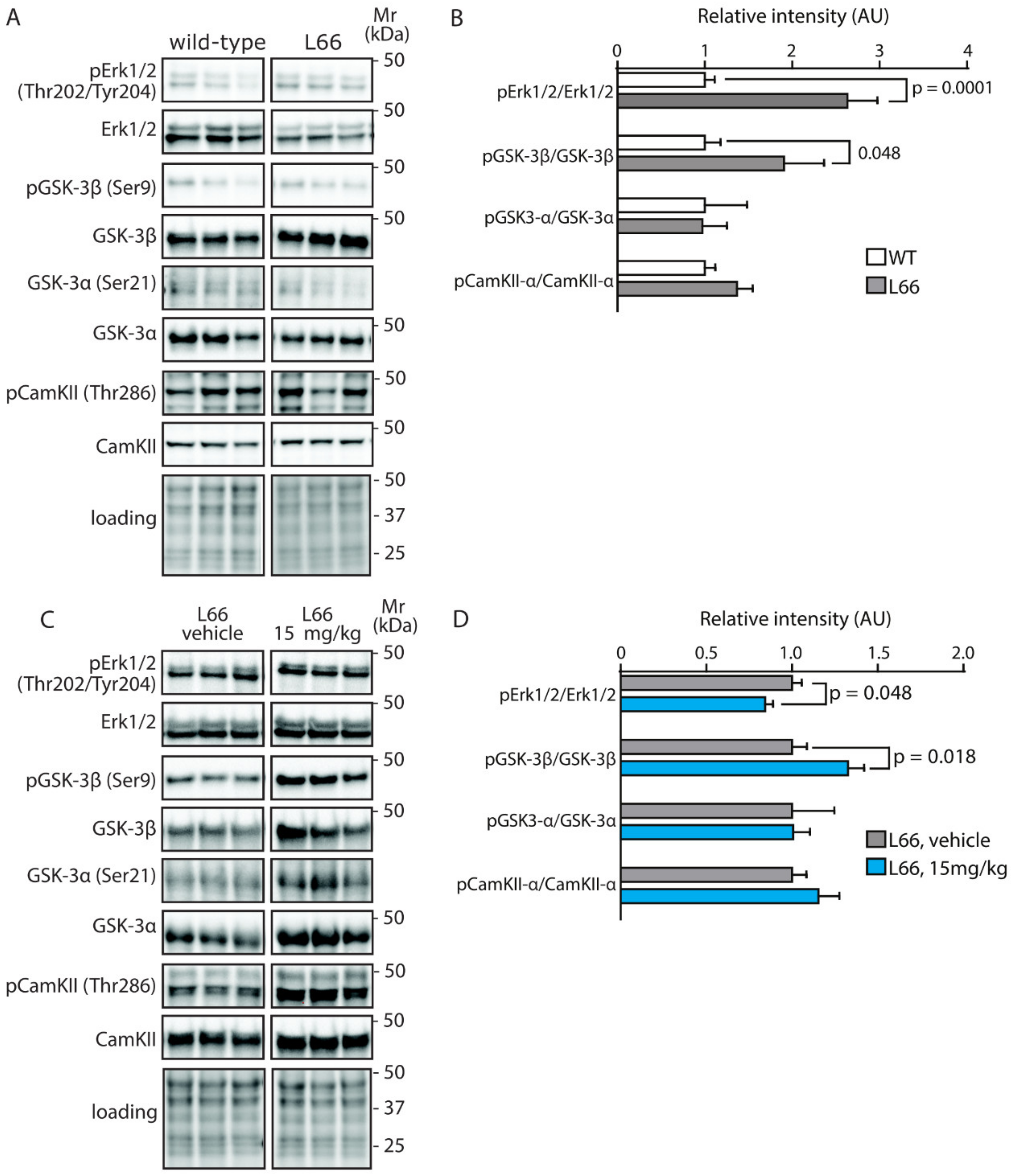
4.3. LMTM Corrects Metabolic Dysfunction, Synaptic Transmission and Stress Responses in L66 Mice
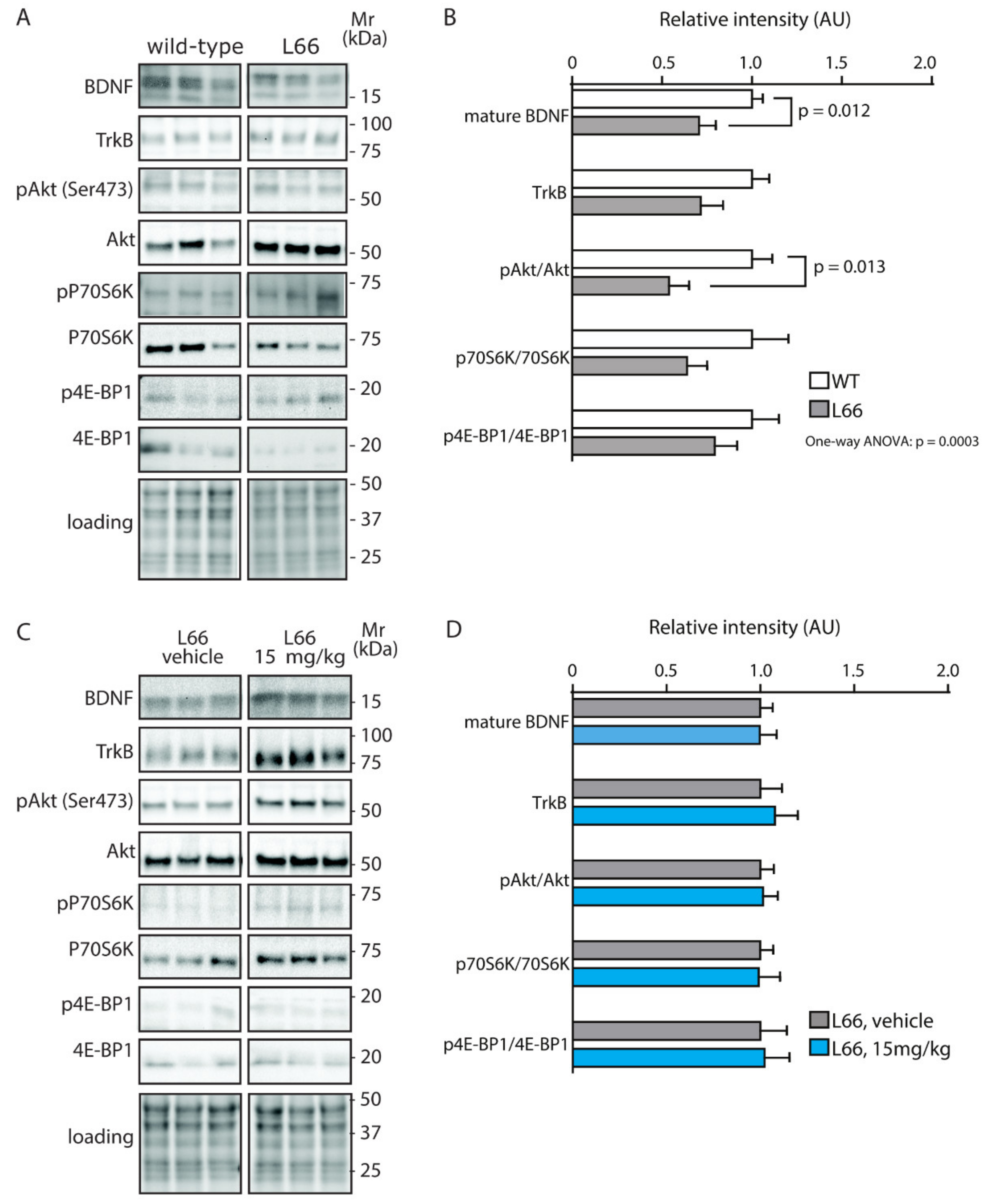
4.4. No Effect of LMTM on Trophic Support Networks
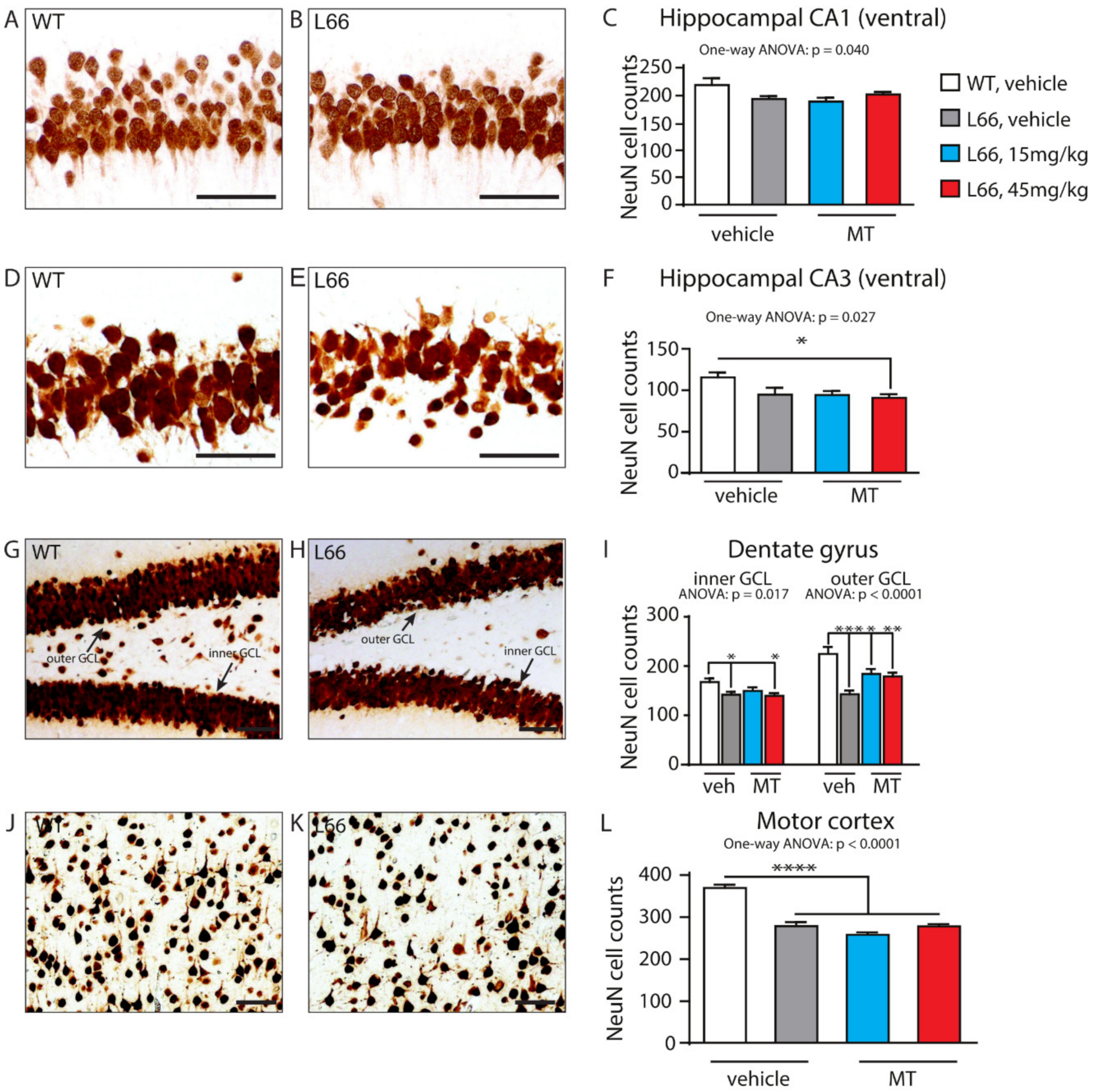
4.5. Tau Compromises Presynaptic Structures and Their Recovery by LMTM
4.6. LMTM Promotes Antioxidant Response via NRF-2 Activation in a Tau-Independent Fashion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Goedert, M.; Jakes, R.; Spillantini, M.G.; Hasegawa, M.; Smith, M.J.; Crowther, R.A. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 1996, 383, 550–553. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M. Pinning down phosphorylated tau. Nature 1999, 399, 739–740. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Ghetti, B.; Spillantini, M.G. Frontotemporal dementia: Implications for understanding Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 4, a006254. [Google Scholar] [CrossRef] [PubMed]
- Conde, C.; Cáceres, A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat. Rev. Neurosci. 2009, 10, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.A.; Neumann, M. Molecular neuropathology of frontotemporal dementia: Insights into disease mechanisms from postmortem studies. J. Neurochem. 2016, 138, 54–70. [Google Scholar] [CrossRef]
- Mietelska-Porowska, A.; Wasik, U.; Goras, M.; Filipek, A.; Niewiadomska, G. Tau protein modifications and interactions: Their role in function and dysfunction. Int. J. Mol. Sci. 2014, 15, 4671–4713. [Google Scholar] [CrossRef]
- Bugiani, O.; Murrell, J.R.; Giaccone, G.; Hasegawa, M.; Ghigo, G.; Tabaton, M.; Morbin, M.; Primavera, A.; Carella, F.; Solaro, C.; et al. Frontotemporal dementia and corticobasal degeneration in a family with a P301S mutation in tau. J. Neuropathol. Exp. Neurol. 1999, 58, 667–677. [Google Scholar] [CrossRef]
- Goedert, M.; Crowther, R.A.; Spillantini, M.G. Tau mutations cause frontotemporal dementias. Neuron 1998, 21, 955–958. [Google Scholar] [CrossRef]
- Hutton, M.; Lendon, C.L.; Rizzu, P.; Baker, M.; Froelich, S.; Houlden, H.H.; Pickering-Brown, S.; Chakraverty, S.; Isaacs, A.; Grover, A.; et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998, 393, 702–704. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Crowther, R.A.; Kamphorst, W.; Heutink, P.; van Swieten, J.C. Tau pathology in two Dutch families with mutations in the microtubule-binding region of tau. Am. J. Pathol. 1998, 153, 1359–1363. [Google Scholar] [CrossRef]
- Ghidoni, R.; Paterlini, A.; Benussi, L. Translational proteomics in Alzheimer’s disease and related disorders. Clin. Biochem. 2013, 46, 480–486. [Google Scholar] [CrossRef]
- Zellner, M.; Veitinger, M.; Umlauf, E. The role of proteomics in dementia and Alzheimer’s disease. Acta Neuropathol. 2009, 118, 181–195. [Google Scholar] [CrossRef]
- Zetterberg, H.; Rüetschi, U.; Portelius, E.; Brinkmalm, G.; Andreasson, U.; Blennow, K.; Brinkmalm, A. Clinical proteomics in neurodegenerative disorders. Acta Neurol. Scand. 2008, 118, 1–11. [Google Scholar] [CrossRef]
- Korolainen, M.A.; Nyman, T.A.; Aittokallio, T.; Pirttilä, T. An update on clinical proteomics in Alzheimer’s research. J. Neurochem. 2010, 112, 1386–1414. [Google Scholar] [CrossRef]
- Brinkmalm, A.; Portelius, E.; Öhrfelt, A.; Brinkmalm, G.; Andreasson, U.; Gobom, J.; Blennow, K.; Zetterberg, H. Explorative and targeted neuroproteomics in Alzheimer’s disease. Biochim. Biophys. Acta Proteins Proteom. 2015, 1854, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Baird, A.L.; Westwood, S.; Lovestone, S. Blood-based proteomic biomarkers of Alzheimer’s disease pathology. Front. Neurol. 2015, 6, 236. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.P.; Sathe, G. Proteomics landscape of Alzheimer’s disease. Proteomes 2021, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Lavery, R.; Varma, V.; Fantoni, G.; Colpo, M.; Thambisetty, M.; Candia, J.; Resnick, S.M.; Bennett, D.A.; Biancotto, A.; et al. Plasma proteomic signatures predict dementia and cognitive impairment. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2020, 6, e12018. [Google Scholar] [CrossRef]
- Bader, J.M.; Geyer, P.E.; Müller, J.B.; Strauss, M.T.; Koch, M.; Leypoldt, F.; Koertvelyessy, P.; Bittner, D.; Schipke, C.G.; Incesoy, E.I.; et al. Proteome profiling in cerebrospinal fluid reveals novel biomarkers of Alzheimer’s disease. Mol. Syst. Biol. 2020, 16, e9356. [Google Scholar] [CrossRef] [PubMed]
- Whelan, C.D.; Mattsson, N.; Nagle, M.W.; Vijayaraghavan, S.; Hyde, C.; Janelidze, S.; Stomrud, E.; Lee, J.; Fitz, L.; Samad, T.A.; et al. Multiplex proteomics identifies novel CSF and plasma biomarkers of early Alzheimer’s disease. Acta Neuropathol. Commun. 2019, 7, 169. [Google Scholar] [CrossRef] [PubMed]
- Agresta, A.M.; de Palma, A.; Bardoni, A.; Salvini, R.; Iadarola, P.; Mauri, P.L. Proteomics as an innovative tool to investigate frontotemporal disorders. Proteom. Clin. Appl. 2016, 10, 457–469. [Google Scholar] [CrossRef]
- Oeckl, P.; Steinacker, P.; Feneberg, E.; Otto, M. Cerebrospinal fluid proteomics and protein biomarkers in frontotemporal lobar degeneration: Current status and future perspectives. Biochim. Biophys. Acta Proteins Proteom. 2015, 1854, 757–768. [Google Scholar] [CrossRef]
- Iridoy, M.O.; Zubiri, I.; Zelaya, M.V.; Martinez, L.; Ausín, K.; Lachen-Montes, M.; Santamaría, E.; Fernandez-Irigoyen, J.; Jericó, I. Neuroanatomical quantitative proteomics reveals common pathogenic biological routes between amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). Int. J. Mol. Sci. 2019, 20, 4. [Google Scholar] [CrossRef]
- Hedl, T.J.; Gil, R.S.; Cheng, F.; Rayner, S.L.; Davidson, J.M.; de Luca, A.; Villalva, M.D.; Ecroyd, H.; Walker, A.K.; Lee, A. Proteomics approaches for biomarker and drug target discovery in ALS and FTD. Front. Neurosci. 2019, 13, 548. [Google Scholar] [CrossRef] [PubMed]
- Swift, I.J.; Sogorb-Esteve, A.; Heller, C.; Synofzik, M.; Otto, M.; Graff, C.; Galimberti, D.; Todd, E.; Heslegrave, A.J.; van der Ende, E.L.; et al. Fluid biomarkers in frontotemporal dementia: Past, present and future. J. Neurol. Neurosurg. Psychiatry 2021, 92, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.T.; Chen-Plotkin, A.; Grossman, M.; Arnold, S.E.; Clark, C.M.; Shaw, L.M.; McCluskey, L.; Elman, L.; Hurtig, H.I.; Siderowf, A.; et al. Novel CSF biomarkers for frontotemporal lobar degenerations. Neurology 2010, 75, 2079–2086. [Google Scholar] [CrossRef] [PubMed]
- Teunissen, C.E.; Elias, N.; Koel-Simmelink, M.J.A.; Durieux-Lu, S.; Malekzadeh, A.; Pham, T.V.; Piersma, S.R.; Beccari, T.; Meeter, L.H.H.; Dopper, E.G.P.; et al. Novel diagnostic cerebrospinal fluid biomarkers for pathologic subtypes of frontotemporal dementia identified by proteomics. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2016, 2, 86–94. [Google Scholar] [CrossRef]
- Simonsen, A.H.; McGuire, J.; Podust, V.N.; Hagnelius, N.O.; Nilsson, T.K.; Kapaki, E.; Vassilopoulos, D.; Waldemar, G. A novel panel of cerebrospinal fluid biomarkers for the differential diagnosis of Alzheimer’s disease versus normal aging and frontotemporal dementia. Dement. Geriatr. Cogn. Disord. 2007, 24, 434–440. [Google Scholar] [CrossRef]
- Puchades, M.; Hansson, S.F.; Nilsson, C.L.; Andreasen, N.; Blennow, K.; Davidsson, P. Proteomic studies of potential cerebrospinal fluid protein markers for Alzheimer’s disease. Mol. Brain Res. 2003, 118, 140–146. [Google Scholar] [CrossRef]
- Hansson, S.F.; Puchades, M.; Blennow, K.; Sjögren, M.; Davidsson, P. Validation of a prefractionation method followed by two-dimensional electrophoresis—Applied to cerebrospinal fluid proteins from frontotemporal dementia patients. Proteome Sci. 2004, 2, 7. [Google Scholar] [CrossRef][Green Version]
- Davidsson, P.; Sjögren, M. The use of proteomics in biomarker discovery in neurodegenerative diseases. Dis. Markers 2005, 21, 81–92. [Google Scholar] [CrossRef]
- Gozal, Y.M.; Dammer, E.B.; Duong, D.M.; Cheng, D.; Gearing, M.; Rees, H.D.; Peng, J.; Lah, J.J.; Levey, A.I. Proteomic analysis of hippocampal dentate granule cells in frontotemporal lobar degeneration: Application of laser capture technology. Front. Neurol. 2011, 2, 24. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, K.; Decker, E.; Zhu, L.; Miller, R.E.; Mirra, S.S.; Spina, S.; Ghetti, B.; Wang, M.; Murrell, J. Aberrantly regulated proteins in frontotemporal dementia. Biochem. Biophys. Res. Commun. 2006, 348, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Gunawardana, C.G.; Mehrabian, M.; Wang, X.; Mueller, I.; Lubambo, I.B.; Jonkman, J.E.N.; Wang, H.; Schmitt-Ulms, G. The human tau interactome: Binding to the ribonucleoproteome, and impaired binding of the proline-to-leucine mutant at position 301 (P301L) to chaperones and the proteasome. Mol. Cell. Proteom. 2015, 14, 3000–3014. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.H.; Jung, I.S.; Han, G.Y.; Kim, N.H.; Kim, H.J.; Kim, C.W. Proteomic profiling of brain cortex tissues in a Tau transgenic mouse model of Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2013, 430, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Tramutola, A.; Sharma, N.; Barone, E.; Lanzillotta, C.; Castellani, A.; Iavarone, F.; Vincenzoni, F.; Castagnola, M.; Butterfield, D.A.; Gaetani, S.; et al. Proteomic identification of altered protein O-GlcNAcylation in a triple transgenic mouse model of Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3309–3321. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Lin, X.; Wang, D.; Zhang, Z.; Guo, Y.; Ren, X.; Xu, B.; Yuan, J.; Liu, J.; Spencer, P.S.; et al. Mitochondrial molecular abnormalities revealed by proteomic analysis of hippocampal organelles of mice triple transgenic for Alzheimer disease. Front. Mol. Neurosci. 2018, 11. [Google Scholar] [CrossRef]
- Yata, K.; Oikawa, S.; Sasaki, R.; Shindo, A.; Yang, R.; Murata, M.; Kanamaru, K.; Tomimoto, H. Astrocytic neuroprotection through induction of cytoprotective molecules; A proteomic analysis of mutant P301S tau-transgenic mouse. Brain Res. 2011, 1410, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Maziuk, B.F.; Apicco, D.J.; Cruz, A.L.; Jiang, L.; Ash, P.E.A.; da Rocha, E.L.; Zhang, C.; Yu, W.H.; Leszyk, J.; Abisambra, J.F.; et al. RNA binding proteins co-localize with small tau inclusions in tauopathy. Acta Neuropathol. Commun. 2018, 6, 71. [Google Scholar] [CrossRef]
- Melis, V.; Zabke, C.; Stamer, K.; Magbagbeolu, M.; Schwab, K.; Marschall, P.; Veh, R.W.; Bachmann, S.; Deiana, S.; Moreau, P.H.; et al. Different pathways of molecular pathophysiology underlie cognitive and motor tauopathy phenotypes in transgenic models for Alzheimer’s disease and frontotemporal lobar degeneration. Cell. Mol. Life Sci. 2015, 72, 2199–2222. [Google Scholar] [CrossRef]
- Lemke, N.; Melis, V.; Lauer, D.; Magbagbeolu, M.; Neumann, B.; Harrington, C.R.; Riedel, G.; Wischik, C.M.; Theuring, F.; Schwab, K. Differential compartmental processing and phosphorylation of pathogenic human tau and native mouse tau in the line 66 model of frontotemporal dementia. J. Biol. Chem. 2020, 295, 18508–18523. [Google Scholar] [CrossRef] [PubMed]
- Cranston, A.L.; Wysocka, A.; Steczkowska, M.; Zadrożny, M.; Palasz, E.; Harrington, C.R.; Theuring, F.; Wischik, C.M.; Riedel, G.; Niewiadomska, G. Cholinergic and inflammatory phenotypes in transgenic tau mouse models of Alzheimer’s disease and frontotemporal lobar degeneration. Brain Commun. 2020, 2, fcaa033. [Google Scholar] [CrossRef] [PubMed]
- Cranston, A.L.; Kraev, I.; Stewart, M.G.; Horsley, D.; Robinson, L.; Dreesen, E.; Armstrong, P.; Harrington, C.R.; Wischik, C.M.; Riedel, G. Rescue of synaptosomal glutamate release defects in tau transgenic mice by the tau aggregation inhibitor hydromethylthionine. 2021. Submitted. [Google Scholar]
- Sperfeld, A.D.; Collatz, M.B.; Baier, H.; Palmbach, M.; Storch, A.; Schwarz, J.; Tatsch, K.; Reske, S.; Joosse, M.; Heutink, P.; et al. FTDP-17: An early-onset phenotype with parkinsonism and epileptic seizures caused by a novel mutation. Ann. Neurol. 1999, 46, 708–715. [Google Scholar] [CrossRef]
- Tagawa, K.; Homma, H.; Saito, A.; Fujita, K.; Chen, X.; Imoto, S.; Oka, T.; Ito, H.; Motoki, K.; Yoshida, C.; et al. Comprehensive phosphoproteome analysis unravels the core signaling network that initiates the earliest synapse pathology in preclinical Alzheimer’s disease brain. Hum. Mol. Genet. 2015, 24, 540–558. [Google Scholar] [CrossRef]
- Harrington, C.R.; Storey, J.M.D.; Clunas, S.; Harrington, K.A.; Horsley, D.; Ishaq, A.; Kemp, S.J.; Larch, C.P.; Marshall, C.; Nicoll, S.L.; et al. Cellular models of aggregation-dependent template-directed proteolysis to characterize Tau aggregation inhibitors for treatment of Alzheimer disease. J. Biol. Chem. 2015, 290, 10862–10875. [Google Scholar] [CrossRef]
- Melis, V.; Magbagbeolu, M.; Rickard, J.E.; Horsley, D.; Davidson, K.; Harrington, K.A.; Goatman, K.; Goatman, E.A.; Deiana, S.; Close, S.P.; et al. Effects of oxidized and reduced forms of methylthioninium in two transgenic mouse tauopathy models. Behav. Pharmacol. 2015, 26, 353–368. [Google Scholar] [CrossRef]
- Schelter, B.O.; Shiells, H.; Baddeley, T.C.; Rubino, C.M.; Ganesan, H.; Hammel, J.; Vuksanovic, V.; Staff, R.T.; Murray, A.D.; Bracoud, L.; et al. Concentration-dependent activity of hydromethylthionine on cognitive decline and brain atrophy in mild to moderate Alzheimer’s disease. J. Alzheimer’s Dis. 2019, 72, 931–946. [Google Scholar] [CrossRef] [PubMed]
- Shiells, H.; Schelter, B.O.; Bentham, P.; Baddeley, T.C.; Rubino, C.M.; Ganesan, H.; Hammel, J.; Vuksanovic, V.; Staff, R.T.; Murray, A.D.; et al. Concentration-dependent activity of hydromethylthionine on clinical decline and brain atrophy in a randomized controlled trial in behavioral variant frontotemporal dementia. J. Alzheimer’s Dis. 2020, 75, 501–519. [Google Scholar] [CrossRef] [PubMed]
- Tucker, D.; Lu, Y.; Zhang, Q. From mitochondrial function to neuroprotection—An emerging role for methylene blue. Mol. Neurobiol. 2018, 55, 5137–5153. [Google Scholar] [CrossRef] [PubMed]
- Gureev, A.P.; Syromyatnikov, M.Y.; Gorbacheva, T.M.; Starkov, A.A.; Popov, V.N. Methylene blue improves sensorimotor phenotype and decreases anxiety in parallel with activating brain mitochondria biogenesis in mid-age mice. Neurosci. Res. 2016, 113, 19–27. [Google Scholar] [CrossRef]
- Wen, Y.; Li, W.; Poteet, E.C.; Xie, L.; Tan, C.; Yan, L.J.; Ju, X.; Liu, R.; Qian, H.; Marvin, M.A.; et al. Alternative mitochondrial electron transfer as a novel strategy for neuroprotection. J. Biol. Chem. 2011, 286, 16504–16515. [Google Scholar] [CrossRef]
- Lee, K.K.; Boelsterli, U.A. Bypassing the compromised mitochondrial electron transport with methylene blue alleviates efavirenz/isoniazid-induced oxidant stress and mitochondria-mediated cell death in mouse hepatocytes. Redox Biol. 2014, 2, 599–609. [Google Scholar] [CrossRef]
- Atamna, H.; Mackey, J.; Dhahbi, J.M. Mitochondrial pharmacology: Electron transport chain bypass as strategies to treat mitochondrial dysfunction. BioFactors 2012, 38, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Atamna, H.; Nguyen, A.; Schultz, C.; Boyle, K.; Newberry, J.; Kato, H.; Ames, B.N. Methylene blue delays cellular senescence and enhances key mitochondrial biochemical pathways. FASEB J. 2008, 22, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Riedel, G.; Klein, J.; Niewiadomska, G.; Kondak, C.; Schwab, K.; Lauer, D.; Magbagbeolu, M.; Steczkowska, M.; Zadrozny, M.; Wydrych, M.; et al. Mechanisms of anticholinesterase interference with tau aggregation inhibitor activity in a tau-transgenic mouse model. Curr. Alzheimer Res. 2020, 17, 285–296. [Google Scholar] [CrossRef]
- Stack, C.; Jainuddin, S.; Elipenahli, C.; Gerges, M.; Starkova, N.; Starkov, A.A.; Jové, M.; Portero-Otin, M.; Launay, N.; Pujol, A.; et al. Methylene blue upregulates Nrf2/ARE genes and prevents tau-related neurotoxicity. Hum. Mol. Genet. 2014, 23, 3716–3732. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Liang, F.; Xu, H.; Yan, W.; Zhang, J. Methylene blue exerts a neuroprotective effect against traumatic brain injury by promoting autophagy and inhibiting microglial activation. Mol. Med. Rep. 2016, 13, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Hochgräfe, K.; Sydow, A.; Matenia, D.; Cadinu, D.; Könen, S.; Petrova, O.; Pickhardt, M.; Goll, P.; Morellini, F.; Mandelkow, E.; et al. Preventive methylene blue treatment preserves cognition in mice expressing full-length pro-aggregant human Tau. Acta Neuropathol. Commun. 2015, 3, 25. [Google Scholar] [CrossRef] [PubMed]
- Congdon, E.E.; Wu, J.W.; Myeku, N.; Figueroa, Y.H.; Herman, M.; Marinec, P.S.; Gestwicki, J.E.; Dickey, C.A.; Yu, W.H.; Duff, K.E. Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo. Autophagy 2012, 8, 609–622. [Google Scholar] [CrossRef]
- Percie du Sert, N.; Hurst, V.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biol. 2020, 18, e3000410. [Google Scholar] [CrossRef]
- Paxinos, G.; Franklin, K. The Mouse Brain in Stereotaxic Coordinates, Compact, 5th ed.; Academic Press: Cambridge, MA, USA, 2019; ISBN 9780128161593. [Google Scholar]
- Schwab, K.; Neumann, B.; Vignon-Zellweger, N.; Fischer, A.; Stein, R.; Jungblut, P.R.; Scheler, C.; Theuring, F. Dietary phytoestrogen supplementation induces sex differences in the myocardial protein pattern of mice: A comparative proteomics study. Proteomics 2011, 11, 3887–3904. [Google Scholar] [CrossRef] [PubMed]
- Heukeshoven, J.; Dernick, R. Simplified method for silver staining of proteins in polyacrylamide gels and the mechanism of silver staining. Electrophoresis 1985, 6, 103–112. [Google Scholar] [CrossRef]
- Nebrich, G.; Herrmann, M.; Sagi, D.; Klose, J.; Giavalisco, P. High MS-compatibility of silver nitrate-stained protein spots from 2-DE gels using ZipPlates and AnchorChips for successful protein identification. Electrophoresis 2007, 28, 1607–1614. [Google Scholar] [CrossRef]
- Jungblut, P.R.; Holzhütter, H.G.; Apweiler, R.; Schlüter, H. The speciation of the proteome. Chem. Cent. J. 2008, 2, 16. [Google Scholar] [CrossRef]
- Takei, N.; Nawa, H. mTOR signaling and its roles in normal and abnormal brain development. Front. Mol. Neurosci. 2014, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR signaling in growth, metabolism, and disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Vicario-Abejón, C.; Owens, D.; McKay, R.; Segal, M. Role of neurotrophins in central synapse formation and stabilization. Nat. Rev. Neurosci. 2002, 3, 965–974. [Google Scholar] [CrossRef]
- Miranda, M.; Morici, J.F.; Zanoni, M.B.; Bekinschtein, P. Brain-derived neurotrophic factor: A key molecule for memory in the healthy and the pathological brain. Front. Cell. Neurosci. 2019, 13, 363. [Google Scholar] [CrossRef]
- Sun, J.; Nan, G. The extracellular signal-regulated kinase 1/2 pathway in neurological diseases: A potential therapeutic target. Int. J. Mol. Med. 2017, 39, 1338–1346. [Google Scholar] [CrossRef]
- Jaworski, T.; Banach-Kasper, E.; Gralec, K. GSK-3β at the intersection of neuronal plasticity and neurodegeneration. Neural Plast. 2019, 2019, 4209475. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, I.G.; Kwak, M.K. Regulatory crosstalk between the oxidative stress-related transcription factor Nfe2l2/Nrf2 and mitochondria. Toxicol. Appl. Pharmacol. 2018, 359, 24–33. [Google Scholar] [CrossRef]
- Herculano-Houzel, S.; Ribeiro, P.; Campos, L.; da Silva, A.V.; Torres, L.B.; Catania, K.C.; Kaas, J.H. Updated neuronal scaling rules for the brains of glires (rodents/lagomorphs). Brain. Behav. Evol. 2011, 78, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.D.; Chan, G.; Stefanoska, K.; Au, C.; Bi, M.; Müller, J.; Przybyla, M.; Feiten, A.; Prikas, E.; Halliday, G.M.; et al. CNS cell type-specific gene profiling of P301S tau transgenic mice identifies genes dysregulated by progressive tau accumulation. J. Biol. Chem. 2019, 294, 14149–14162. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.C.; Nana, A.L.; Hepker, M.; Hwang, J.H.L.; Gaus, S.E.; Spina, S.; Cosme, C.G.; Gan, L.; Grinberg, L.T.; Geschwind, D.H.; et al. Preferential tau aggregation in von Economo neurons and fork cells in frontotemporal lobar degeneration with specific MAPT variants. Acta Neuropathol. Commun. 2019, 7, 159. [Google Scholar] [CrossRef]
- Dupree, E.J.; Jayathirtha, M.; Yorkey, H.; Mihasan, M.; Petre, B.A.; Darie, C.C. A critical review of bottom-up proteomics: The good, the bad, and the future of this field. Proteomes 2020, 8, 14. [Google Scholar] [CrossRef]
- Ning, F.; Wu, X.; Wang, W. Exploiting the potential of 2DE in proteomics analyses. Expert Rev. Proteomics 2016, 13, 901–903. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Li, N.; Zhan, X.; Qian, S. Revival of 2DE-LC/MS in proteomics and its potential for large-scale study of human proteoforms. Med. One 2018, 3, e180008. [Google Scholar] [CrossRef]
- Zhan, X.; Li, B.; Zhan, X.; Schlüter, H.; Jungblut, P.R.; Coorssen, J.R. Innovating the concept and practice of two-dimensional gel electrophoresis in the analysis of proteomes at the proteoform level. Proteomes 2019, 7, 36. [Google Scholar] [CrossRef]
- Lachén-Montes, M.; González-Morales, A.; Schvartz, D.; Zelaya, M.V.; Ausin, K.; Fernández-Irigoyen, J.; Sánchez, J.C.; Santamaría, E. The olfactory bulb proteotype differs across frontotemporal dementia spectrum. J. Proteom. 2019, 201, 37–47. [Google Scholar] [CrossRef]
- Umoh, M.E.; Dammer, E.B.; Dai, J.; Duong, D.M.; Lah, J.J.; Levey, A.I.; Gearing, M.; Glass, J.D.; Seyfried, N.T. A proteomic network approach across the ALS-FTD disease spectrum resolves clinical phenotypes and genetic vulnerability in human brain. EMBO Mol. Med. 2018, 10, 48–62. [Google Scholar] [CrossRef]
- David, D.C.; Ittner, L.M.; Gehrig, P.; Nergenau, D.; Shepherd, C.; Halliday, G.; Götz, J. β-amyloid treatment of two complementary P301L tau-expressing Alzheimer’s disease models reveals similar deregulated cellular processes. Proteomics 2006, 6, 6566–6577. [Google Scholar] [CrossRef]
- David, D.C.; Hauptmann, S.; Scherping, I.; Schuessel, K.; Keil, U.; Rizzu, P.; Ravid, R.; Dröse, S.; Brandt, U.; Müller, W.E.; et al. Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J. Biol. Chem. 2005, 280, 23802–23814. [Google Scholar] [CrossRef] [PubMed]
- De Jesús-Cortés, H.J.; Nogueras-Ortiz, C.J.; Gearing, M.; Arnold, S.E.; Vega, I.E. Amphiphysin-1 protein level changes associated with tau-mediated neurodegeneration. Neuroreport 2012, 23, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Decker, J.M.; Krüger, L.; Sydow, A.; Zhao, S.; Frotscher, M.; Mandelkow, E.; Mandelkow, E.M. Pro-aggregant Tau impairs mossy fiber plasticity due to structural changes and Ca++ dysregulation. Acta Neuropathol. Commun. 2015, 3, 23. [Google Scholar] [CrossRef] [PubMed]
- McInnes, J.; Wierda, K.; Snellinx, A.; Bounti, L.; Wang, Y.C.; Stancu, I.C.; Apóstolo, N.; Gevaert, K.; Dewachter, I.; Spires-Jones, T.L.; et al. Synaptogyrin-3 mediates presynaptic dysfunction induced by Tau. Neuron 2018, 97, 823–835. [Google Scholar] [CrossRef]
- Zhou, L.; McInnes, J.; Wierda, K.; Holt, M.; Herrmann, A.G.; Jackson, R.J.; Wang, Y.C.; Swerts, J.; Beyens, J.; Miskiewicz, K.; et al. Tau association with synaptic vesicles causes presynaptic dysfunction. Nat. Commun. 2017, 8, 15295. [Google Scholar] [CrossRef]
- Altmann, A.; Cash, D.M.; Bocchetta, M.; Heller, C.; Reynolds, R.; Moore, K.; Convery, R.S.; Thomas, D.L.; van Swieten, J.C.; Moreno, F.; et al. Analysis of brain atrophy and local gene expression in genetic frontotemporal dementia. Brain Commun. 2020, 2, fcaa122. [Google Scholar] [CrossRef]
- Wischik, C.M.; Schelter, B.O.; Wischik, D.J.; Storey, J.M.D.; Harrington, C.R. Modeling prion-like processing of tau protein in Alzheimer’s disease for pharmaceutical development. J. Alzheimer’s Dis. 2018, 62, 1287–1303. [Google Scholar] [CrossRef]
- Wilcock, G.K.; Gauthier, S.; Frisoni, G.B.; Jia, J.; Hardlund, J.H.; Moebius, H.J.; Bentham, P.; Kook, K.A.; Schelter, B.O.; Wischik, D.J.; et al. Potential of low dose leuco-methylthioninium bis(hydromethanesulphonate) (LMTM) monotherapy for treatment of mild Alzheimer’s disease: Cohort analysis as modified primary outcome in a phase III clinical trial. J. Alzheimer’s Dis. 2018, 61, 435–457. [Google Scholar] [CrossRef]
- Al-Hilaly, Y.K.; Pollack, S.J.; Rickard, J.E.; Simpson, M.; Raulin, A.C.; Baddeley, T.; Schellenberger, P.; Storey, J.M.D.; Harrington, C.R.; Wischik, C.M.; et al. Cysteine-independent inhibition of Alzheimer’s disease-like paired helical filament assembly by leuco-methylthioninium (LMT). J. Mol. Biol. 2018, 430, 4119–4131. [Google Scholar] [CrossRef]
- Benussi, L.; Binetti, G.; Ghidoni, R. Loss of neuroprotective factors in neurodegenerative dementias: The end or the starting point? Front. Neurosci. 2017, 11, 672. [Google Scholar] [CrossRef]
- Mazzaro, N.; Barini, E.; Spillantini, M.G.; Goedert, M.; Medini, P.; Gasparini, L. Tau-driven neuronal and neurotrophic dysfunction in a mouse model of early tauopathy. J. Neurosci. 2016, 36, 2086–2100. [Google Scholar] [CrossRef]
- Bhurtel, S.; Katila, N.; Neupane, S.; Srivastav, S.; Park, P.H.; Choi, D.Y. Methylene blue protects dopaminergic neurons against MPTP-induced neurotoxicity by upregulating brain-derived neurotrophic factor. Ann. N. Y. Acad. Sci. 2018, 1431, 58–71. [Google Scholar] [CrossRef]
- Ryskalin, L.; Limanaqi, F.; Frati, A.; Busceti, C.L.; Fornai, F. mTOR-related brain dysfunctions in neuropsychiatric disorders. Int. J. Mol. Sci. 2018, 19, 2226. [Google Scholar] [CrossRef] [PubMed]
- Yates, S.C.; Zafar, A.; Hubbard, P.; Nagy, S.; Durant, S.; Bicknell, R.; Wilcock, G.; Christie, S.; Esiri, M.M.; Smith, A.D.; et al. Dysfunction of the mTOR pathway is a risk factor for Alzheimer’s disease. Acta Neuropathol. Commun. 2014, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Bockaert, J.; Marin, P. mTOR in brain physiology and pathologies. Physiol. Rev. 2015, 95, 1157–1187. [Google Scholar] [CrossRef]
- Switon, K.; Kotulska, K.; Janusz-Kaminska, A.; Zmorzynska, J.; Jaworski, J. Molecular neurobiology of mTOR. Neuroscience 2017, 341, 112–153. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.S.; Wong-Riley, M.T.T. Coupling of energy metabolism and synaptic transmission at the transcriptional level: Role of nuclear respiratory factor 1 in regulating both cytochrome c oxidase and NMDA glutamate receptor subunit genes. J. Neurosci. 2009, 29, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.S.; Liang, H.L.; Wong-Riley, M.T.T. Nuclear respiratory factor 1 co-regulates AMPA glutamate receptor subunit 2 and cytochrome c oxidase: Tight coupling of glutamatergic transmission and energy metabolism in neurons. J. Neurochem. 2009, 108, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Devine, M.J.; Kittler, J.T. Mitochondria at the neuronal presynapse in health and disease. Nat. Rev. Neurosci. 2018, 19, 63–80. [Google Scholar] [CrossRef]
- Cieri, D.; Vicario, M.; Vallese, F.; D’Orsi, B.; Berto, P.; Grinzato, A.; Catoni, C.; de Stefani, D.; Rizzuto, R.; Brini, M.; et al. Tau localises within mitochondrial sub-compartments and its caspase cleavage affects ER-mitochondria interactions and cellular Ca2+ handling. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3247–3256. [Google Scholar] [CrossRef] [PubMed]
- Wischik, C.; Lai, R.; Harrington, C. Modelling prion-like processing of tau protein in Alzheimer’s disease for pharmaceutical development. In Brain Microtubule Associated Proteins: Modifications in Disease; Harwood Academic Publishers: Amsterdam, The Netherlands, 1997; pp. 185–241. [Google Scholar]
- Lin, A.L.; Poteet, E.; Du, F.; Gourav, R.C.; Liu, R.; Wen, Y.; Bresnen, A.; Huang, S.; Fox, P.T.; Yang, S.H.; et al. Methylene blue as a cerebral metabolic and hemodynamic enhancer. PLoS ONE 2012, 7, e46585. [Google Scholar] [CrossRef] [PubMed]
- Atamna, H.; Atamna, W.; Al-Eyd, G.; Shanower, G.; Dhahbi, J.M. Combined activation of the energy and cellular-defense pathways may explain the potent anti-senescence activity of methylene blue. Redox Biol. 2015, 6, 426–435. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| p-Value | ||||
|---|---|---|---|---|
| FUNCTIONS | L66 vs. | L66 (15mg/kg MT) | L66 (45mg/kg MT) | |
| Wild-Type Controls | vs. L66 (Vehicle Control) | vs. L66 (Vehicle Control) | ||
| Genotype | Patterning of dendrites | 3.63 × 10−4 | - | - |
| Abnormal quantity of synaptic vesicles | 7.08 × 10−4 | - | - | |
| Long-term potentiation | 6.31 × 10−3 | - | - | |
| Quantity of dendrites | 7.08 × 10−3 | - | - | |
| Excitation of spiny neurons | 1.20 × 10−2 | - | - | |
| Quantity of postsynaptic density | 1.20 × 10−2 | - | - | |
| Elongation of axons | 1.70 × 10−2 | - | - | |
| Synthesis of dopamine | 1.82 × 10−2 | - | - | |
| Synthesis of l-glutamic acid | 1.82 × 10−2 | - | - | |
| Loss of hippocampal neurons | 3.63 × 10−2 | - | - | |
| Maturation of synaptic vesicles | 3.63 × 10−2 | - | - | |
| Release of neurotransmitter | 4.57 × 10−2 | - | - | |
| Genotype and LMTM | Endocytosis of synaptic vesicles | 6.31E x 10−7 | 7.94 × 10−7 | 8.71 × 10−8 |
| Transport of synaptic vesicles | 2.82 × 10−6 | 3.47 × 10−6 | 8.91 × 10−7 | |
| Morphology of presynaptic terminals | 7.41 × 10−5 | 1.86 × 10−2 | 2.09 × 10−2 | |
| Recycling of synaptic vesicles | 3.47 × 10−4 | 6.92 × 10−5 | 7.24 × 10−6 | |
| Density of dendritic spines | 1.20 × 10−2 | 2.95 × 10−3 | 4.79 × 10−3 | |
| Synaptic transmission of neurons | 1.45 × 10−2 | 6.76 × 10−4 | 1.20 × 10−3 | |
| Size of postsynaptic density | 1.82 × 10−2 | 3.47 × 10−4 | 4.27 × 10−4 | |
| Neurotransmission | 2.82 × 10−2 | 7.41 × 10−6 | 2.29 × 10−5 | |
| Loss of dendrites | 3.02 × 10−2 | 3.39 × 10−3 | 4.07 × 10−3 | |
| Synaptic depression | 4.17 × 10−2 | 5.62 × 10−4 | 1.12 × 10−3 | |
| Synaptic transmission | 4.27 × 10−2 | 3.39 × 10−6 | 1.00 × 10−5 | |
| LMTM | Size of neurons | - | 1.74 × 10−5 | 3.80 × 10−5 |
| Association of synaptic vesicles | - | 3.47 × 10−4 | 4.27 × 10−4 | |
| Formation of cellular inclusion bodies | - | 6.61 × 10−4 | 1.15 × 10−4 | |
| Neurodegeneration of axons | - | 9.77 × 10−4 | 1.74 × 10−3 | |
| Swelling of mitochondria | - | 2.04 × 10−3 | 2.45 × 10−3 | |
| Microtubule dynamics | - | 2.51 × 10−3 | 5.01 × 10−3 | |
| Size of axons | - | 3.39 × 10−3 | 4.07 × 10−3 | |
| Depolarisation of mitochondria | - | 3.39 × 10−3 | 4.07 × 10−3 | |
| Abnormal morphology of axons | - | 6.03 × 10−3 | 3.55 × 10−2 | |
| Production of reactive oxygen species | - | 8.71 × 10−3 | 1.15 × 10−2 | |
| Formation of amyloid-beta plaques | - | 1.15 × 10−2 | 1.38 × 10−2 | |
| Formation of tau filament | - | 1.86 × 10−2 | 2.09 × 10−2 | |
| Ubiquitination of protein | - | 1.86 × 10−2 | 2.09 × 10−2 | |
| p-Value | ||||
|---|---|---|---|---|
| PATHWAYS | L66 vs. | L66 (15mg/kg MT) | L66 (45mg/kg MT) | |
| Wild-Type Controls | vs. L66 (Vehicle Control) | vs. L66 (Vehicle Control) | ||
| Genotype | Protein kinase A signalling | 3.89 × 10−4 | - | - |
| GnRH signalling | 5.50 × 10−3 | - | - | |
| Gap junction signalling | 1.07 × 10−2 | - | - | |
| Aspartate biosynthesis | 1.82 × 10−2 | - | - | |
| IGF-1 signalling | 2.04 × 10−2 | - | - | |
| Cell cycle | 2.34 × 10−2 | - | - | |
| Synaptic long term potentiation | 2.45 × 10−2 | - | - | |
| Wnt/Ca2+ pathway | 2.45 × 10−2 | - | - | |
| fMLP signalling in neutrophils | 2.95 × 10−2 | - | - | |
| ERK5 signalling | 4.68 × 10−2 | - | - | |
| Genotype and LMTM | Gluconeogenesis I | 3.63 × 10−6 | 4.90 × 10−10 | 2.45 × 10−11 |
| Glycolysis I | 1.66 × 10−4 | 1.62 × 10−13 | 4.47 × 10−7 | |
| TCA cycle II (eukaryotic) | 1.66 × 10−4 | 1.00 × 10−11 | 2.45 × 10−11 | |
| P70S6K signalling | 5.13 × 10−4 | 9.55 × 10−3 | 1.51 × 10−2 | |
| Mitochondrial function | 1.48 × 10−3 | 9.55 × 10−12 | 5.50 × 10−11 | |
| Huntington’s disease signalling | 1.86 × 10−3 | 1.12 × 10−6 | 8.91 × 10−7 | |
| Unfolded protein response | 2.69 × 10−3 | 1.05 × 10−2 | 1.48 × 10−2 | |
| Clathrin-mediated endocytosis signalling | 2.82 × 10−3 | 6.76 × 10−7 | 6.46 × 10−8 | |
| 14-3-3-mediated signalling | 4.07 × 10−3 | 1.86 × 10−3 | 3.31 × 10−3 | |
| Glutamate biosynthesis II | 1.20 × 10−2 | 3.72 × 10−2 | 4.07 × 10−2 | |
| NRF2-mediated oxidative stress response | 1.66 × 10−2 | 3.98 × 10−6 | 1.00 × 10−8 | |
| Glutamate degradation II | 1.82 × 10−2 | 1.02 × 10−3 | 1.26 × 10−3 | |
| PI3K/AKT signalling | 2.51 × 10−2 | 1.26 × 10−2 | 5.50 × 10−3 | |
| Regulation of EIF4 and P70S6K signalling | 4.07 × 10−2 | 2.00 × 10−2 | 3.09 × 10−2 | |
| Tight junction signalling | 5.00 × 10−2 | 4.27 × 10−2 | 2.29 × 10−2 | |
| LMTM | Aspartate degradation II | - | 5.89 × 10−7 | 8.71 × 10−7 |
| Oxidative phosphorylation | - | 3.55 × 10−6 | 8.51 × 10−6 | |
| Protein ubiquitination pathway | - | 9.33 × 10−5 | 6.61 × 10−5 | |
| RhoA signalling | - | 9.55 × 10−5 | 3.63 × 10−5 | |
| Parkinson’s signalling | - | 1.82 × 10−4 | 2.63 × 10−4 | |
| Fatty acid α-oxidation | - | 6.61 × 10−4 | 1.00 × 10−2 | |
| NADH repair | - | 1.02 × 10−3 | 1.26 × 10−3 | |
| Signalling by Rho family GTPases | - | 1.20 × 10−3 | 2.24 × 10−4 | |
| PPARA/RXRA activation | - | 1.78 × 10−3 | 1.12 × 10−2 | |
| Glutathione-mediated detoxification | - | 3.02 × 10−3 | 4.92 × 10−7 | |
| Telomerase signalling | - | 4.57 × 10−3 | 7.41 × 10−3 | |
| Super pathway of methionine degradation | - | 6.76 × 10−3 | 8.71 × 10−3 | |
| Regulation of actin-based motility by Rho | - | 1.12 × 10−2 | 3.63 × 10−3 | |
| Semaphorin signalling in neurons | - | 1.32 × 10−2 | 1.82 × 10−2 | |
| VEGF signalling | - | 1.32 × 10−2 | 4.47 × 10−3 | |
| Neuregulin signalling | - | 1.38 × 10−2 | 2.04 × 10−2 | |
| Axonal guidance signalling | - | 1.70 × 10−2 | 1.66 × 10−2 | |
| PPAR signalling | - | 1.70 × 10−2 | 2.45 × 10−2 | |
| Super pathway of citrulline metabolism | - | 1.70 × 10−2 | 2.09 × 10−2 | |
| Rho-GDI signalling | - | 2.14 × 10−2 | 1.00 × 10−3 | |
| GABA receptor signalling | - | 2.88 × 10−2 | 3.89 × 10−2 | |
| Xenobiotic metabolism signalling | - | 3.09 × 10−2 | 2.51 × 10−3 | |
| EIF2 signalling | - | 3.89 × 10−2 | 5.75 × 10−2 | |
| Actin cytoskeleton signalling | - | 4.07 × 10−2 | 8.71 × 10−3 | |
| FAK signalling | - | 4.57 × 10−2 | 1.66 × 10−2 | |
| Integrin signalling | - | 8.13 × 10−2 | 5.01 × 10−2 | |
| RAC signalling | - | 9.55 × 10−2 | 4.37 × 10−2 | |
| p-Value | ||||
|---|---|---|---|---|
| DISEASES | L66 vs. | L66 (15mg/kg MT) | L66 (45mg/kg MT) | |
| Wild-Type Controls | vs. L66 (Vehicle Control) | vs. L66 (Vehicle Control) | ||
| Genotype | Memory | 3.63 × 10−3 | - | - |
| Behaviour | 2.95 × 10−2 | - | - | |
| Genotype and LMTM | Alzheimer’s disease | 3.31 × 10−4 | 3.39 × 10−3 | 2.24 × 10−3 |
| Degeneration of brain | 5.50 × 10−3 | 5.89 × 10−4 | 1.20 × 10−3 | |
| LMTM | Disorder of basal ganglia | - | 2.63 × 10−8 | 2.00 × 10−7 |
| Progressive motor neuropathy | - | 9.12 × 10−8 | 5.37 × 10−8 | |
| Neuromuscular disease | - | 4.37 × 10−7 | 2.63 × 10−6 | |
| Movement disorders | - | 6.17 × 10−7 | 1.10 × 10−6 | |
| Parkinson’s disease | - | 8.71 × 10−7 | 2.29 × 10−6 | |
| Neurodegeneration of hippocampus | - | 9.12 × 10−3 | 1.10 × 10−2 | |
| Cognitive impairment | - | 2.75 × 10−2 | 2.69 × 10−3 | |
| UPSTREAM | L66 vs. | L66 (15mg/kg MT) | L66 (45mg/kg MT) | ||||
|---|---|---|---|---|---|---|---|
| REGULATORS | Wild-Type Controls | vs. L66 (Vehicle Control) | vs. L66 (Vehicle Control) | ||||
| p-Value | Activation z-Score | p-Value | Activation z-Score | p-Value | Activation z-Score | ||
| Genotype | PLCB4 | 5.89 × 10−3 | - | - | - | - | - |
| Sept5 | 1.17 × 10−2 | - | - | - | - | - | |
| ZNF746 | 1.74 × 10−2 | - | - | - | - | - | |
| AKT1 | 2.34 × 10−2 | - | - | - | - | - | |
| SLC18A3 | 2.34 × 10−2 | - | - | - | - | - | |
| AGRP | 2.88 × 10−2 | - | - | - | - | - | |
| CDK5 | 3.47 × 10−2 | - | - | - | - | - | |
| CDK5R1 | 3.47 × 10−2 | - | - | - | - | - | |
| NOS2 | 3.98 × 10−2 | - | - | - | - | - | |
| MAPT | 1.35 × 10−14 | - | 4.90 × 10−38 | - | 2.75 × 10−37 | - | |
| Genotype and LMTM | PSEN1 | 1.05 × 10−11 | - | 9.12 × 10−32 | - | 5.75 × 10−33 | - |
| APP | 6.31 × 10−11 | - | 2.14 × 10−31 | - | 1.45 × 10−30 | - | |
| HTT | 1.41 × 10−7 | - | 1.10 × 10−9 | - | 3.89 × 10−8 | - | |
| SNCA | 2.95 × 10−4 | - | 9.12 × 10−10 | - | 2.63 × 10−9 | - | |
| YWHAG | 5.01 × 10−4 | - | 4.57 × 10−3 | - | 5.50 × 10−3 | - | |
| FMR1 | 1.51 × 10−3 | - | 3.39 × 10−6 | - | 6.61 × 10−6 | - | |
| RTN4 | 1.91 × 10−3 | - | 6.46 × 10−3 | - | 9.12 × 10−3 | - | |
| MKNK1 | 1.48 × 10−2 | - | 3.16 × 10−6 | - | 7.76 × 10−6 | - | |
| HSF1 | 3.47 × 10−2 | - | 4.57 × 10−3 | - | 5.50 × 10−3 | - | |
| BDNF | 3.89 × 10−2 | −1.12 | 1.66 × 10−4 | −0.92 | 4.37 × 10−4 | −0.92 | |
| LMTM | MTOR | - | - | 2.04 × 10−8 | 2.1 (active) | 5.01 × 10−9 | 1.7 (active) |
| ADORA2A | - | - | 3.63 × 10−8 | −0.58 | 1.17 × 10−9 | −1.1 | |
| SOD1 | - | - | 2.04 × 10−5 | - | 2.45 × 10−4 | - | |
| E2F1 | - | - | 2.24 × 10−3 | - | 3.47 × 10−3 | - | |
| PARK2 | - | - | 1.05 × 10−2 | - | 1.26 × 10−2 | - | |
| ARX | - | - | 1.78 × 10−2 | - | 1.95 × 10−2 | - | |
| EIF2B2 | - | - | 1.78 × 10−2 | - | 1.95 × 10−2 | - | |
| MARK2 | - | - | 1.78 × 10−2 | - | 1.95 × 10−2 | - | |
| MYO5A | - | - | 1.78 × 10−2 | - | 1.95 × 10−2 | - | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schwab, K.; Melis, V.; Harrington, C.R.; Wischik, C.M.; Magbagbeolu, M.; Theuring, F.; Riedel, G. Proteomic Analysis of Hydromethylthionine in the Line 66 Model of Frontotemporal Dementia Demonstrates Actions on Tau-Dependent and Tau-Independent Networks. Cells 2021, 10, 2162. https://doi.org/10.3390/cells10082162
Schwab K, Melis V, Harrington CR, Wischik CM, Magbagbeolu M, Theuring F, Riedel G. Proteomic Analysis of Hydromethylthionine in the Line 66 Model of Frontotemporal Dementia Demonstrates Actions on Tau-Dependent and Tau-Independent Networks. Cells. 2021; 10(8):2162. https://doi.org/10.3390/cells10082162
Chicago/Turabian StyleSchwab, Karima, Valeria Melis, Charles R. Harrington, Claude M. Wischik, Mandy Magbagbeolu, Franz Theuring, and Gernot Riedel. 2021. "Proteomic Analysis of Hydromethylthionine in the Line 66 Model of Frontotemporal Dementia Demonstrates Actions on Tau-Dependent and Tau-Independent Networks" Cells 10, no. 8: 2162. https://doi.org/10.3390/cells10082162
APA StyleSchwab, K., Melis, V., Harrington, C. R., Wischik, C. M., Magbagbeolu, M., Theuring, F., & Riedel, G. (2021). Proteomic Analysis of Hydromethylthionine in the Line 66 Model of Frontotemporal Dementia Demonstrates Actions on Tau-Dependent and Tau-Independent Networks. Cells, 10(8), 2162. https://doi.org/10.3390/cells10082162