
Implications of Innate Immunity in Post-Acute Sequelae of Non-Persistent Viral Infections


Abstract
:1. Introduction
1.1. Acute Viral Infections Trigger and Modulate the Innate Immune Responses
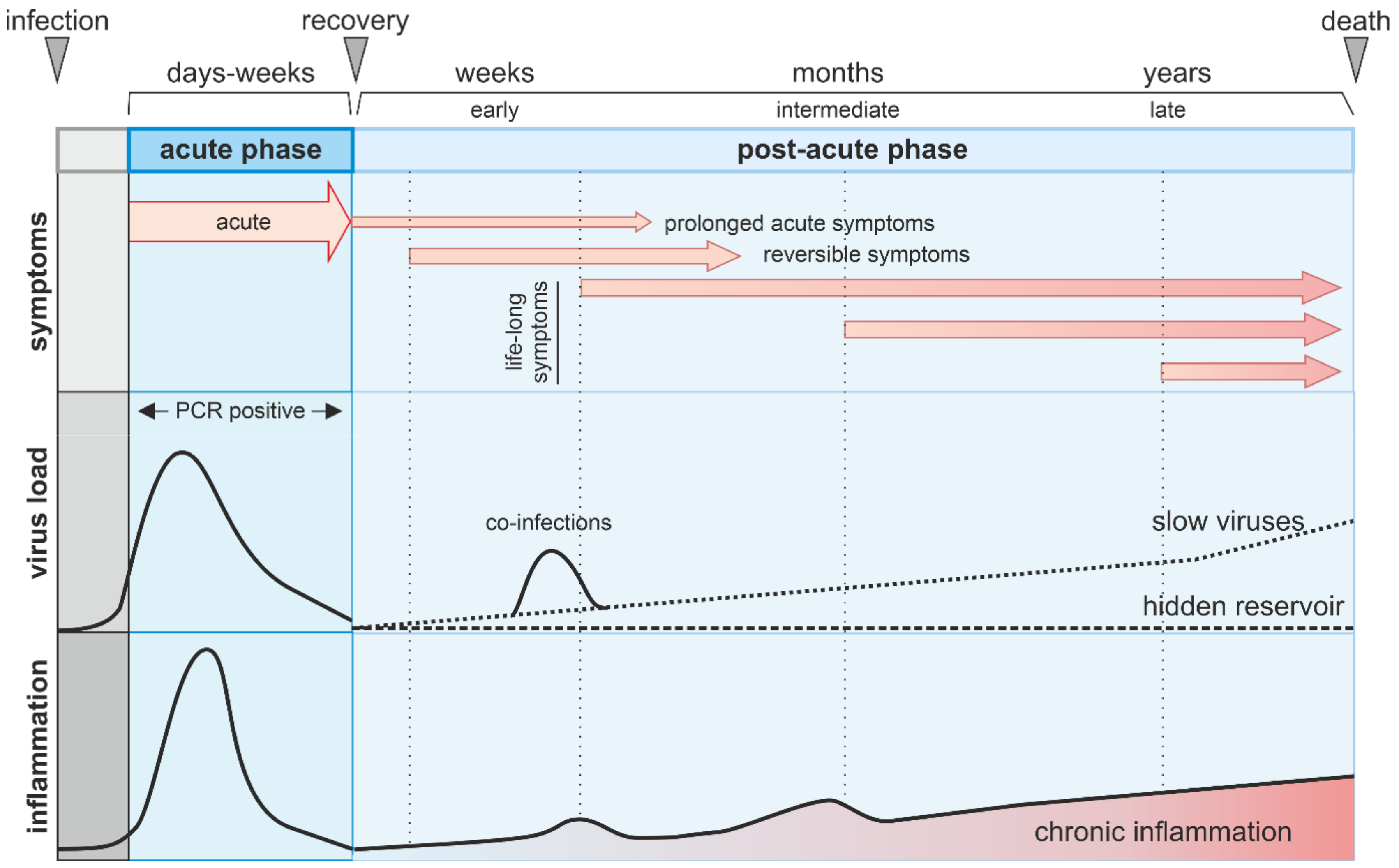
1.2. What Are Post-Acute Sequelae of Viral Infections?
2. Post-Acute Sequelae of Non-Persistent Viruses
2.1. DNA Viruses
2.2. dsRNA Viruses
2.3. (−) RNA Viruses
2.4. (+) ssRNA Viruses
2.5. Post-Acute Sequelae of COVID19 (PASC)
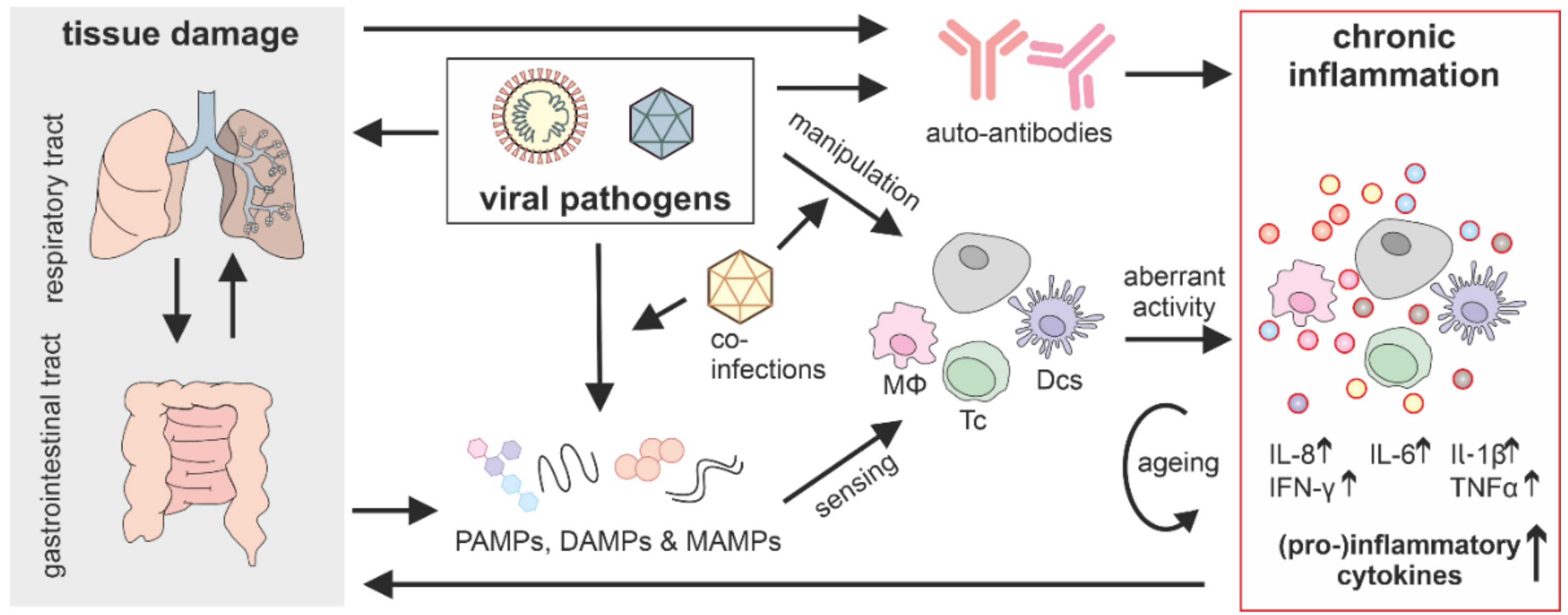
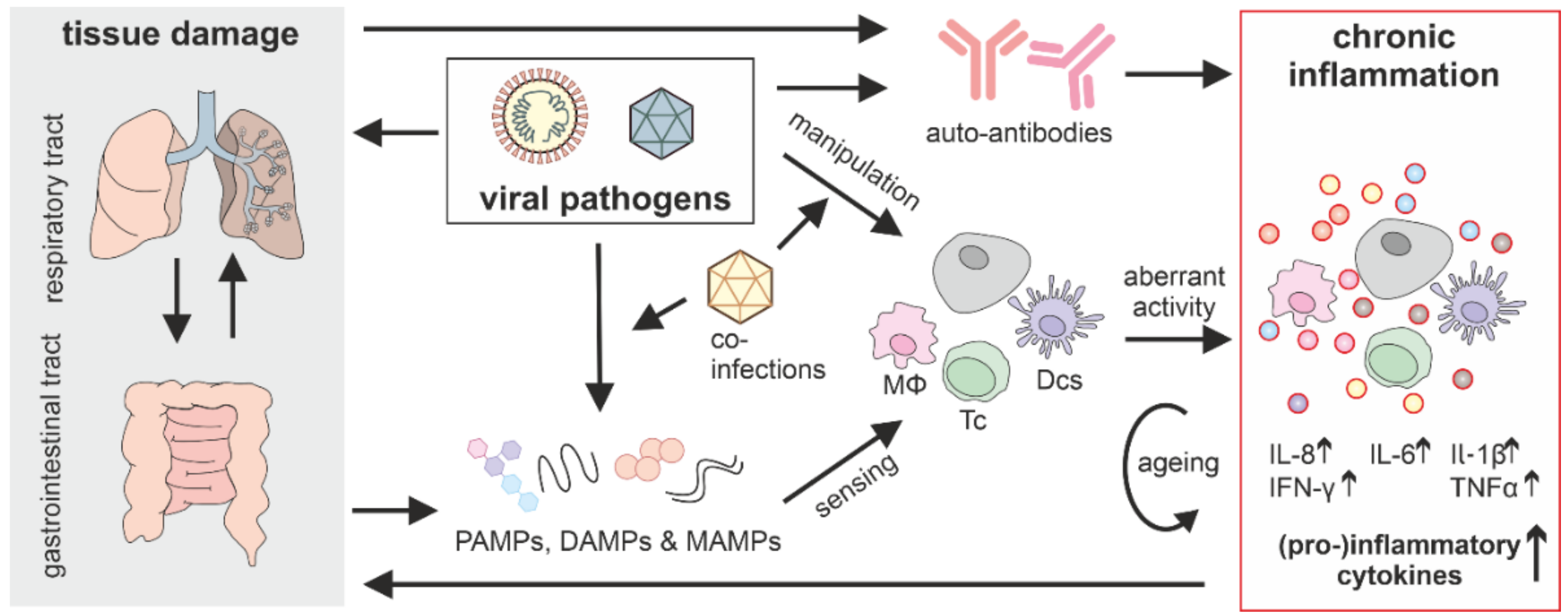
3. Aberrant Innate Immune Activation in Post-Acute Sequelae
3.1. Innate Immune Responses in Post-Acute Conditions of Persistent Viruses
3.2. Chronic Inflammation Induction by Non-Persistent Viruses
4. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Takeuchi, O.; Akira, S. Innate immunity to virus infection. Immunol. Rev. 2008, 227, 75–86. [Google Scholar] [CrossRef]
- Iwasaki, A.; Medzhitov, R. Regulation of adaptive immunity by the innate immune system. Science 2010, 327, 291–295. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, K.A.; Kagan, J.C. Toll-like receptors and the control of immunity. Cell 2020, 180, 1044–1066. [Google Scholar] [CrossRef]
- Sparrer, K.; Gack, M.U. Intracellular detection of viral nucleic acids. Curr. Opin. Microbiol. 2015, 26, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Hale, B.G.; Albrecht, R.; García-Sastre, A. Innate immune evasion strategies of influenza viruses. Futur. Microbiol. 2010, 5, 23–41. [Google Scholar] [CrossRef] [Green Version]
- Kikkert, M. Innate immune evasion by human respiratory RNA viruses. J. Innate Immun. 2019, 12, 4–20. [Google Scholar] [CrossRef]
- Hayn, M.; Hirschenberger, M.; Koepke, L.; Nchioua, R.; Straub, J.H.; Klute, S.; Hunszinger, V.; Zech, F.; Bozzo, C.P.; Aftab, W.; et al. Systematic functional analysis of SARS-CoV-2 proteins uncovers viral innate immune antagonists and remaining vulnerabilities. Cell Rep. 2021, 35, 109126. [Google Scholar] [CrossRef] [PubMed]
- Mangalmurti, N.; Hunter, C.A. Cytokine storms: Understanding COVID-19. Immunity 2020, 53, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Fajgenbaum, D.C.; June, C.H. Cytokine storm. N. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, L.; Fritz, R.S.; Straus, S.E.; Gubareva, L.; Hayden, F.G. Symptom pathogenesis during acute influenza: Interleukin-6 and other cytokine responses. J. Med. Virol. 2001, 64, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Wong, C.K.; Chan, P.; Chan, M.C.-W.; Wong, R.Y.K.; Lun, S.W.M.; Ngai, K.L.K.; Lui, C.Y.G.; Wong, B.C.K.; Lee, S.K.W.; et al. Cytokine response patterns in severe pandemic 2009 H1N1 and seasonal influenza among hospitalized adults. PLoS ONE 2011, 6, e26050. [Google Scholar] [CrossRef] [PubMed]
- Cohen, L.; Fiore-Gartland, A.; Randolph, A.G.; Panoskaltsis-Mortari, A.; Wong, S.-S.; Ralston, J.; Wood, T.; Seeds, R.; Huang, Q.S.; Webby, R.J.; et al. A modular cytokine analysis method reveals novel associations with clinical phenotypes and identifies sets of co-signaling cytokines across influenza natural infection cohorts and healthy controls. Front. Immunol. 2019, 10, 1338. [Google Scholar] [CrossRef]
- Vogel, A.J.; Harris, S.; Marsteller, N.; Condon, S.A.; Brown, D.M. Early cytokine dysregulation and viral replication are associated with mortality during lethal influenza infection. Viral Immunol. 2014, 27, 214–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Q.; Wang, B.; Mao, J. The pathogenesis and treatment of the “Cytokine Storm” in COVID-19. J. Infect. 2020, 80, 607–613. [Google Scholar] [CrossRef]
- Islam, M.F.; Cotler, J.; Jason, L.A. Post-viral fatigue and COVID-19: Lessons from past epidemics. Fatigue Biomed. Health Behav. 2020, 8, 61–69. [Google Scholar] [CrossRef]
- Al-Aly, Z.; Xie, Y.; Bowe, B. High-dimensional characterization of post-acute sequelae of COVID-19. Nat. Cell Biol. 2021, 594, 259–264. [Google Scholar] [CrossRef]
- Poole-Wright, K.; Gaughran, F.; Evans, R.; Chalder, T. Fatigue outcomes following coronavirus or influenza virus infection: A systematic review and meta-analysis. medRxiv 2020. [Google Scholar] [CrossRef]
- Archer, M. The post-viral syndrome: A review. J. R. Coll. Gen. Pr. 1987, 37, 212–214. [Google Scholar]
- Marshall, M. The lasting misery of coronavirus long-haulers. Nat. Cell Biol. 2020, 585, 339–341. [Google Scholar] [CrossRef]
- Dalakas, M.C. The post-polio syndrome as an evolved clinical entity. Ann. N. Y. Acad. Sci. 1995, 753, 68–80. [Google Scholar] [CrossRef]
- Ramlow, J.; Alexander, M.; Laporte, R.; Kaufmann, C.; Kuller, L. Epidemiology of the post-polio syndrome. Am. J. Epidemiol. 1992, 136, 769–786. [Google Scholar] [CrossRef]
- Nalbandian, A.; Sehgal, K.; Gupta, A.; Madhavan, M.V.; McGroder, C.; Stevens, J.S.; Cook, J.R.; Nordvig, A.S.; Shalev, D.; Sehrawat, T.S.; et al. Post-acute COVID-19 syndrome. Nat. Med. 2021, 27, 601–615. [Google Scholar] [CrossRef] [PubMed]
- Proal, A.D.; VanElzakker, M.B. Long COVID or post-acute sequelae of COVID-19 (PASC): An overview of biological factors that may contribute to persistent symptoms. Front. Microbiol. 2021, 12, 698169. [Google Scholar] [CrossRef] [PubMed]
- Rasa, S.; Nora-Krukle, Z.; Henning, N.; Eliassen, E.; Shikova, E.; Harrer, T.; Scheibenbogen, C.; Murovska, M.; Prusty, B.K. Chronic Viral Infections in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS); BioMed Central Ltd.: London, UK, 2018. [Google Scholar]
- Sotzny, F.; Blanco, J.; Capelli, E.; Castro-Marrero, J.; Steiner, S.; Murovska, M.; Scheibenbogen, C. Myalgic encephalomyelitis/chronic fatigue syndrome—Evidence for an autoimmune disease. Autoimmun. Rev. 2018, 17, 601–609. [Google Scholar] [CrossRef]
- Kühl, U.; Pauschinger, M.; Noutsias, M.; Seeberg, B.; Bock, T.; Lassner, D.; Poller, W.; Kandolf, R.; Schultheiss, H.-P. High prevalence of viral genomes and multiple viral infections in the myocardium of adults with “idiopathic” left ventricular dysfunction. Circulation 2005, 111, 887–893. [Google Scholar] [CrossRef] [Green Version]
- Tschöpe, C.; Ammirati, E.; Bozkurt, B.; Caforio, A.L.P.; Cooper, L.T.; Felix, S.B.; Hare, J.M.; Heidecker, B.; Heymans, S.; Hübner, N.; et al. Myocarditis and inflammatory cardiomyopathy: Current evidence and future directions. Nat. Rev. Cardiol. 2020, 18, 169–193. [Google Scholar] [CrossRef]
- Van Bijsterveld, O.P.; Van Hemel, O.L. Inflammatory sequelae after adenovirus infection. J. Fr. D’ophtalmol. 1988, 11, 25–29. [Google Scholar]
- Green, P.H.R.; Cellier, C. Celiac disease. N. Engl. J. Med. 2007, 357, 1731–1743. [Google Scholar] [CrossRef] [PubMed]
- Harrison, L.C.; Perrett, K.P.; Jachno, K.; Nolan, T.M.; Honeyman, M.C. Does rotavirus turn on type 1 diabetes? PLoS Pathog. 2019, 15, e1007965. [Google Scholar] [CrossRef] [PubMed]
- Blomqvist, M.; Juhela, S.; Erkkilä, S.; Korhonen, S.; Simell, T.; Kupila, A.; Vaarala, O.; Simell, O.; Knip, M.; Ilonen, J. Rotavirus infections and development of diabetes-associated autoantibodies during the first 2 years of life. Clin. Exp. Immunol. 2002, 128, 511–515. [Google Scholar] [CrossRef]
- Bouziat, R.; Hinterleitner, R.; Brown, J.J.; Stencel-Baerenwald, J.E.; Ikizler, M.; Mayassi, T.; Meisel, M.; Kim, S.M.; Discepolo, V.; Pruijssers, A.J.; et al. Reovirus infection triggers inflammatory responses to dietary antigens and development of celiac disease. Science 2017, 356, 44–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vries, R.; McQuaid, S.; Van Amerongen, G.; Yüksel, S.; Verburgh, R.J.; Osterhaus, A.; Duprex, W.P.; De Swart, R. Measles immune suppression: Lessons from the macaque model. PLoS Pathog. 2012, 8, e1002885. [Google Scholar] [CrossRef]
- Griffin, D.E.; Lin, W.-H.W.; Nelson, A.N. Understanding the causes and consequences of measles virus persistence. F1000Research 2018, 7, 237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mina, M.J.; Metcalf, C.E.; De Swart, R.; Osterhaus, A.; Grenfell, B. Long-term measles-induced immunomodulation increases overall childhood infectious disease mortality. Science 2015, 348, 694–699. [Google Scholar] [CrossRef] [Green Version]
- Griffin, D. Measles virus persistence and its consequences. Curr. Opin. Virol. 2020, 41, 46–51. [Google Scholar] [CrossRef]
- Riddell, M.; Moss, W.J.; Hauer, D.; Monze, M.; Griffin, D.E. Slow clearance of measles virus RNA after acute infection. J. Clin. Virol. 2007, 39, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.K.; Mahadevan, A.; Malhotra, H.S.; Rizvi, I.; Kumar, N.; Uniyal, R. Subacute sclerosing panencephalitis. Rev. Med. Virol. 2019, 29, e2058. [Google Scholar] [CrossRef]
- Patterson, J.B.; Cornu, T.I.; Redwine, J.; Dales, S.; Lewicki, H.; Holz, A.; Thomas, D.; Billeter, M.A.; Oldstone, M.B. Evidence that the hypermutated m protein of a subacute sclerosing panencephalitis measles virus actively contributes to the chronic progressive CNS disease. Virolgy 2001, 291, 215–225. [Google Scholar] [CrossRef] [Green Version]
- Ng, B.-Y.; Lim, C.T.; Yeoh, A.; Lee, W. Neuropsychiatric sequelae of nipah virus encephalitis. J. Neuropsychiatry Clin. Neurosci. 2004, 16, 500–504. [Google Scholar] [CrossRef]
- Sejvar, J.J.; Hossain, J.; Saha, S.K.; Gurley, E.S.; Banu, S.; Hamadani, J.; Faiz, M.A.; Siddiqui, F.M.; Mohammad, Q.D.; Mollah, A.H.; et al. Long-term neurological and functional outcome in Nipah virus infection. Ann. Neurol. 2007, 62, 235–242. [Google Scholar] [CrossRef]
- Tan, C.T.; Goh, K.J.; Wong, K.T.; Sarji, S.A.; Chua, K.B.; Chew, N.K.; Murugasu, P.; Loh, Y.L.; Chong, H.T.; Tan, K.S.; et al. Relapsed and late-onset nipah encephalitis. Ann. Neurol. 2002, 51, 703–708. [Google Scholar] [CrossRef]
- Clark, D.V.; Kibuuka, H.; Millard, M.; Wakabi, S.; Lukwago, L.; Taylor, A.; Eller, M.; Eller, L.A.; Michael, N.L.; Honko, A.; et al. Long-term sequelae after Ebola virus disease in Bundibugyo, Uganda: A retrospective cohort study. Lancet Infect. Dis. 2015, 15, 905–912. [Google Scholar] [CrossRef] [Green Version]
- Diallo, M.S.K.; Toure, A.; Sow, M.S.; Kpamou, C.; Keita, A.K.; Taverne, B.; Peeters, M.; Msellati, P.; Barry, T.A.; Etard, J.-F.; et al. Understanding the long-term evolution and predictors of sequelae of Ebola virus disease survivors in Guinea: A 48-month prospective, longitudinal cohort study (PostEboGui). Clin. Infect. Dis. 2021. [Google Scholar] [CrossRef] [PubMed]
- Keita, M.; Diallo, B.; Mesfin, S.; Marega, A.; Nebie, K.Y.; Magassouba, N.; Barry, A.; Coulibaly, S.; Barry, B.; Baldé, M.O.; et al. Subsequent mortality in survivors of Ebola virus disease in Guinea: A nationwide retrospective cohort study. Lancet Infect. Dis. 2019, 19, 1202–1208. [Google Scholar] [CrossRef] [Green Version]
- Wiedemann, A.; Foucat, E.; Hocini, H.; Lefebvre, C.; Hejblum, B.P.; Durand, M.; Krüger, M.; Keita, A.K.; Ayouba, A.; Mély, S.; et al. Long-lasting severe immune dysfunction in Ebola virus disease survivors. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Fausther-Bovendo, H.; Qiu, X.; McCorrister, S.; Westmacott, G.; Sandstrom, P.; Castilletti, C.; Di Caro, A.; Ippolito, G.; Kobinger, G.P. Ebola virus infection induces autoimmunity against dsDNA and HSP60. Sci. Rep. 2017, 7, srep42147. [Google Scholar] [CrossRef] [PubMed]
- Jacob, S.; Crozier, I.; Ii, W.A.F.; Hewlett, A.; Kraft, C.S.; De La Vega, M.-A.; Soka, M.J.; Wahl, V.; Griffiths, A.; Bollinger, L.; et al. Ebola virus disease. Nat. Rev. Dis. Prim. 2020, 6, 1–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tozay, S.; Fischer, W.; Wohl, D.; Kilpatrick, K.; Zou, F.; Reeves, E.; Pewu, K.; Demarco, J.; Loftis, A.J.; King, K.; et al. Long-term complications of Ebola virus disease: Prevalence and predictors of major symptoms and the role of inflammation. Clin. Infect. Dis. 2019, 71, 1749–1755. [Google Scholar] [CrossRef] [PubMed]
- Martini, G.A. Marburg virus disease. Postgrad. Med. J. 1973, 49, 542–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shifflett, K.; Marzi, A. Marburg virus pathogenesis—Differences and similarities in humans and animal models. Virol. J. 2019, 16, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Ezeomah, C.; Adoga, A.; Ihekweazu, C.; Paessler, S.; Cisneros, I.; Tomori, O.; Walker, D. Sequelae of lassa fever: Postviral cerebellar ataxia. Open Forum Infect. Dis. 2019, 6. [Google Scholar] [CrossRef]
- Ficenec, S.C.; Schieffelin, J.S.; Emmett, S.D. A review of hearing loss associated with zika, Ebola, and Lassa fever. Am. J. Trop. Med. Hyg. 2019, 101, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Li, A.L.; Grant, D.; Gbakie, M.; Kanneh, L.; Mustafa, I.; Bond, N.; Engel, E.; Schieffelin, J.; Vandy, M.J.; Yeh, S.; et al. Ophthalmic manifestations and vision impairment in Lassa fever survivors. PLoS ONE 2020, 15, e0243766. [Google Scholar] [CrossRef] [PubMed]
- Jartti, T.; Bønnelykke, K.; Elenius, V.; Feleszko, W. Role of viruses in asthma. Semin. Immunopathol. 2020, 42, 61–74. [Google Scholar] [CrossRef] [Green Version]
- Bønnelykke, K.; Vissing, N.H.; Sevelsted, A.; Johnston, S.L.; Bisgaard, H. Association between respiratory infections in early life and later asthma is independent of virus type. J. Allergy Clin. Immunol. 2015, 136, 81.e4–86.e4. [Google Scholar] [CrossRef]
- Krammer, F.; Smith, G.J.D.; Fouchier, R.; Peiris, J.S.M.; Kedzierska, K.; Doherty, P.C.; Palese, P.; Shaw, M.L.; Treanor, J.; Webster, R.G.; et al. Influenza. Nat. Rev. Dis. Prim. 2018, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Fauroux, B.; Simões, E.A.F.; Checchia, P.A.; Paes, B.; Figueras-Aloy, J.; Manzoni, P.; Bont, L.; Carbonell-Estrany, X. The burden and long-term respiratory morbidity associated with respiratory syncytial virus infection in early childhood. Infect. Dis. Ther. 2017, 6, 173–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, T.; Ooi, Y.; Zaw, E.M.; Utjesanovic, N.; Campbell, H.; Cunningham, S.; Bont, L.; Nair, H.; Zhang, S.; Li, Y.; et al. Association between respiratory syncytial virus-associated acute lower respiratory infection in early life and recurrent wheeze and asthma in later childhood. J. Infect. Dis. 2019, 222, S628–S633. [Google Scholar] [CrossRef] [Green Version]
- Souza, I.N.O.; Barros-Aragão, F.G.Q.; Frost, P.S.; Figueiredo, C.P.; Clarke, J.R. Late neurological consequences of zika virus infection: Risk factors and pharmaceutical approaches. Pharmaceuticals 2019, 12, 60. [Google Scholar] [CrossRef] [Green Version]
- Koppolu, V.; Raju, T.S. Zika virus outbreak: A review of neurological complications, diagnosis, and treatment options. J. Neurovirol. 2018, 24, 255–272. [Google Scholar] [CrossRef]
- Leonhard, S.E.; Bresani-Salvi, C.C.; Batista, J.D.L.; Cunha, S.; Jacobs, B.C.; Ferreira, M.L.B.; Militão de Albuquerque, M.D.F.P. Guillain-Barré syndrome related to Zika virus infection: A systematic review and meta-analysis of the clinical and electrophysiological phenotype. PLoS Negl. Trop. Dis. 2020, 14, e0008264. [Google Scholar] [CrossRef] [PubMed]
- Grijalva, I.; Grajales-Muñiz, C.; González-Bonilla, C.; Borja-Aburto, V.H.; Paredes-Cruz, M.; Guerrero-Cantera, J.; González-Ibarra, J.; Vallejos-Parás, A.; Rojas-Mendoza, T.; Santacruz-Tinoco, C.E.; et al. Zika and dengue but not chikungunya are associated with Guillain-Barré syndrome in Mexico: A case-control study. PLoS Negl. Trop. Dis. 2020, 14, e0008032. [Google Scholar] [CrossRef]
- Ferreira, M.L.B.; Albuquerque, M.D.F.P.M.D.; de Brito, C.A.A.; França, R.F.D.O.; Moreira, J.P.; Machado, M.D.M.; Melo, R.D.P.; Medialdea-Carrera, R.; Mesquita, S.D.; Santos, M.L.; et al. Neurological disease in adults with Zika and chikungunya virus infection in Northeast Brazil: A prospective observational study. Lancet Neurol. 2020, 19, 826–839. [Google Scholar] [CrossRef]
- Acevedo, N.; Waggoner, J.; Rodriguez, M.; Rivera, L.; Landivar, J.; Pinsky, B.; Zambrano, H. Zika virus, chikungunya virus, and dengue virus in cerebrospinal fluid from adults with neurological manifestations, Guayaquil, Ecuador. Front. Microbiol. 2017, 8, 42. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, L.D.A.S.; Nogueira, N.G.A.; Nascentes, G.A.N. Prospective study of patients with persistent symptoms of dengue in Brazil. Rev. Inst. Med. Trop. São Paulo 2017, 59, e65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.-H.; Ning, Z.-J.; Liu, Y.-M.; Li, X.-H. Neurological manifestations of dengue infection. Front. Cell. Infect. Microbiol. 2017, 7, 449. [Google Scholar] [CrossRef] [Green Version]
- García, G.; González, N.; Pérez, A.B.; Sierra, B.; Aguirre, E.; Rizo, D.; Izquierdo, A.; Sánchez, L.; Díaz, D.; Lezcay, M.; et al. Long-term persistence of clinical symptoms in dengue-infected persons and its association with immunological disorders. Int. J. Infect. Dis. 2011, 15, e38–e43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, H.; Sander, B.; Nelder, M.P. Long-term sequelae of West Nile virus-related illness: A systematic review. Lancet 2015, 15. [Google Scholar] [CrossRef]
- Murray, K.; Walker, C.; Herrington, E.; Lewis, J.A.; McCormick, J.; Beasley, D.W.C.; Tesh, R.B.; Fisher-Hoch, S. Persistent infection with West Nile virus years after initial infection. J. Infect. Dis. 2010, 201, 2–4. [Google Scholar] [CrossRef] [Green Version]
- Garcia, M.N.; Hasbun, R.; Murray, K.O. Persistence of West Nile virus. Microbes Infect. 2015, 17, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Vittor, A.Y.; Long, M.; Chakrabarty, P.; Aycock, L.; Kollu, V.; DeKosky, S.T. West Nile virus-induced neurologic sequelae—Relationship to neurodegenerative cascades and dementias. Curr. Trop. Med. Rep. 2020, 7, 25–36. [Google Scholar] [CrossRef]
- Haglund, M.; Günther, G. Tick-borne encephalitis—Pathogenesis, clinical course and long-term follow-up. Vaccine 2003, 21, S11–S18. [Google Scholar] [CrossRef]
- Solomon, T.; Dung, N.M.; Kneen, R.; Gainsborough, M.; Vaughn, D.W.; Khanh, V.T. Neurological aspects of tropical disease: Japanese encephalitis. J. Neurol. Neurosurg. Psychiatry 2000, 68, 405–415. [Google Scholar] [CrossRef]
- Suhrbier, A. Rheumatic manifestations of chikungunya: Emerging concepts and interventions. Nat. Rev. Rheumatol. 2019, 15, 597–611. [Google Scholar] [CrossRef]
- Mehta, R.; Gerardin, P.; De Brito, C.A.A.; Soares, C.N.; Ferreira, M.L.B.; Solomon, T. The neurological complications of chikungunya virus: A systematic review. Rev. Med. Virol. 2018, 28, e1978. [Google Scholar] [CrossRef] [Green Version]
- Ronca, S.E.; Dineley, K.T.; Paessler, S. Neurological sequelae resulting from encephalitic alphavirus infection. Front. Microbiol. 2016, 7, 959. [Google Scholar] [CrossRef]
- Selden, S.M.; Cameron, S. Changing epidemiology of Ross River virus disease in South Australia. Med. J. Aust. 1996, 165, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Potter, M.C.; Baxter, V.K.; Mathey, R.W.; Alt, J.; Rojas, C.; Griffin, D.E.; Slusher, B.S. Neurological sequelae induced by alphavirus infection of the CNS are attenuated by treatment with the glutamine antagonist 6-diazo-5-oxo-l-norleucine. J. Neurovirol. 2015, 21, 159–173. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.-Y.; Lin, H.-Y.; Gau, S.S.-F.; Lu, C.-Y.; Hsia, S.-H.; Huang, Y.-C.; Huang, L.-M.; Lin, T.-Y. Enterovirus A71 neurologic complications and long-term sequelae. J. Biomed. Sci. 2019, 26, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, L.; Cheng, Y.; Li, Y.; Shang, Q.; Huang, J.; Ma, C.; Fang, S.; Long, L.; Zhou, C.; Chen, Z.; et al. Long-term neurodevelopment outcomes of hand, foot and mouth disease inpatients infected with EV-A71 or CV-A16, a retrospective cohort study. Emerg. Microbes Infect. 2021, 10, 1–37. [Google Scholar] [CrossRef] [PubMed]
- Aw-Yong, K.L.; NikNadia, N.M.N.; Tan, C.W.; Sam, I.; Chan, Y.F. Immune responses against enterovirus A71 infection: Implications for vaccine success. Rev. Med. Virol. 2019, 29, e2073. [Google Scholar] [CrossRef] [PubMed]
- Tee, H.K.; Zainol, M.I.; Sam, I.-C.; Chan, Y.F. Recent advances in the understanding of enterovirus A71 infection: A focus on neuropathogenesis. Expert Rev. Anti-Infect. Ther. 2021, 19, 733–747. [Google Scholar] [CrossRef]
- Trojan, D.A.; Cashman, N.R. Post-poliomyelitis syndrome. Muscle Nerve 2004, 31, 6–19. [Google Scholar] [CrossRef]
- Baj, A.; Monaco, S.; Zanusso, G.; Dall’Ora, E.; Bertolasi, L.; Toniolo, A. Virology of the post-polio syndrome. Futur. Virol. 2007, 2, 183–192. [Google Scholar] [CrossRef]
- Farbu, E.; Rekand, T.; Tysnes, O.-B.; Aarli, J.; Gilhus, N.; Vedeler, C. GM1 antibodies in post-polio syndrome and previous paralytic polio. J. Neuroimmunol. 2003, 139, 141–144. [Google Scholar] [CrossRef]
- Ginsberg, A.H.; Gale, M.J.; Rose, L.M.; Clark, E.A. T-cell alterations in late postpoliomyelitis. Arch. Neurol. 1989, 46, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Stolwijk-Swüste, J.M.; Beelen, A.; Lankhorst, G.J.; Nollet, F. The course of functional status and muscle strength in patients with late-onset sequelae of poliomyelitis: A systematic review. Arch. Phys. Med. Rehabil. 2005, 86, 1693–1701. [Google Scholar] [CrossRef]
- Logue, J.K.; Franko, N.M.; McCulloch, D.J.; McDonald, D.; Magedson, A.; Wolf, C.R.; Chu, H.Y. Sequelae in adults at 6 months after COVID-19 infection. JAMA Netw. Open 2021, 4, e210830. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.A.; Groß, R.; Conzelmann, C.; Krüger, J.; Merle, U.; Steinhart, J.; Weil, T.; Koepke, L.; Bozzo, C.P.; Read, C.; et al. SARS-CoV-2 infects and replicates in cells of the human endocrine and exocrine pancreas. Nat. Metab. 2021, 3, 149–165. [Google Scholar] [CrossRef]
- Lim, S.; Bae, J.H.; Kwon, H.-S.; Nauck, M.A. COVID-19 and diabetes mellitus: From pathophysiology to clinical management. Nat. Rev. Endocrinol. 2020, 17, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-T.; Lidsky, P.V.; Xiao, Y.; Lee, I.T.; Cheng, R.; Nakayama, T.; Jiang, S.; Demeter, J.; Bevacqua, R.J.; Chang, C.A.; et al. SARS-CoV-2 infects human pancreatic β cells and elicits β cell impairment. Cell Metab. 2021, 33, 1565.e5–1576.e5. [Google Scholar] [CrossRef]
- Augustin, M.; Schommers, P.; Stecher, M.; Dewald, F.; Gieselmann, L.; Gruell, H.; Horn, C.; Vanshylla, K.; Di Cristanziano, V.; Osebold, L.; et al. Post-COVID syndrome in non-hospitalised patients with COVID-19: A longitudinal prospective cohort study. Lancet Reg. Health Eur. 2021, 6, 100122. [Google Scholar] [CrossRef]
- Davis, H.E.; Assaf, G.S.; McCorkell, L.; Wei, H.; Low, R.J.; Re’Em, Y.; Redfield, S.; Austin, J.P.; Akrami, A. Characterizing long COVID in an international cohort: 7 months of symptoms and their impact. EClinicalMedicine 2021. [Google Scholar] [CrossRef] [PubMed]
- Brodin, P. Immune determinants of COVID-19 disease presentation and severity. Nat. Med. 2021, 27, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Licciardi, F.; Pruccoli, G.; Denina, M.; Parodi, E.; Taglietto, M.; Rosati, S.; Montin, D. SARS-CoV-2-induced Kawasaki-like hyperinflammatory syndrome: A novel COVID phenotype in children. Pediatrics 2020, 146, e20201711. [Google Scholar] [CrossRef]
- Moldofsky, H.; Patcai, J. Chronic widespread musculoskeletal pain, fatigue, depression and disordered sleep in chronic post-SARS syndrome; a case-controlled study. BMC Neurol. 2011, 11, 37. [Google Scholar] [CrossRef] [Green Version]
- Lam, M.H.-B.; Wing, Y.-K.; Yu, M.W.-M.; Leung, C.-M.; Ma, R.C.W.; Kong, A.P.S.; So, W.; Fong, S.Y.-Y.; Lam, S.-P. Mental morbidities and chronic fatigue in severe acute respiratory syndrome survivors. Arch. Intern. Med. 2009, 169, 2142–2147. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, O. Long-Term sequelae following previous coronavirus epidemics. Clin. Med. 2021, 21, e68–e70. [Google Scholar] [CrossRef]
- Wirth, K.; Scheibenbogen, C. A unifying hypothesis of the pathophysiology of myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS): Recognitions from the finding of autoantibodies against ß2-adrenergic receptors. Autoimmun. Rev. 2020, 19, 102527. [Google Scholar] [CrossRef] [PubMed]
- Hemming-Harlo, M.; Lähdeaho, M.-L.; Mäki, M.; Vesikari, T. Rotavirus vaccination does not increase type 1 diabetes and may decrease celiac disease in children and adolescents. Pediatr. Infect. Dis. J. 2019, 38, 539–541. [Google Scholar] [CrossRef]
- Perrett, K.; Jachno, K.; Nolan, T.M.; Harrison, L.C. Association of rotavirus vaccination with the incidence of type 1 diabetes in children. JAMA Pediatr. 2019, 173, 280–282. [Google Scholar] [CrossRef]
- Ishizaki, Y.; Takemoto, M.; Kira, R.; Kusuhara, K.; Torisu, H.; Sakai, Y.; Sanefuji, M.; Yukaya, N.; Hara, T. Association of toll-like receptor 3 gene polymorphism with subacute sclerosing panencephalitis. J. Neurovirol. 2008, 14, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Coffin, K.M.; Johnston, S.C.; Babka, A.M.; Bell, T.M.; Long, S.Y.; Honko, A.N.; Kuhn, J.H.; Zeng, X. Nipah virus persists in the brains of nonhuman primate survivors. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Ebola Virus Disease. 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/ebola-virus-disease (accessed on 12 August 2021).
- Reisler, R.B.; Zeng, X.; Schellhase, C.W.; Bearss, J.J.; Warren, T.K.; Trefry, J.C.; Christopher, G.W.; Kortepeter, M.G.; Bavari, S.; Cardile, A.P. Ebola virus causes intestinal tract architectural disruption and bacterial invasion in non-human primates. Viruses 2018, 10, 513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cashman, K.A.; Wilkinson, E.R.; Zeng, X.; Cardile, A.P.; Facemire, P.R.; Bell, T.M.; Bearss, J.J.; Shaia, C.; Schmaljohn, C.S. Immune-mediated systemic vasculitis as the proposed cause of sudden-onset sensorineural hearing loss following Lassa virus exposure in cynomolgus macaques. mBio 2018, 9, e01896-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keeler, S.P.; Agapov, E.V.; Hinojosa, M.E.; Letvin, A.N.; Wu, K.; Holtzman, M.J. Influenza a virus infection causes chronic lung disease linked to sites of active viral RNA remnants. J. Immunol. 2018, 201, 2354–2368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Driscoll, A.J.; Arshad, S.H.; Bont, L.; Brunwasser, S.M.; Cherian, T.; Englund, J.A.; Fell, D.B.; Hammitt, L.L.; Hartert, T.V.; Innis, B.L.; et al. Does respiratory syncytial virus lower respiratory illness in early life cause recurrent wheeze of early childhood and asthma? Critical review of the evidence and guidance for future studies from a World Health Organization-sponsored meeting. Vaccine 2020, 38, 2435–2448. [Google Scholar] [CrossRef]
- Lehners, N.; Tabatabai, J.; Prifert, C.; Wedde, M.; Puthenparambil, J.; Weissbrich, B.; Biere, B.; Schweiger, B.; Egerer, G.; Schnitzler, P. Long-term shedding of influenza virus, parainfluenza virus, respiratory syncytial virus and nosocomial epidemiology in patients with hematological disorders. PLoS ONE 2016, 11, e0148258. [Google Scholar] [CrossRef]
- Rane, C.K.; Jackson, S.R.; Pastore, C.F.; Zhao, G.; Weiner, A.I.; Patel, N.N.; Herbert, D.R.; Cohen, N.A.; Vaughan, A.E. Development of solitary chemosensory cells in the distal lung after severe influenza injury. Am. J. Physiol. Cell. Mol. Physiol. 2019, 316, L1141–L1149. [Google Scholar] [CrossRef]
- Heaton, N.S.; Langlois, R.A.; Sachs, D.; Lim, J.K.; Palese, P.; Tenoever, B.R. Long-term survival of influenza virus infected club cells drives immunopathology. J. Exp. Med. 2014, 211, 1707–1714. [Google Scholar] [CrossRef] [Green Version]
- Garcia, G.L.; Valenzuela, A.; Manzoni, T.; Vaughan, A.E.; López, C.B. Distinct chronic post-viral lung diseases upon infection with influenza or parainfluenza viruses differentially impact superinfection outcome. Am. J. Pathol. 2020, 190, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Willison, H.J.; Jacobs, B.C.; van Doorn, P. Guillain-Barré syndrome. Lancet 2016, 388, 717–727. [Google Scholar] [CrossRef] [Green Version]
- Silva-Filho, J.L.; Oliveira, L.G.; Monteiro, L.; Parise, P.L.; Zanluqui, N.G.; Polonio, C.M.; Freitas, C.L.; Toledo-Teixeira, D.A.; Souza, W.M.; Bittencourt, N.; et al. Gas6 drives zika virus-induced neurological complications in humans and congenital syndrome in immunocompetent mice. bioRxiv 2021. [Google Scholar] [CrossRef]
- Bhatt, S.; Gething, P.; Brady, O.; Messina, J.P.; Farlow, A.W.; Moyes, C.; Drake, J.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O.; et al. The global distribution and burden of dengue. Nat. Cell Biol. 2013, 496, 504–507. [Google Scholar] [CrossRef]
- Fulton, C.D.; Beasley, D.W.; Bente, D.A.; Dineley, K.T. Long-term, West Nile virus-induced neurological changes: A comparison of patients and rodent models. Brain Behav. Immun. Health 2020, 7, 100105. [Google Scholar] [CrossRef]
- Garber, C.; Soung, A.; Vollmer, L.L.; Kanmogne, M.; Last, A.; Brown, J.; Klein, R.S. T cells promote microglia-mediated synaptic elimination and cognitive dysfunction during recovery from neuropathogenic flaviviruses. Nat. Neurosci. 2019, 22, 1276–1288. [Google Scholar] [CrossRef]
- Van Aalst, M.; Nelen, C.M.; Goorhuis, A.; Stijnis, C.; Grobusch, M.P. Long-Term Sequelae of Chikungunya Virus Disease: A Systematic Review; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Poo, Y.S.; Rudd, P.A.; Gardner, J.; Wilson, J.A.C.; Larcher, T.; Colle, M.-A.; Le, T.T.; Nakaya, H.; Warrilow, D.; Allcock, R.; et al. Multiple immune factors are involved in controlling acute and chronic chikungunya virus infection. PLoS Negl. Trop. Dis. 2014, 8, e3354. [Google Scholar] [CrossRef]
- Bantle, C.M.; Phillips, A.T.; Smeyne, R.J.; Rocha, S.M.; Olson, K.E.; Tjalkens, R.B. Infection with mosquito-borne alphavirus induces selective loss of dopaminergic neurons, neuroinflammation and widespread protein aggregation. NPJ Parkinson’s Dis. 2019, 5, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herzum, M.; Ruppert, V.; Küytz, B.; Jomaa, H.; Nakamura, I.; Maisch, B. Coxsackievirus B3 infection leads to cell death of cardiac myocytes. J. Mol. Cell. Cardiol. 1994, 26, 907–913. [Google Scholar] [CrossRef]
- Triantafilou, K.; Orthopoulos, G.; Vakakis, E.; Ahmed, M.A.E.; Golenbock, D.T.; Lepper, P.M.; Triantafilou, M. Human cardiac inflammatory responses triggered by Coxsackie B viruses are mainly toll-like receptor (TLR) 8-dependent. Cell. Microbiol. 2005, 7, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Bickerstaffe, A.; Beelen, A.; Lutter, R.; Nollet, F. Elevated plasma inflammatory mediators in post-polio syndrome: No association with long-term functional decline. J. Neuroimmunol. 2015, 289, 162–167. [Google Scholar] [CrossRef]
- Tatematsu, M.; Nishikawa, F.; Seya, T.; Matsumoto, M. Toll-like receptor 3 recognizes incomplete stem structures in single-stranded viral RNA. Nat. Commun. 2013, 4, 1833. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, T.; Krammer, F.; Iwasaki, A. The first 12 months of COVID-19: A timeline of immunological insights. Nat. Rev. Immunol. 2021, 21, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Vanderbeke, L.; Van Mol, P.; Van Herck, Y.; De Smet, F.; Humblet-Baron, S.; Martinod, K.; Antoranz, A.; Arijs, I.; Boeckx, B.; Bosisio, F.M.; et al. Monocyte-driven atypical cytokine storm and aberrant neutrophil activation as key mediators of COVID-19 disease severity. Nat. Commun. 2021, 12, 4117. [Google Scholar] [CrossRef] [PubMed]
- Peluso, M.J.; Lu, S.; Tang, A.F.; Durstenfeld, M.S.; Ho, H.; Goldberg, S.A.; Forman, C.A.; Munter, S.E.; Hoh, R.; Tai, V.; et al. Markers of immune activation and inflammation in individuals with post-acute sequelae of SARS-CoV-2 infection. medRxiv 2021. [Google Scholar] [CrossRef]
- Gaebler, C.; Wang, Z.; Lorenzi, J.C.C.; Muecksch, F.; Finkin, S.; Tokuyama, M.; Cho, A.; Jankovic, M.; Schaefer-Babajew, D.; Oliveira, T.Y.; et al. Evolution of antibody immunity to SARS-CoV-2. Nat. Cell Biol. 2021, 591, 639–644. [Google Scholar] [CrossRef]
- Ceulemans, L.J.; Khan, M.; Yoo, S.-J.; Zapiec, B.; Van Gerven, L.; Van Slambrouck, J.; Vanstapel, A.; Van Raemdonck, D.; Vos, R.; Wauters, E.; et al. Persistence of SARS-CoV-2 RNA in lung tissue after mild COVID-19. Lancet Respir. Med. 2021. [Google Scholar] [CrossRef]
- Ramlall, V.; Thangaraj, P.M.; Meydan, C.; Foox, J.; Butler, D.; Kim, J.; May, B.; De Freitas, J.K.; Glicksberg, B.S.; Mason, C.E.; et al. Immune complement and coagulation dysfunction in adverse outcomes of SARS-CoV-2 infection. Nat. Med. 2020, 26, 1609–1615. [Google Scholar] [CrossRef] [PubMed]
- Java, A.; Apicelli, A.J.; Liszewski, M.K.; Coler-Reilly, A.; Atkinson, J.P.; Kim, A.H.; Kulkarni, H.S. The complement system in COVID-19: Friend and foe? JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.-H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Béziat, V.; et al. Auto-antibodies against type I IFNs in patients with life-threatening COVID-19. Science 2020, 370, eabd4585. [Google Scholar] [CrossRef]
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.K.D.; Hodeib, S.; Korol, C.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370, eabd4570. [Google Scholar] [CrossRef] [PubMed]
- Bhadelia, N.; Belkina, A.C.; Olson, A.; Winters, T.; Urick, P.; Lin, N.; Rifkin, I.; Kataria, Y.; Yuen, R.R.; Sagar, M.; et al. Distinct autoimmune antibody signatures between hospitalized acute COVID-19 patients, SARS-CoV-2 convalescent individuals, and unexposed pre-pandemic controls. medRxiv 2021. [Google Scholar] [CrossRef]
- Zuo, Y.; Estes, S.K.; Ali, R.A.; Gandhi, A.A.; Yalavarthi, S.; Shi, H.; Sule, G.; Gockman, K.; Madison, J.A.; Zuo, M.; et al. Prothrombotic autoantibodies in serum from patients hospitalized with COVID-19. Sci. Transl. Med. 2020, 12, eabd3876. [Google Scholar] [CrossRef]
- Bennett, J.M.; Reeves, G.; Billman, G.E.; Sturmberg, J.P. Inflammation-nature’s way to efficiently respond to all types of challenges: Implications for understanding and managing “the epidemic” of chronic diseases. Front. Med. 2018, 5, 316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T.; Gilroy, D. Chronic inflammation: A failure of resolution? Int. J. Exp. Pathol. 2006, 88, 85–94. [Google Scholar] [CrossRef]
- Boasso, A.; Shearer, G.M. Chronic innate immune activation as a cause of HIV-1 immunopathogenesis. Clin. Immunol. 2008, 126, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, A. Innate immune recognition of HIV-1. Immunity 2012, 37, 389–398. [Google Scholar] [CrossRef] [Green Version]
- Siliciano, R.F.; Greene, W.C. HIV latency. Harb. Perspect. Med. 2011, 1, a007096. [Google Scholar] [CrossRef] [Green Version]
- Cary, D.C.; Fujinaga, K.; Peterlin, B.M. Molecular mechanisms of HIV latency. J. Clin. Investig. 2016, 126, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Meås, H.Z.; Haug, M.; Beckwith, M.S.; Louet, C.; Ryan, L.; Hu, Z.; Landskron, J.; Nordbø, S.A.; Tasken, K.; Yin, H.; et al. Sensing of HIV-1 by TLR8 activates human T cells and reverses latency. Nat. Commun. 2020, 11, 147. [Google Scholar] [CrossRef] [PubMed]
- Olson, A.; Coote, C.; Snyder-Cappione, J.; Lin, N.; Sagar, M. HIV-1 transcription but not intact provirus levels are associated with systemic inflammation. J. Infect. Dis. 2021, 223, 1934–1942. [Google Scholar] [CrossRef] [PubMed]
- Von Sydow, M.; Sönnerborg, A.; Gaines, H. Strannegård, Interferon-alpha and tumor necrosis factor-alpha in serum of patients in various stages of HIV-1 infection. AIDS Res. Hum. Retrovir. 1991, 7, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G.; Tracy, R.; Douek, D.C. Systemic effects of inflammation on health during chronic HIV infection. Immunity 2013, 39, 633–645. [Google Scholar] [CrossRef] [Green Version]
- Brenchley, J.M.; Price, D.; Schacker, T.W.; Asher, T.; Silvestri, G.; Rao, S.; Kazzaz, Z.; Bornstein, E.; Lambotte, O.; Altmann, D.; et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 2006, 12, 1365–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, C.Y.; Alden, S.L.; Funderburg, N.; Fu, P.; Levine, A.D. Progressive proximal-to-distal reduction in expression of the tight junction complex in colonic epithelium of virally-suppressed HIV+ individuals. PLoS Pathog. 2014, 10, e1004198. [Google Scholar] [CrossRef]
- Nazli, A.; Chan, O.; Dobson-Belaire, W.N.; Ouellet, M.; Tremblay, M.J.; Gray-Owen, S.D.; Arsenault, A.L.; Kaushic, C. Exposure to HIV-1 directly impairs mucosal epithelial barrier integrity allowing microbial translocation. PLoS Pathog. 2010, 6, e1000852. [Google Scholar] [CrossRef]
- Crakes, K.R.; Jiang, G. Gut microbiome alterations during HIV/SIV infection: Implications for HIV cure. Front. Microbiol. 2019, 10, 1104. [Google Scholar] [CrossRef]
- Libby, P.; Kobold, S. Inflammation: A common contributor to cancer, aging, and cardiovascular diseases, expanding the concept of cardio-oncology. Cardiovasc. Res. 2019, 115, 824–829. [Google Scholar] [CrossRef] [PubMed]
- Zapata, H.J.; Shaw, A.C. Aging of the human innate immune system in HIV infection. Curr. Opin. Immunol. 2014, 29, 127–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedaghat, A.R.; German, J.; Teslovich, T.M.; Cofrancesco, J.; Jie, C.C.; Talbot, C.C.; Siliciano, R.F. Chronic CD4+ T-cell activation and depletion in human immunodeficiency virus type 1 infection: Type I interferon-mediated disruption of T-cell dynamics. J. Virol. 2008, 82, 1870–1883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rempel, H.; Sun, B.; Calosing, C.; Pillai, S.K.; Pulliam, L. Interferon-α drives monocyte gene expression in chronic unsuppressed HIV-1 infection. AIDS 2010, 24, 1415–1423. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, S.; Tanaskovic, S.; Helbig, K.; Rajasuriar, R.; Kramski, M.; Murray, J.; Beard, M.; Purcell, A.; Lewin, S.R.; Price, P.; et al. CD4+ T-cell deficiency in HIV patients responding to antiretroviral therapy is associated with increased expression of interferon-stimulated genes in CD4+ T cells. J. Infect. Dis. 2011, 204, 1927–1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Megahed, F.A.K.; Zhou, X.; Sun, P. The interactions between HBV and the innate immunity of hepatocytes. Viruses 2020, 12, 285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Meng, Z.; Jiang, M.; Pei, R.; Trippler, M.; Broering, R.; Bucchi, A.; Sowa, J.-P.; Dittmer, U.; Yang, D.; et al. Hepatitis B virus suppresses toll-like receptor-mediated innate immune responses in murine parenchymal and nonparenchymal liver cells. Hepatology 2008, 49, 1132–1140. [Google Scholar] [CrossRef] [PubMed]
- Hösel, M.; Quasdorff, M.; Wiegmann, K.; Webb, D.; Zedler, U.; Broxtermann, M.; Tedjokusumo, R.; Esser, K.; Arzberger, S.; Kirschning, C.J.; et al. Not interferon, but interleukin-6 controls early gene expression in hepatitis B virus infection. Hepatology 2009, 50, 1773–1782. [Google Scholar] [CrossRef]
- Wieland, S.; Thimme, R.; Purcell, R.H.; Chisari, F.V. Genomic analysis of the host response to hepatitis B virus infection. Proc. Natl. Acad. Sci. USA 2004, 101, 6669–6674. [Google Scholar] [CrossRef] [Green Version]
- Rehermann, B.; Nascimbeni, M. Immunology of hepatitis B virus and hepatitis C virus infection. Nat. Rev. Immunol. 2005, 5, 215–229. [Google Scholar] [CrossRef]
- Venook, A.P.; Papandreou, C.; Furuse, J.; De Guevara, L.L. The incidence and epidemiology of hepatocellular carcinoma: A global and regional perspective. Oncologist 2010, 15, 5–13. [Google Scholar] [CrossRef]
- Bertoletti, A.; Kennedy, P.T. The immune tolerant phase of chronic HBV infection: New perspectives on an old concept. Cell. Mol. Immunol. 2014, 12, 258–263. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Gong, R.; Mu, Y.; Chen, Y.; Zhu, C.; Sun, Z.; Chen, M.; Liu, Y.; Zhu, Y.; Wu, J. Hepatitis B virus induces a novel inflammation network involving three inflammatory factors, IL-29, IL-8, and cyclooxygenase-2. J. Immunol. 2011, 187, 4844–4860. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Tian, Z. HBV-induced immune imbalance in the development of HCC. Front. Immunol. 2019, 10, 2048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balmasova, I.P.; Yushchuk, N.D.; Mynbaev, O.; Alla, N.R.; Malova, E.; Shi, Z.; Gao, C.-L. Immunopathogenesis of chronic hepatitis B. World J. Gastroenterol. 2014, 20, 14156–14171. [Google Scholar] [CrossRef]
- Bertoletti, A. Kinetics of the immune response during HBV and HCV infection. Hepatology 2003, 38, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Wieland, S.F.; Chisari, F.V. Stealth and cunning: Hepatitis B and Hepatitis C viruses. J. Virol. 2005, 79, 9369–9380. [Google Scholar] [CrossRef] [Green Version]
- Vanlandschoot, P.; Van Houtte, F.; Roobrouck, A.; Farhoudi, A.; Stelter, F.; Peterson, D.L.; Gómez-Gutiérrez, J.; Gavilanes, F.; Leroux-Roels, G. LPS-binding protein and CD14-dependent attachment of hepatitis B surface antigen to monocytes is determined by the phospholipid moiety of the particles. J. Gen. Virol. 2002, 83, 2279–2289. [Google Scholar] [CrossRef]
- Song, H.; Tan, G.; Yang, Y.; Cui, A.; Li, H.; Li, T.; Wu, Z.; Yang, M.; Lv, G.; Chi, X.; et al. Hepatitis B virus-induced imbalance of inflammatory and antiviral signaling by differential phosphorylation of STAT1 in human monocytes. J. Immunol. 2019, 202, 2266–2275. [Google Scholar] [CrossRef] [Green Version]
- Lebossé, F.; Testoni, B.; Fresquet, J.; Facchetti, F.; Galmozzi, E.; Fournier, M.; Hervieu, V.; Berthillon, P.; Berby, F.; Bordes, I.; et al. Intrahepatic innate immune response pathways are downregulated in untreated chronic hepatitis B. J. Hepatol. 2016, 66, 897–909. [Google Scholar] [CrossRef]
- Marcocci, M.E.; Napoletani, G.; Protto, V.; Kolesova, O.; Piacentini, R.; Puma, D.D.L.; Lomonte, P.; Grassi, C.; Palamara, A.T.; De Chiara, G. Herpes simplex virus-1 in the brain: The dark side of a sneaky infection. Trends Microbiol. 2020, 28, 808–820. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.C.; Goldstein, D.; Montgomery, R. Age-dependent dysregulation of innate immunity. Nat. Rev. Immunol. 2013, 13, 875–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kayed, R. Infectious etiology and amyloidosis in Alzheimer’s Disease: The puzzle continues. J. Biol. Chem. 2021, 297, 100936. [Google Scholar] [CrossRef]
- Itzhaki, R. Overwhelming evidence for a major role for herpes simplex virus type 1 (HSV1) in Alzheimer’s Disease (AD), underwhelming evidence against. Vaccines 2021, 9, 679. [Google Scholar] [CrossRef]
- Duarte, L.F.; Farías, M.A.; Álvarez, D.M.; Bueno, S.M.; Riedel, C.; González, P.A. Herpes simplex virus type 1 infection of the central nervous system: Insights into proposed interrelationships with neurodegenerative disorders. Front. Cell. Neurosci. 2019, 13, 46. [Google Scholar] [CrossRef] [Green Version]
- Hait, A.S.; Olagnier, D.; Sancho-Shimizu, V.; Skipper, K.A.; Helleberg, M.; Larsen, S.M.; Bodda, C.; Moldovan, L.I.; Ren, F.; Andersen, N.-S.B.; et al. Defects in LC3B2 and ATG4A underlie HSV2 meningitis and reveal a critical role for autophagy in antiviral defense in humans. Sci. Immunol. 2020, 5, eabc2691. [Google Scholar] [CrossRef] [PubMed]
- Wright, W.F.; Palisoc, K.; Baghli, S. Mollaret meningitis. J. Neurol. Sci. 2019, 396, 148–149. [Google Scholar] [CrossRef] [PubMed]
- Monk, B.J.; Tewari, K.S. The spectrum and clinical sequelae of human papillomavirus infection. Gynecol. Oncol. 2007, 107, S6–S13. [Google Scholar] [CrossRef] [Green Version]
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [Green Version]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-C.; Yeh, W.-C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M. Inflammasomes in the gastrointestinal tract: Infection, cancer and gut microbiota homeostasis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 721–737. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.R.; Blumenschein, W.; Murphy, E.; Diveu, C.; Wiekowski, M.; Abbondanzo, S.; Lucian, L.; Geissler, R.; Brodie, S.; Kimball, A.; et al. IL-23 stimulates epidermal hyperplasia via TNF and IL-20R2–dependent mechanisms with implications for psoriasis pathogenesis. J. Exp. Med. 2006, 203, 2577–2587. [Google Scholar] [CrossRef]
- Herold, S.; Becker, C.; Ridge, K.M.; Budinger, G.S. Influenza virus-induced lung injury: Pathogenesis and implications for treatment. Eur. Respir. J. 2015, 45, 1463–1478. [Google Scholar] [CrossRef] [Green Version]
- Coates, B.M.; Staricha, K.L.; Koch, C.M.; Cheng, Y.; Shumaker, D.K.; Budinger, G.R.S.; Perlman, H.; Misharin, A.V.; Ridge, K.M. Inflammatory monocytes drive influenza a virus-mediated lung injury in juvenile mice. J. Immunol. 2018, 200, 2391–2404. [Google Scholar] [CrossRef] [Green Version]
- Enaud, R.; Prevel, R.; Ciarlo, E.; Beaufils, F.; Wieërs, G.; Guery, B.; Delhaes, L. The gut-lung axis in health and respiratory diseases: A place for inter-organ and inter-kingdom crosstalks. Front. Cell. Infect. Microbiol. 2020, 10, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, J.-K.; Kim, T.S.; Hufford, M.M.; Braciale, T.J. Viral infection of the lung: Host response and sequelae. J. Allergy Clin. Immunol. 2013, 132, 1263–1276. [Google Scholar] [CrossRef] [PubMed]
- Sudre, C.H.; Murray, B.; Varsavsky, T.; Graham, M.S.; Penfold, R.S.; Bowyer, R.C.; Pujol, J.C.; Klaser, K.; Antonelli, M.; Canas, L.S.; et al. Attributes and predictors of long COVID. Nat. Med. 2021, 27, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Stojanović, S.D.; Fiedler, J.; Bauersachs, J.; Thum, T.; Sedding, D.G. Senescence-induced inflammation: An important player and key therapeutic target in atherosclerosis. Eur. Hear. J. 2020, 41, 2983–2996. [Google Scholar] [CrossRef] [Green Version]
- Camell, C.D.; Yousefzadeh, M.J.; Zhu, Y.; Prata, L.G.P.L.; Huggins, M.A.; Pierson, M.; Zhang, L.; O’Kelly, R.D.; Pirtskhalava, T.; Xun, P.; et al. Senolytics reduce coronavirus-related mortality in old mice. Science 2021, 373, eabe4832. [Google Scholar] [CrossRef]
- Karin, O.; Alon, U. Senescent cell accumulation mechanisms inferred from parabiosis. GeroScience 2020, 43, 329–341. [Google Scholar] [CrossRef]
- Seoane, R.; Vidal, S.; Bouzaher, Y.H.; Motiam, A.; Rivas, C. The interaction of viruses with the cellular senescence response. Biology 2020, 9, 455. [Google Scholar] [CrossRef] [PubMed]
- Solana, R.; Pawelec, G.; Tarazona, R. Aging and innate immunity. Immunity 2006, 24, 491–494. [Google Scholar] [CrossRef] [Green Version]
- Licastro, F.; Candore, G.; Lio, D.; Porcellini, E.; Colonna-Romano, G.; Franceschi, C.; Caruso, C. Innate immunity and inflammation in ageing: A key for understanding age-related diseases. Immun. Ageing 2005, 2, 8. [Google Scholar] [CrossRef] [Green Version]
- Feldman, C.; Anderson, R. The role of co-infections and secondary infections in patients with COVID-19. Pneumonia 2021, 13, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Virgin, H.W. The virome in mammalian physiology and disease. Cell 2014, 157, 142–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virgin, H.W.; Wherry, E.J.; Ahmed, R. Redefining chronic viral infection. Cell 2009, 138, 30–50. [Google Scholar] [CrossRef] [Green Version]
- Sherwani, S.; Khan, M.A.; SulimanAlmogbel, M. Autoantibodies in Viral Infections; IntechOpen: London, UK, 2018. [Google Scholar]
- Cusick, M.F.; Libbey, J.E.; Fujinami, R.S. Molecular mimicry as a mechanism of autoimmune disease. Clin. Rev. Allergy Immunol. 2011, 42, 102–111. [Google Scholar] [CrossRef]
- Sousa, C.; Germain, R.N. Analysis of adjuvant function by direct visualization of antigen presentation in vivo: Endotoxin promotes accumulation of antigen-bearing dendritic cells in the T cell areas of lymphoid tissue. J. Immunol. 1999, 162, 6552–6561. [Google Scholar]
- Cella, M.; Engering, A.; Pinet, V.; Pieters, J.; Lanzavecchia, A. Inflammatory stimuli induce accumulation of MHC class II complexes on dendritic cells. Nat. Cell Biol. 1997, 388, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Garza, K.M.; Chan, S.M.; Suri, R.; Nguyen, L.T.; Odermatt, B.; Schoenberger, S.P.; Ohashi, P.S. Role of antigen-presenting cells in mediating tolerance and autoimmunity. J. Exp. Med. 1999, 191, 2021–2028. [Google Scholar] [CrossRef] [PubMed]
- Vella, A.T.; McCormack, J.E.; Linsley, P.S.; Kappler, J.W.; Marrack, P. Lipopolysaccharide interferes with the induction of peripheral T cell death. Immunity 1995, 2, 261–270. [Google Scholar] [CrossRef] [Green Version]
- Ehl, S.; Hombach, J.; Aichele, P.; Rülicke, T.; Odermatt, B.; Hengartner, H.; Zinkernagel, R.; Pircher, H. Viral and bacterial infections interfere with peripheral tolerance induction and activate CD8+ T cells to cause immunopathology. J. Exp. Med. 1998, 187, 763–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, R.J.; Vanhoorelbeke, K.; Leypoldt, F.; Kaya, Z.; Bieber, K.; McLachlan, S.M.; Komorowski, L.; Luo, J.; Marques, O.C.; Hammers, C.; et al. Mechanisms of autoantibody-induced pathology. Front. Immunol. 2017, 8, 603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkon, K.; Casali, P. Nature and functions of autoantibodies. Nat. Clin. Pr. Rheumatol. 2008, 4, 491–498. [Google Scholar] [CrossRef] [Green Version]
- Carroll, K.C.; Hobden, J.A.; Miller, S.; Morse, S.A.; Mietzner, T.A.; Detrick, B.; Mitchell, T.G.; McKerrow, J.H.; Sakanari, J.A. Rabies, slow virus infections, and prion diseases. In Jawetz, Melnick and Adelberg’s Medical Microbiology; McGraw-Hill Education: New York, NY, USA, 2019. [Google Scholar]
- Perelygina, L.; Plotkin, S.; Russo, P.; Hautala, T.; Bonilla, F.; Ochs, H.D.; Joshi, A.; Routes, J.; Patel, K.; Wehr, C.; et al. Rubella persistence in epidermal keratinocytes and granuloma M2 macrophages in patients with primary immunodeficiencies. J. Allergy Clin. Immunol. 2016, 138, 1436.e11–1439.e11. [Google Scholar] [CrossRef] [Green Version]
- Thorson, A.E.; Deen, G.F.; Bernstein, K.T.; Liu, W.J.; Yamba, F.; Habib, N.; Sesay, F.R.; Gaillard, P.; Massaquoi, T.A.; McDonald, S.L.R.; et al. Persistence of Ebola virus in semen among Ebola virus disease survivors in Sierra Leone: A cohort study of frequency, duration, and risk factors. PLoS Med. 2021, 18, e1003273. [Google Scholar] [CrossRef] [PubMed]
- Whitmer, S.L.; Ladner, J.T.; Wiley, M.; Patel, K.; Dudas, G.; Rambaut, A.; Sahr, F.; Prieto, K.; Shepard, S.S.; Carmody, E.; et al. Active Ebola virus replication and heterogeneous evolutionary rates in EVD survivors. Cell Rep. 2018, 22, 1159–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shantha, J.G.; Crozier, I.; Yeh, S. An update on ocular complications of Ebola virus disease. Curr. Opin. Ophthalmol. 2017, 28, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Heeney, J.L. Hidden reservoirs. Nature 2015, 527, 453–455. [Google Scholar] [CrossRef]
- Norrby, E.; Kristensson, K. Measles virus in the brain. Brain Res. Bull. 1997, 44, 213–220. [Google Scholar] [CrossRef]
- Takahashi, T.; Iwasaki, A. Sex differences in immune responses. Science 2021, 371, 347–348. [Google Scholar] [CrossRef] [PubMed]
- Peckham, H.; de Gruijter, N.M.; Raine, C.; Radziszewska, A.; Ciurtin, C.; Wedderburn, L.R.; Rosser, E.C.; Webb, K.; Deakin, C.T. Male sex identified by global COVID-19 meta-analysis as a risk factor for death and ITU admission. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- Takahashi, T.; Yale IMPACT Research Team; Ellingson, M.K.; Wong, P.; Israelow, B.; Lucas, C.; Klein, J.; Silva, J.; Mao, T.; Oh, J.E.; et al. Sex differences in immune responses that underlie COVID-19 disease outcomes. Nat. Cell Biol. 2020, 588, 315–320. [Google Scholar] [CrossRef]
- Mahmud, R.; Rahman, M.; Rassel, M.A.; Monayem, F.B.; Sayeed, S.K.J.B.; Islam, S.; Islam, M.M. Post-COVID-19 syndrome among symptomatic COVID-19 patients: A prospective cohort study in a tertiary care center of Bangladesh. PLoS ONE 2021, 16, e0249644. [Google Scholar] [CrossRef]
- Wang, E.Y.; Team, Y.I.; Mao, T.; Klein, J.; Dai, Y.; Huck, J.D.; Jaycox, J.R.; Liu, F.; Zhou, T.; Israelow, B.; et al. Diverse functional autoantibodies in patients with COVID-19. Nat. Cell Biol. 2021, 1–6. [Google Scholar] [CrossRef]
- Wallukat, G.; Hohberger, B.; Wenzel, K.; Fürst, J.; Schulze-Rothe, S.; Wallukat, A.; Hönicke, A.-S.; Müller, J. Functional autoantibodies against G-protein coupled receptors in patients with persistent Long-COVID-19 symptoms. J. Transl. Autoimmun. 2021, 4, 100100. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, R.K.; Kashour, T.; Hamid, Q.; Halwani, R.; Tleyjeh, I.M. Unraveling the mystery surrounding post-acute sequelae of COVID-19. Front. Immunol. 2021, 12, 686029. [Google Scholar] [CrossRef] [PubMed]
- Sette, A.; Crotty, S. Adaptive immunity to SARS-CoV-2 and COVID-19. Cell 2021, 184, 861–880. [Google Scholar] [CrossRef] [PubMed]
- Wold, W.S.M.; Toth, K. Adenovirus vectors for gene therapy, vaccination and cancer gene therapy. Curr. Gene Ther. 2013, 13, 421–433. [Google Scholar] [CrossRef]
- Munis, A.M.; Bentley, E.M.; Takeuchi, Y. A tool with many applications: Vesicular stomatitis virus in research and medicine. Expert Opin. Biol. Ther. 2020, 20, 1187–1201. [Google Scholar] [CrossRef]
- Marx, V. Scientists set out to connect the dots on long COVID. Nat. Methods 2021, 18, 449–453. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Virus | Genome | Family | PAS | References |
|---|---|---|---|---|
| PVB19 | linear ssDNA | Parvoviridae | myalgic encephalomyelitis, chronic fatigue syndrome, myocarditis | [24,25,26,27] |
| AdV | linear dsDNA | Adenoviridae | ocular irritations, respiratory complications, myocarditis | [26,27,28] |
| Rotavirus A | dsRNA | Reoviridae | Celiac disease, diabetes mellitus | [29,30,31,32] |
| MeV | (−) ssRNA | Paramyxoviridae | immunosuppression, immune dysregulation/chronic inflammation, SSPE | [33,34,35,36,37,38,39] |
| NiV | (−) ssRNA | Paramyxoviridae | Neurological sequelae, relapsed encephalitis | [40,41,42] |
| EBOV | (−) ssRNA | Filoviridae | Fatigue, musculoskeletal pain, ocular and auditory disorders, neurological problems, renal failure | [43,44,45,46,47,48,49] |
| MARV | (−) ssRNA | Filoviridae | myalgia, arthritis, conjunctivitis, psychosis | [50,51] |
| LASV | (−) ssRNA | Arenaviridae | Eye inflammation, hearing loss, ataxia | [52,53,54] |
| IAV | (−) ssRNA | Orthomyxoviridae | Asthma, reduced lung function, pneumonia | [55,56,57] |
| RSV | (−) ssRNA | Paramyxoviridae | Asthma, reduced lung function | [55,56,58,59] |
| ZIKV | (+) ssRNA | Flaviviridae | Encephalitis, myelitis, GBS | [60,61,62,63,64,65] |
| DENV | (+) ssRNA | Flaviviridae | Fatigue, musculoskeletal pain, memory loss, GBS | [63,65,66,67,68] |
| WNV | (+) ssRNA | Flaviviridae | Fatigue, myalgia, memory loss, motor problems, neurological problems | [69,70,71,72] |
| TBEV | (+) ssRNA | Flaviviridae | neurological complications, cognitive impairment, tremor, aphasia, sleep disorders, vertigo | [73] |
| JEV | (+) ssRNA | Flaviviridae | intellectual disabilities, neurological sequelae, motor problems, convulsions | [74] |
| CHIKV | (+) ssRNA | Togaviridae | arthralgia, arthritis, neurological disorders | [64,65,75,76] |
| VEEV | (+) ssRNA | Togaviridae | psychological changes and intellectual disabilities | [77] |
| EEEV | (+) ssRNA | Togaviridae | psychological changes and intellectual disabilities | [77] |
| WEEV | (+) ssRNA | Togaviridae | psychological changes and intellectual disabilities | [77] |
| RRV | (+) ssRNA | Togaviridae | arthralgia, fatigue, arthritis, joint problems | [78] |
| SINV | (+) ssRNA | Togaviridae | joint problems, arthritis, rheumatological symptoms, arthralgia | [79] |
| EV A 71 | (+) ssRNA | Picornaviridae | ventilatory problems, neurodevelopmental delay, cerebellar dysfunction | [26,27,80,81,82,83] |
| CV | (+) ssRNA | Picornaviridae | ventilatory problems, neurodevelopmental delay, cerebellar dysfunction, myocarditis | [26,27,81] |
| PV | (+) ssRNA | Picornaviridae | decreasing muscular function, acute weakness, pain, fatigue | [84,85,86,87,88] |
| SARS-CoV-2 | (+) ssRNA | Coronaviridae | PASC: fatigue, abnormal thermoregulation, skin diseases, intestinal symptoms, diabetes, reduced respiratory capacity MIS-C: persistent fever, hyperinflammation, gastrointestinal symptoms, muscle pain | [22,23,89,90,91,92,93,94,95,96] |
| SARS-CoV | (+) ssRNA | Coronaviridae | fatigue, reduced lung capacity and ventilation, myalgia, mental health problems | [97,98] |
| MERS-CoV | (+) ssRNA | Coronaviridae | Chronic fatigue, mental health problems, reduced lung function | [99] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hirschenberger, M.; Hunszinger, V.; Sparrer, K.M.J. Implications of Innate Immunity in Post-Acute Sequelae of Non-Persistent Viral Infections. Cells 2021, 10, 2134. https://doi.org/10.3390/cells10082134
Hirschenberger M, Hunszinger V, Sparrer KMJ. Implications of Innate Immunity in Post-Acute Sequelae of Non-Persistent Viral Infections. Cells. 2021; 10(8):2134. https://doi.org/10.3390/cells10082134
Chicago/Turabian StyleHirschenberger, Maximilian, Victoria Hunszinger, and Konstantin Maria Johannes Sparrer. 2021. "Implications of Innate Immunity in Post-Acute Sequelae of Non-Persistent Viral Infections" Cells 10, no. 8: 2134. https://doi.org/10.3390/cells10082134
APA StyleHirschenberger, M., Hunszinger, V., & Sparrer, K. M. J. (2021). Implications of Innate Immunity in Post-Acute Sequelae of Non-Persistent Viral Infections. Cells, 10(8), 2134. https://doi.org/10.3390/cells10082134