
Selective Autophagy in Hyperglycemia-Induced Microvascular and Macrovascular Diseases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
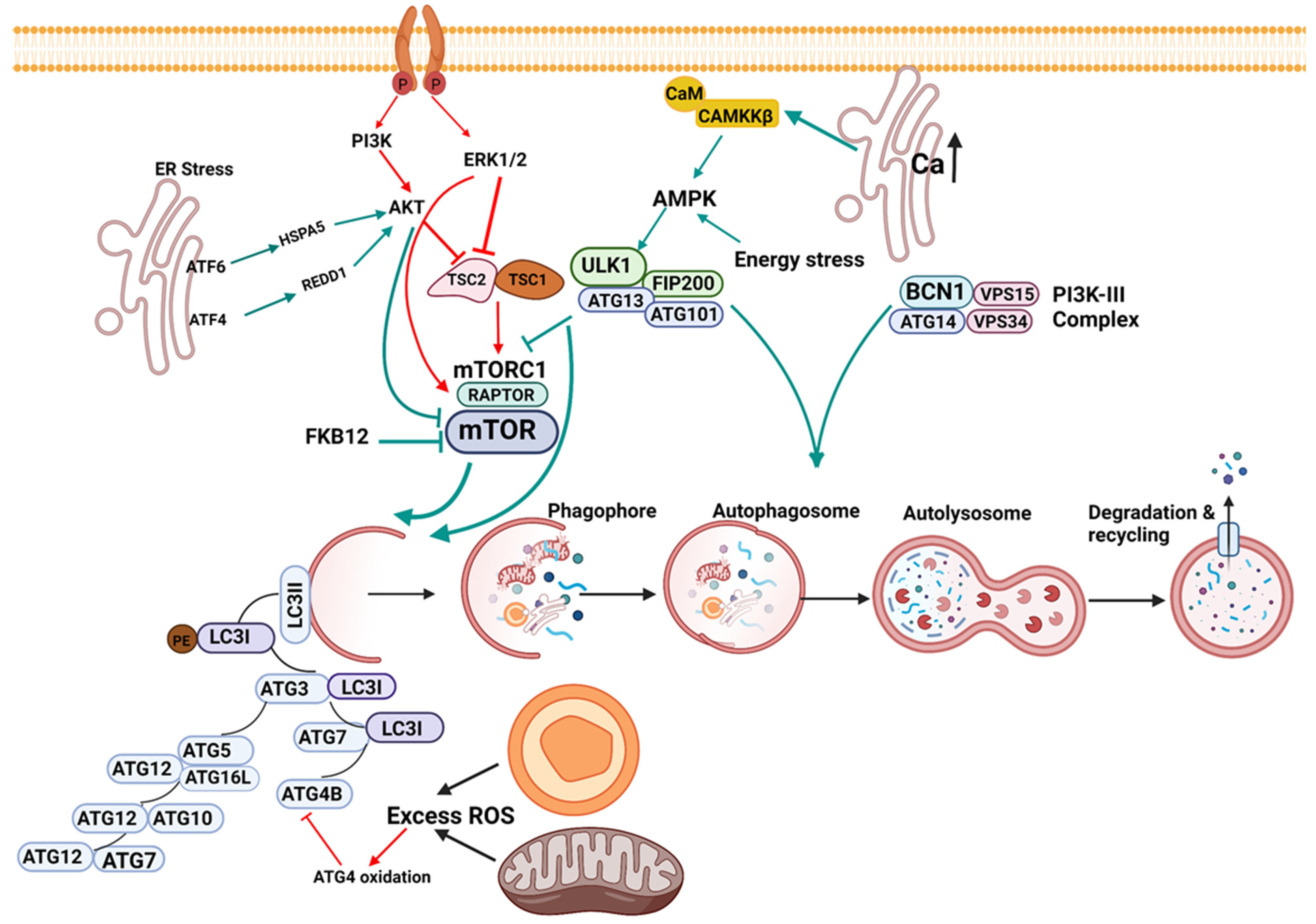
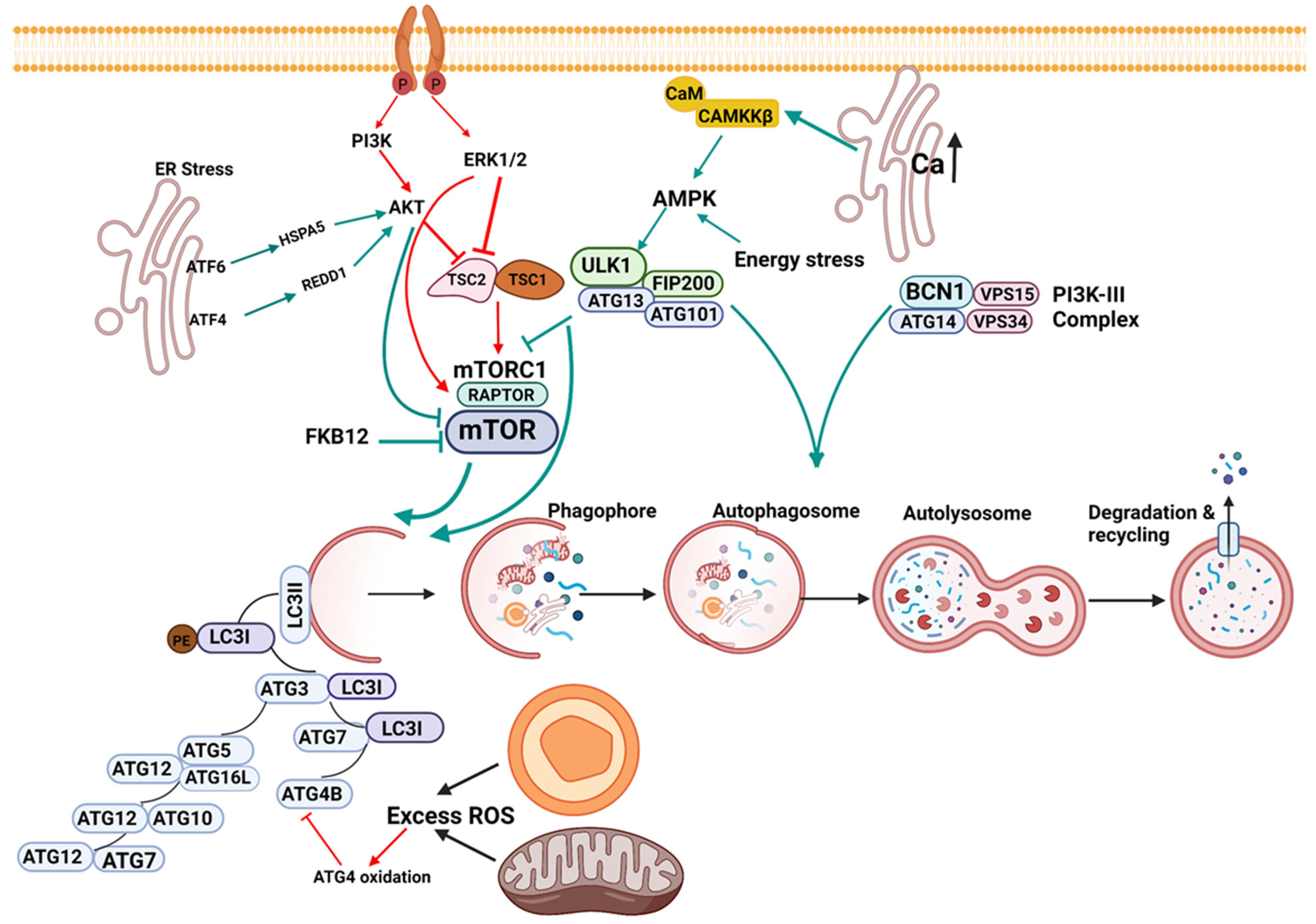
1.1. Initiation of Autophagy
1.2. Nucleation
1.3. Elongation
1.4. Lysosomal Fusion
1.5. Signaling in Macroautophagy
1.6. Selective Autophagy of Organelles
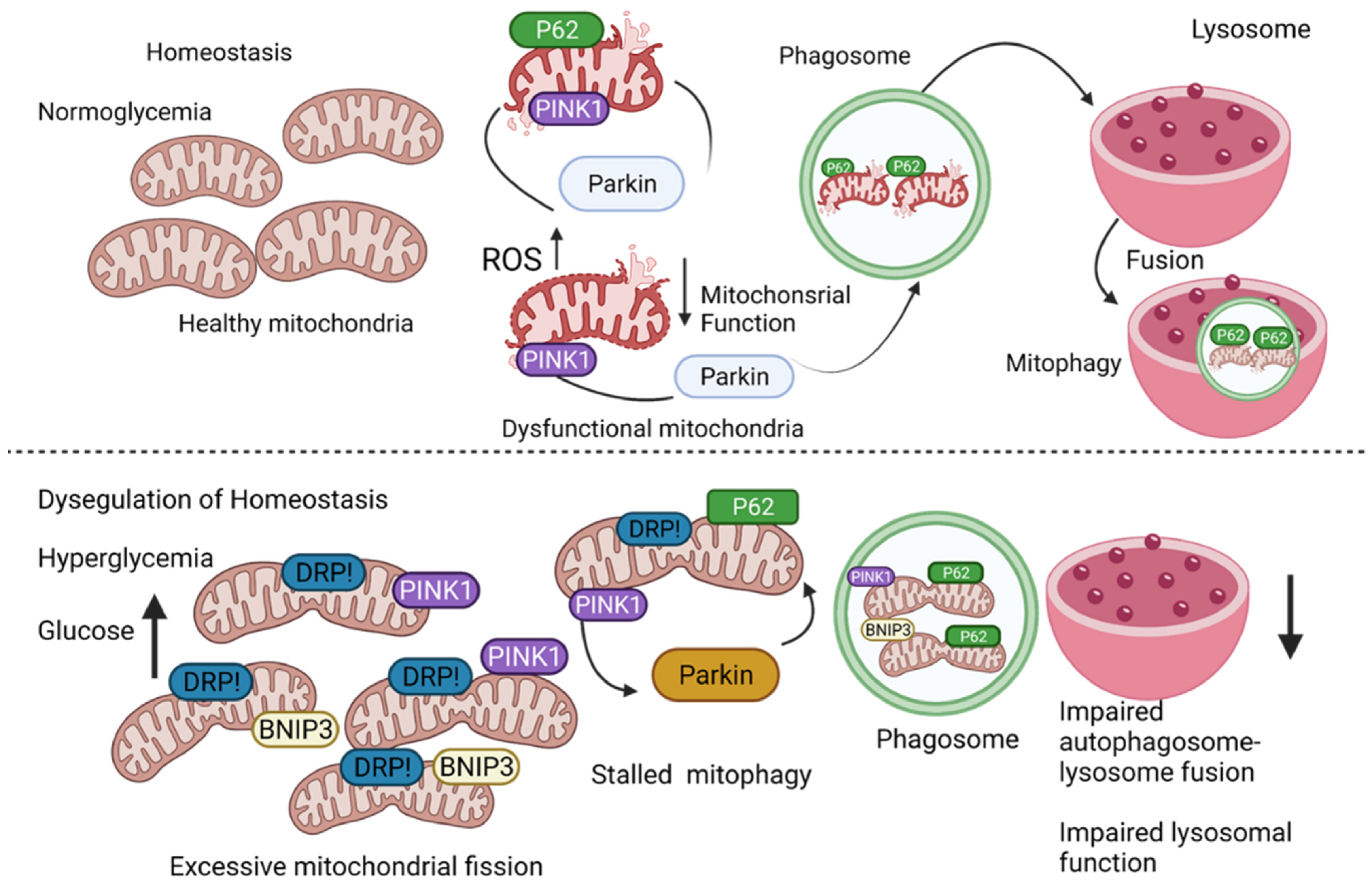
2. Mitophagy
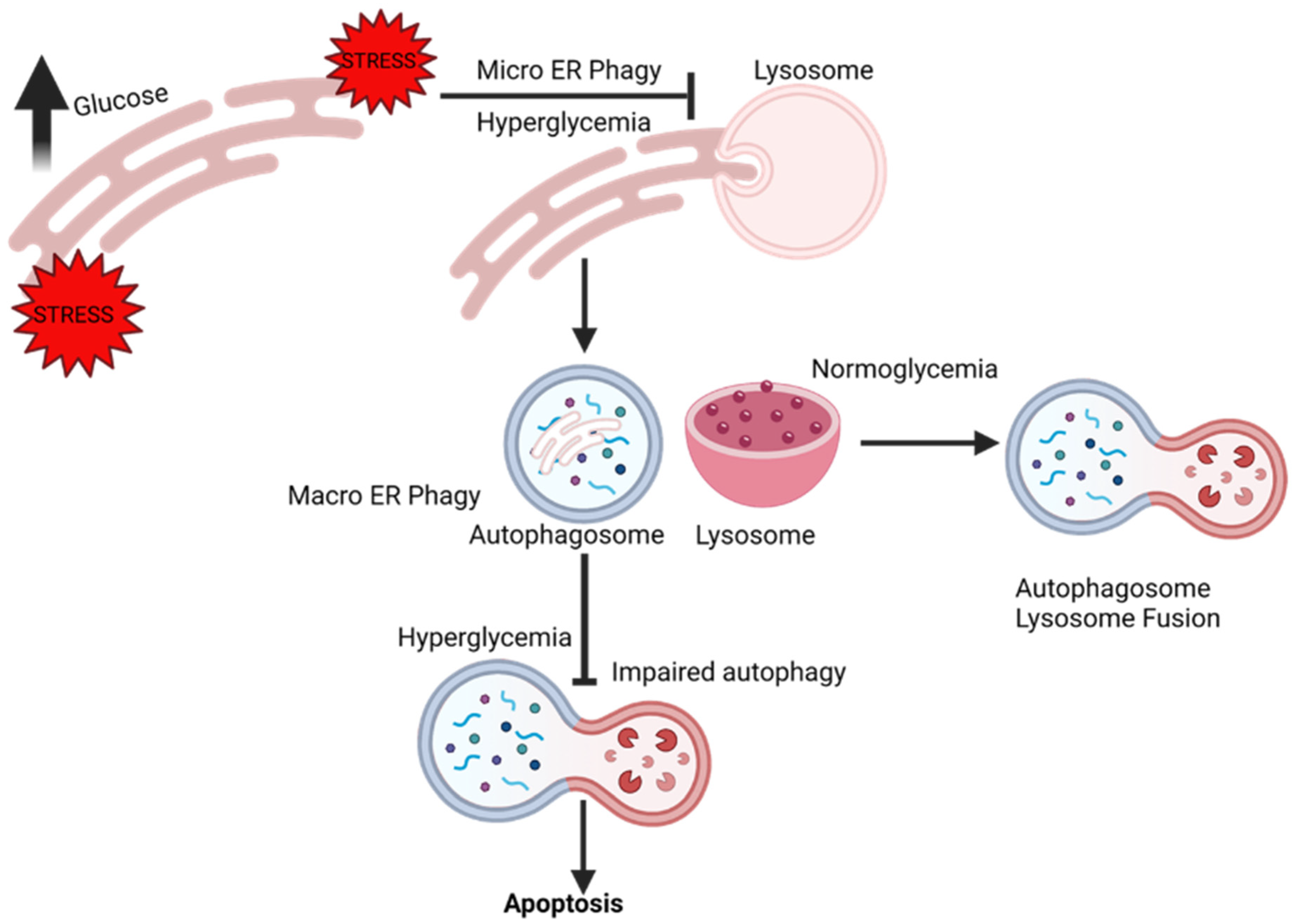
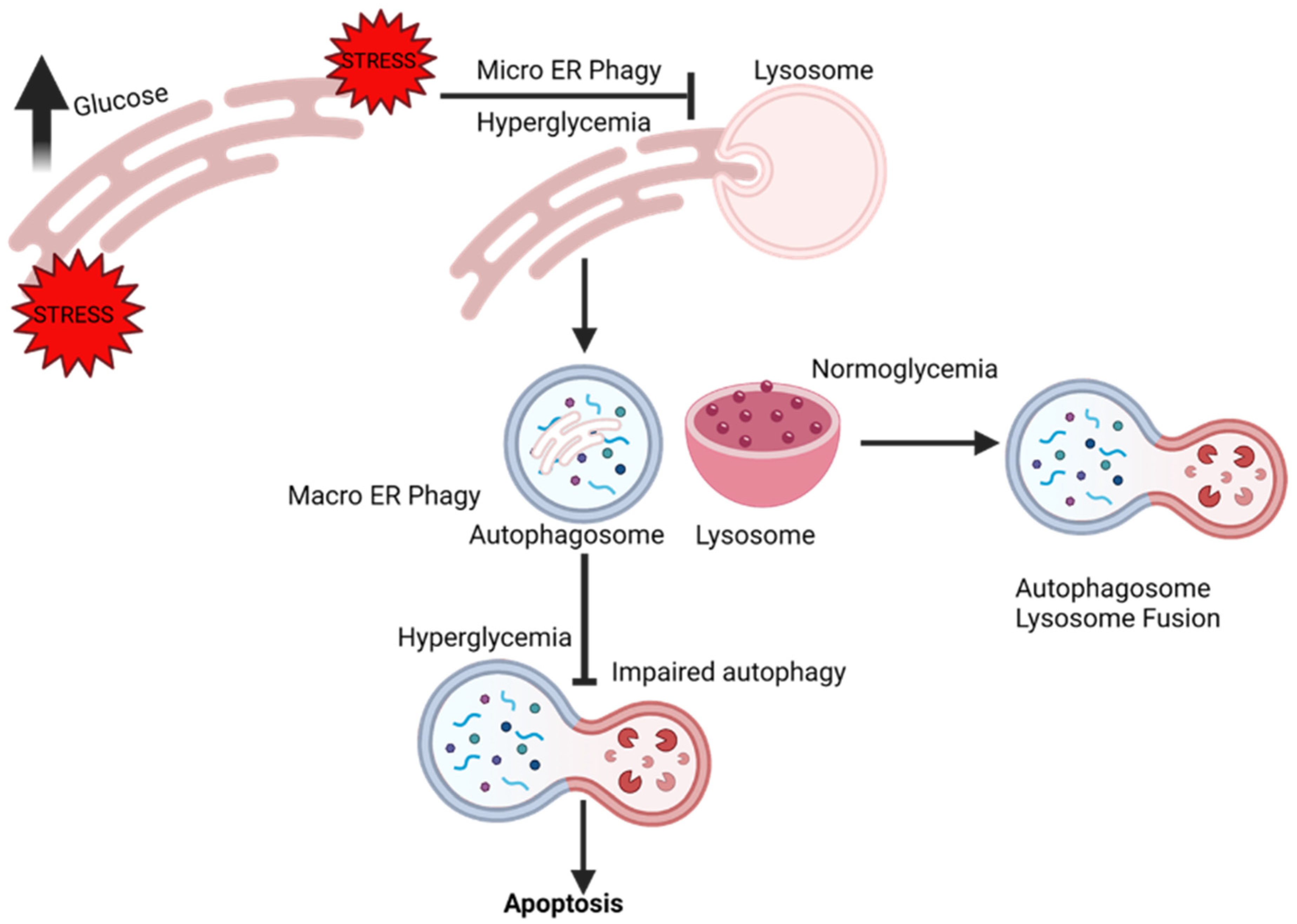
3. ER-Phagy
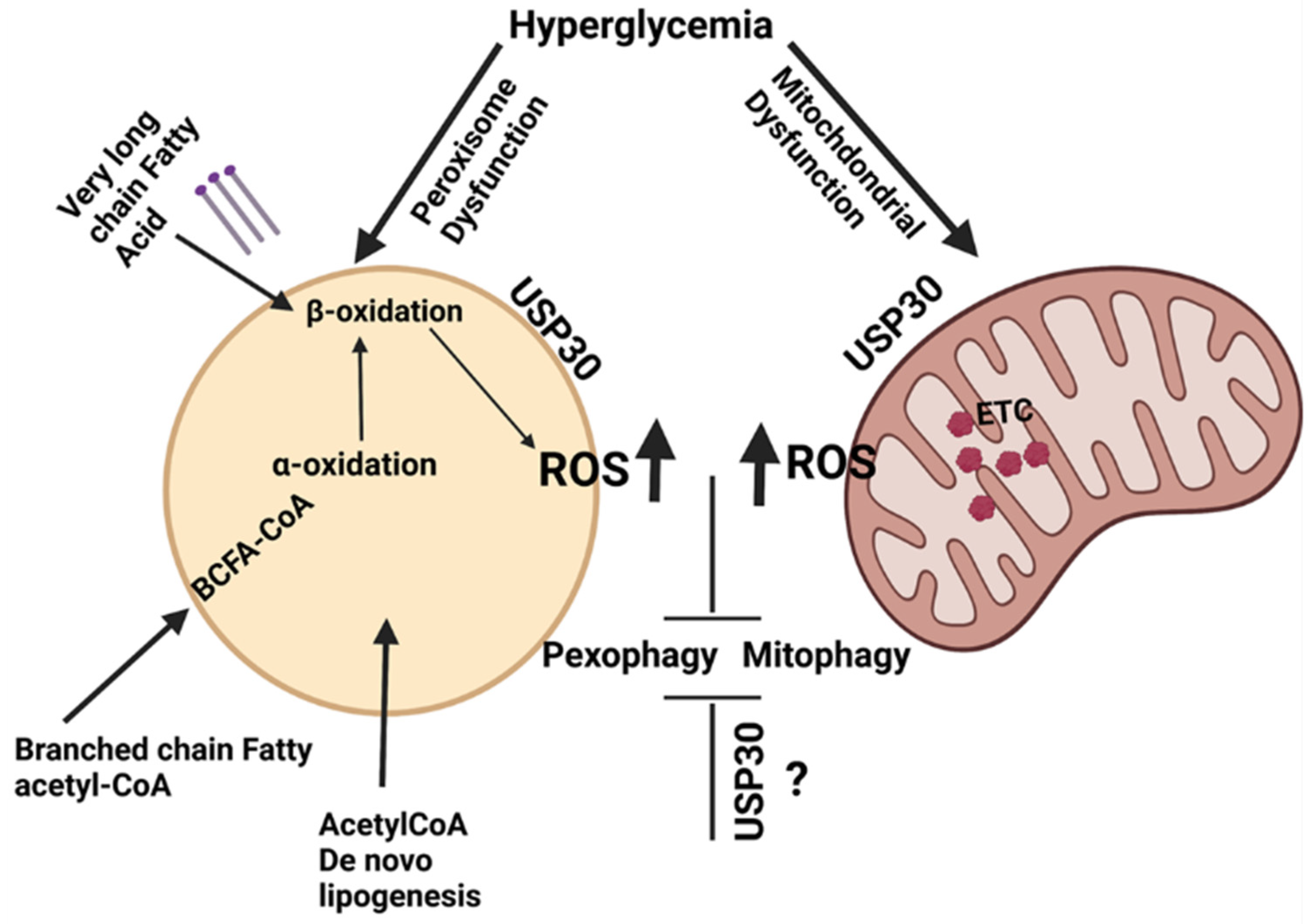
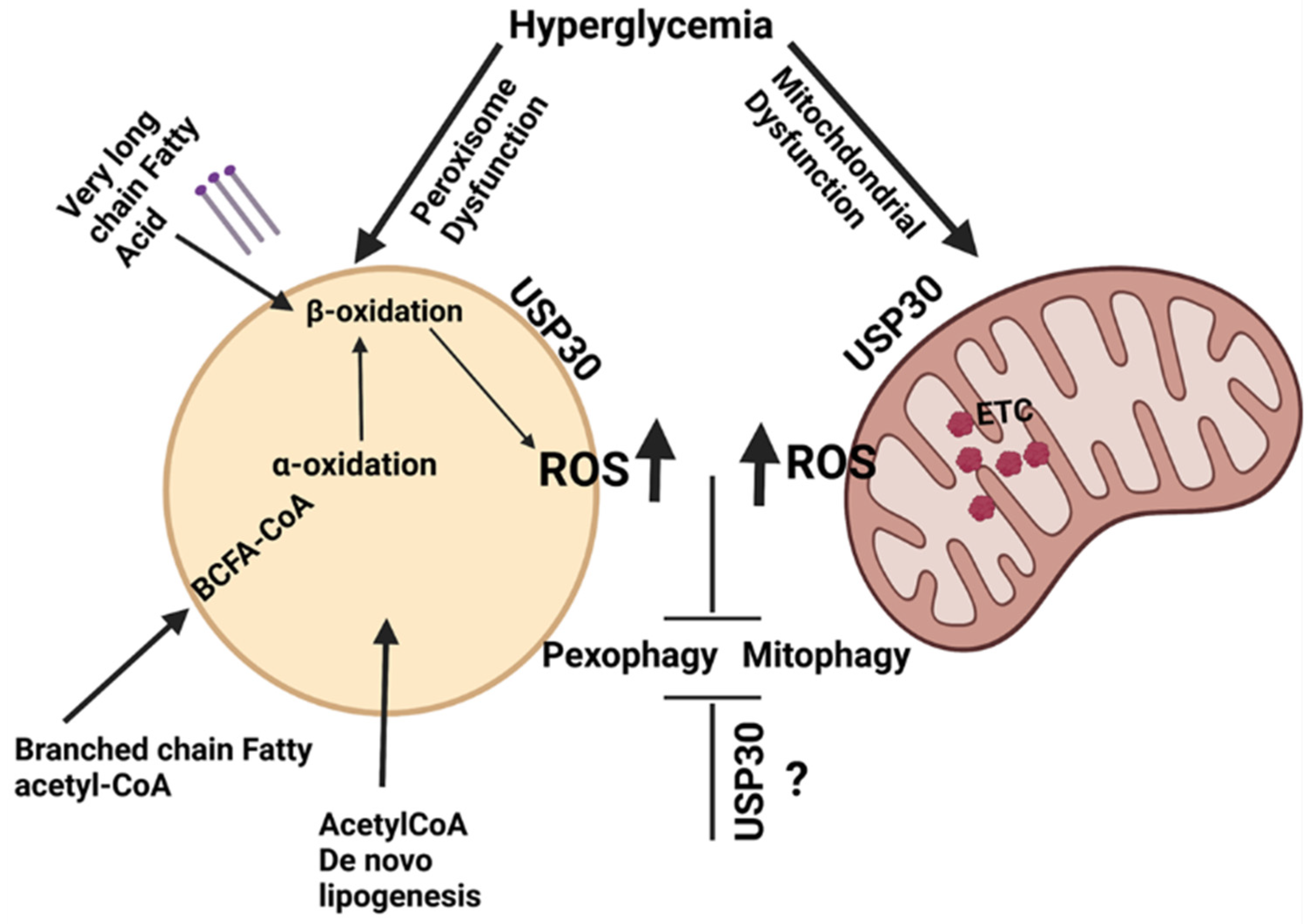
4. Pexophagy
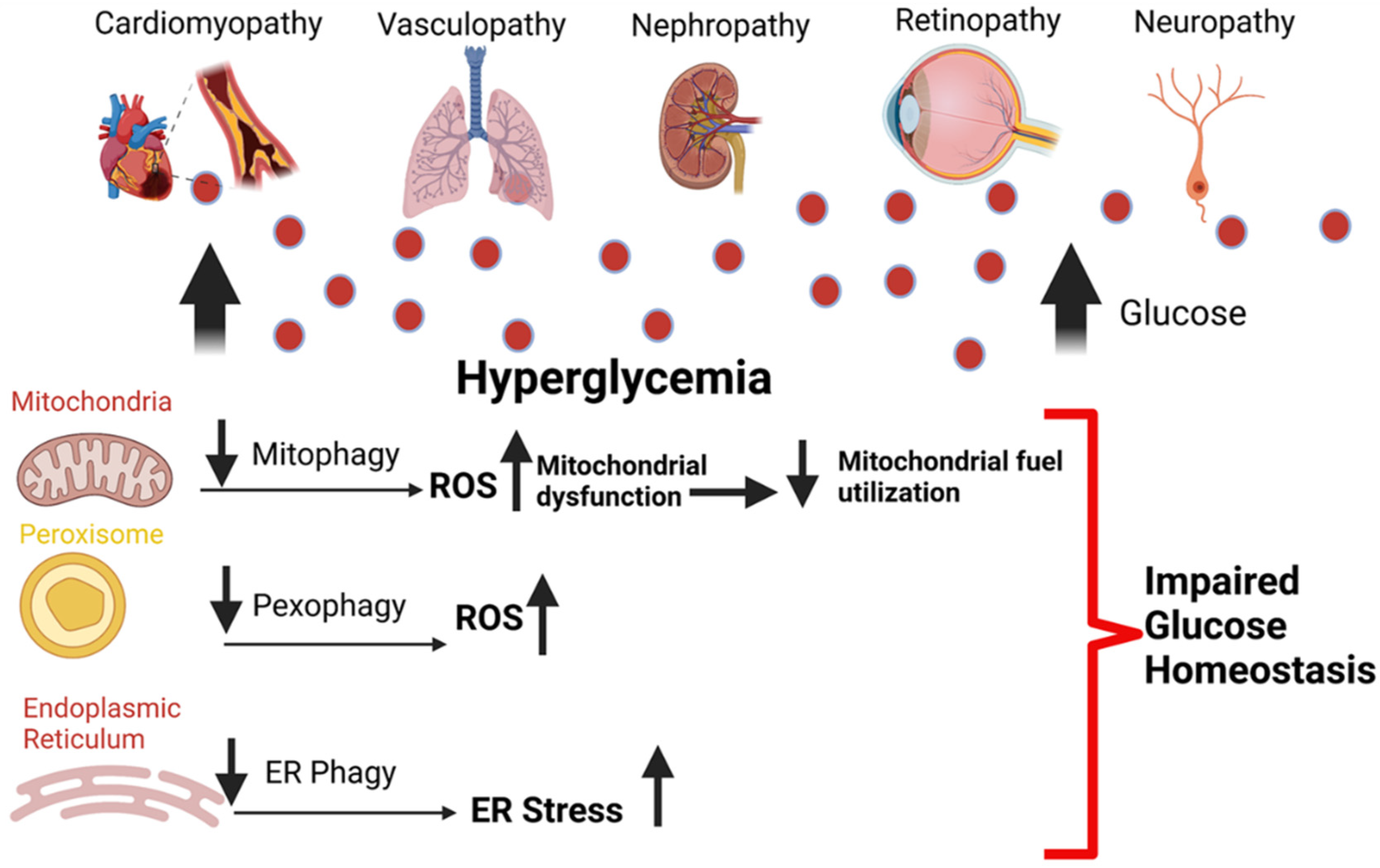
5. An Overview of the Role of Autophagy in Diabetes
6. Autophagy and the Cardiovascular System
7. Mitophagy and Microvascular Diseases
7.1. Diabetic Nephropathy
7.2. Diabetic Retinopathy
7.3. Diabetic Neuropathy
8. Mitophagy and Macrovascular Diseases
8.1. Diabetic Vasculopathy
8.2. Diabetic Cardiomyopathy
9. ER-Phagy and Vascular Complications
10. Pexophagy and Vascular Complications
11. Crosstalk and Organelle Autophagy
12. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviation
| AGEs | Advanced glycation end products |
| AKI | Acute kidney injury |
| Akt | Protein kinase B |
| AMBRA 1 | Activating molecule in BECN1-regulated autophagy protein 1 |
| AMPK | AMP-activated protein kinase |
| AMPKα1 | AMP-activated protein kinase alpha-1 |
| APJ | Apelin receptor |
| ATF4 | Activating Transcription Factor 4 |
| ATF6 | Activating Transcription Factor 6 |
| ATG4 | Autophagy related protein 4 |
| Atg5 | Autophagy related protein 5 |
| Atg7 | Autophagy related protein 7 |
| ATG8/LC3 | Autophagy-related protein 8 |
| ATG9 | Autophagy-related protein 9 |
| ATG13 | autophagy related protein 13 |
| ATG14 | autophagy related protein 14 |
| ATG101 | autophagy related protein 101 |
| ATP | Adenosine triphosphate |
| BCL2 | B-cell lymphoma 2 |
| BCN1 | Beclin 1 |
| BDNF | Brain-derived neurotrophic factor |
| BNIP3 | BCL2 Interacting Protein 3 |
| BRD4 | Bromodomain containing protein 4 |
| Bax | bcl-2-like protein 4 |
| Bip | Binding immunoglobulin protein |
| CAMKK2 | Calcium/Calmodulin Dependent Protein Kinase Kinase 2 |
| cAMP | Cyclic adenosine monophosphate |
| C-caspase-3 | Cleaved caspase-3 |
| CAD | Coronary artery disease |
| CEBPB | CCAAT Enhancer Binding Protein Beta |
| CHOP | C/EBP homologous protein |
| CMA | Chaperon-mediated autophagy |
| CoQ10 | coenzyme Q10 |
| COX IV | mammalian cytochrome c oxidase complex five |
| CRISPR-Cas9 | clustered regularly interspaced short palindromic repeats protein 9 |
| CVD | Cardiovascular disease |
| DCM | Diabetic cardiomyopathy |
| DN | Diabetic nephropathy |
| DR | Diabetic retinopathy |
| Db | Diabetic |
| Drp1 | Dynamin-related protein |
| eIF2α | Eukaryotic Initiation Factor 2 |
| ER | Endoplasmic reticulum |
| ER-phagy | Endoplasmic reticulum reticulophagy |
| ERK1/2 | Extracellular signal-regulated kinases |
| Fis-1 | Mitochondrial fission protein |
| FIP200 | FAK family kinase-interacting protein of 200 kDa |
| FKB12 | FK506-binding protein 12-rapamycin-associated protein 1 |
| FOXO | Forkhead box |
| FOXO3 | Forkhead box O3 |
| GLP-1 | Glucagon-like peptide 1 |
| GTP | Guanosine triphosphate |
| GTPases | GTP binding protein |
| GSNOR | S-nitrosoglutathione reductase |
| H9C2 | Cell line |
| HFD | High fat diet |
| HG | High glucose |
| HIF1α | Hypoxia Inducible Factor 1α |
| HOG- LDL | High oxidized glycated low density lipoprotein |
| HSC70 | Heat shock cognate 71 kDa protein |
| HSPA5 | Heat Shock Protein Family A Member 5 |
| IAPP | Islet amyloid polypeptide |
| IR | Ischemia-reperfusion |
| Ins2 Akita/+ | mouse model |
| JNK | c-Jun N-terminal kinase |
| JQ1 | Bromodomain and extra-terminal domain inhibitor |
| LAMP1 | lysosome-associated membrane glycoprotein 1 |
| LAMP-2A | Lysosome-associated membrane protein 2 |
| LC3 | Microtubule-associated protein 1A/1B-light chain 3 |
| LC3II | LC3-phosphatidylethanolamine conjugate |
| LD | lipid droplet |
| LDL | Low-density lipoprotein |
| Mdivi-1 | Mitochondrial division inhibitor |
| MDV | Mitochondria-derived vesicles |
| MFF | Mitochondrial fission factor |
| MSC | Mesenchymal stem cells |
| mtDNA | Mitochondrial DNA |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| mtROS | Mitochondrial reactive oxygen species |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NOX4 | NADPH oxidase 4 |
| Noxa | Phorbol-12-myristate-13-acetate-induced protein 1 |
| NR4A1 | Nuclear receptor subfamily 4 group A member 1 |
| Nrf/ARE | Nuclear factor erythroid 2-related factor 2/antioxidant responsive element |
| P53 | Tumor protein p53 |
| PAD | Peripheral artery disease |
| PC12 | Cell line derived from pheochromocytoma of the rat adrenal medulla |
| PGC1α | Peroxisome proliferator-activated receptor-γ coactivator 1-α |
| PIK3C3/Vps34 | Phosphatidylinositol 3-kinase catalytic subunit type 3 |
| PIK3R4/p150 | Phosphoinositide 3-kinase regulatory subunit 4 |
| PINK1 | PTEN-induced kinase 1 |
| PKCβ2 | Protein Kinase C-β |
| PLIN2 | Perilipin 2 |
| PRAS40 | Proline-rich Akt substrate of 40 kDa |
| PtdIns3K | Phosphatidylinositol 3-kinase complex |
| PtdIns3P | Phosphatidylinositol 3-phosphate |
| REDD1 | Regulated in development and DNA damage responses 1 |
| ROS | Reactive oxygen species |
| SH3GLB1 | SH3 Domain Containing GRB2 Like, Endophilin B1 |
| SNAP29 | Synaptosome Associated Protein 29 |
| SNARE | SNAP receptor |
| STX17 | Syntaxin 17 |
| T2D | Type-2-diabetes |
| TB1 | Tat-Beclin1 |
| TFAM | Mitochondrial transcription factor A |
| TFEB | Transcription Factor EB |
| TOMM20 | Translocase Of Outer Mitochondrial Membrane 20 |
| TSC1/2 complex | Tuberous sclerosis 1/2 complex |
| TUDCA | Tauroursodeoxycholic acid |
| TXNIP | Thioredoxin-interacting protein |
| ULK1 | Unc-51 Like Autophagy Activating Kinase 1 |
| USP30 | Ubiquitin specific peptidase 30 |
| UVRAG | UV radiation resistance-associated gene protein |
| VPS34 | vacuolar sorting 34 protein |
| VAM7 | Vacuolar morphogenesis protein 7 |
| VAM9 | Vacuolar morphogenesis protein 9 |
| VAMP8 | Vesicle-associated membrane protein 8 |
| vWF | Von Willebrand factor |
| ZKSCAN3 | Zinc Finger with KRAB And SCAN Domains 3 |
References
- Einarson, T.R.; Acs, A.; Ludwig, C.; Panton, U.H. Prevalence of cardiovascular disease in type 2 diabetes: A systematic literature review of scientific evidence from across the world in 2007–2017. Cardiovasc. Diabetol. 2018, 17, 83. [Google Scholar] [CrossRef] [Green Version]
- Mohammedi, K.; Woodward, M.; Hirakawa, Y.; Zoungas, S.; Williams, B.; Lisheng, L.; Rodgers, A.; Mancia, G.; Neal, B.; Harrap, S.; et al. Microvascular and macrovascular disease and risk for major peripheral arterial disease in patients with type 2 diabetes. Diabetes Care 2016, 39, 1796–1803. [Google Scholar] [CrossRef] [Green Version]
- Oku, M.; Sakai, Y. Three distinct types of microautophagy based on membrane dynamics and molecular machineries. BioEssays 2018, 40, 1800008. [Google Scholar] [CrossRef] [PubMed]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutton, M.N.; Huang, G.Y.; Zhou, J.; Mao, W.; Langley, R.; Lu, Z.; Bast, R.C., Jr. Amino acid deprivation-induced autophagy requires upregulation of DIRAS3 throughreduction of E2F1 and E2F4 transcriptional repression. Cancers 2019, 11, 603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Li, J.; Kang, R.; Tang, D. Interplay between lipid metabolism and autophagy. Front. Cell Dev. Biol. 2020, 8, 431. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.W.; Chung, H.Y. The effects of calorie restriction on autophagy: Role on aging intervention. Nutrients 2019, 11, 2923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandt, N.; Gunnarsson, T.P.; Bangsbo, J.; Pilegaard, H. Exercise and exercise training-induced increase in autophagy markers in human skeletal muscle. Physiol. Rep. 2018, 6, e13651. [Google Scholar] [CrossRef]
- Texada, M.J.; Malita, A.; Christensen, C.F.; Dall, K.B.; Faergeman, N.J.; Nagy, S.; Halberg, K.A.; Rewitz, K. Autophagy-mediated cholesterol trafficking controls steroid production. Dev. Cell 2019, 48, 659–671. [Google Scholar] [CrossRef] [Green Version]
- Anding, A.L.; Baehrecke, E.H. Cleaning House: Selective Autophagy of Organelles. Dev. Cell 2017, 41, 10–22. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Q.; Yang, M.; Li, P.; Wu, C. Bacteria exploit autophagy for their own benefit. Infect. Drug Resist. 2019, 12, 3205–3215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burman, C.; Ktistakis, N.T. Regulation of autophagy by phosphatidylinositol 3-phosphate. FEBS Lett. 2010, 584, 1302–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, J.; Mannack, L. The autophagosome: Current understanding of formation and maturation. Res. Rep. Biochem. 2015, 5, 39. [Google Scholar] [CrossRef] [Green Version]
- Geng, J.; Klionsky, D.J. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. “Protein modifications: Beyond the usual suspects” review series. EMBO Rep. 2008, 9, 859–864. [Google Scholar] [CrossRef] [Green Version]
- Young, A.R.J.; Chan, E.Y.W.; Hu, X.W.; Köchl, R.; Crawshaw, S.G.; High, S.; Halley, D.W.; Lippincott-Schwartz, J.; Tooze, S.A. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J. Cell Sci. 2006, 119, 3888–3900. [Google Scholar] [CrossRef] [Green Version]
- Fader, C.M.; Sánchez, D.G.; Mestre, M.B.; Colombo, M.I. TI-VAMP/VAMP7 and VAMP3/cellubrevin: Two v-SNARE proteins involved in specific steps of the autophagy/multivesicular body pathways. Biochim. Biophys. Acta Mol. Cell Res. 2009, 1793, 1901–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef] [Green Version]
- Schmeisser, K.; Parker, J.A. Pleiotropic effects of mTOR and autophagy during development and aging. Front. Cell Dev. Biol. 2019, 7, 192. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S. Regulation of autophagy by mTOR-dependent and mTOR-independent pathways: Autophagy dysfunction in neurodegenerative diseases and therapeutic application of autophagy enhancers. Biochem. Soc. Trans. 2013, 41, 1103–1130. [Google Scholar] [CrossRef] [Green Version]
- Sciarretta, S.; Volpe, M.; Sadoshima, J. mTOR signaling in cardiac physiology and disease. Circ. Res. 2014, 114, 549–564. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, M.; Park, R.; Kim, H.; Namkoong, S.; Jo, D.; Huh, Y.H.; Jang, I.S.; Lee, J.I.; Park, J. AMPK contributes to autophagosome maturation and lysosomal fusion. Sci. Rep. 2018, 8, 12637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pattingre, S.; Bauvy, C.; Codogno, P. Amino acids interfere with the ERK1/2-dependent control of macroautophagy by controlling the activation of Raf-1 in human colon cancer HT-29 cells. J. Biol. Chem. 2003, 278, 16667–16674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Yu, J.; Li, D.; Yu, S.; Ke, J.; Wang, L.; Wang, Y.; Qiu, Y.; Gao, X.; Zhang, J.; et al. Store-operated calcium entry-activated autophagy protects EPC proliferation via the CAMKK2-MTOR pathway in ox-LDL exposure. Autophagy 2017, 13, 82–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashid, H.O.; Yadav, R.K.; Kim, H.R.; Chae, H.J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef] [PubMed]
- Roca-Agujetas, V.; De Dios, C.; Lestón, L.; Marí, M.; Morales, A.; Colell, A. Recent insights into the mitochondrial role in autophagy and its regulation by oxidative stress. Oxid. Med. Cell. Longev. 2019, 2019, 3809308. [Google Scholar] [CrossRef] [Green Version]
- Mrakovcic, M.; Fröhlich, L.F. P53-mediated molecular control of autophagy in tumor cells. Biomolecules 2018, 8, 14. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, S.; Goodwin, J.G.; Chauhan, S.; Manyam, G.; Wang, J.; Kamat, A.M.; Boyd, D.D. ZKSCAN3 (ZNF306) is a master transcriptional repressor of autophagy. Mol. Cell 2013, 50, 16–28. [Google Scholar] [CrossRef] [Green Version]
- Di Malta, C.; Cinque, L.; Settembre, C. Transcriptional regulation of autophagy: Mechanisms and diseases. Front. Cell Dev. Biol. 2019, 7, 114. [Google Scholar] [CrossRef]
- Amm, I.; Sommer, T.; Wolf, D.H. Protein quality control and elimination of protein waste: The role of the ubiquitin-proteasome system. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 182–196. [Google Scholar] [CrossRef] [Green Version]
- Khaminets, A.; Behl, C.; Dikic, I. Ubiquitin-dependent and independent signals in selective autophagy. Trends Cell Biol. 2016, 26, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Zaffagnini, G.; Martens, S. Mechanisms of selective autophagy. J. Mol. Biol. 2016, 428, 1714–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemasters, J.J. Variants of mitochondrial autophagy: Types 1 and 2 mitophagy and micromitophagy (Type 3). Redox Biol. 2014, 2, 749–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chino, H.; Mizushima, N. ER-phagy: Quality control and turnover of endoplasmic reticulum. Trends Cell Biol. 2020, 30, 384–398. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.-h.; Noda, T. Autophagosome formation in relation to the endoplasmic reticulum. J. Biomed. Sci. 2020, 27, 6–11. [Google Scholar] [CrossRef]
- Li, J.; Wang, W. Mechanisms and functions of pexophagy in mammalian cells. Cells 2021, 10, 1094. [Google Scholar] [CrossRef]
- Vasko, R. Peroxisomes and kidney injury. Antioxid. Redox Signal. 2016, 25, 217–231. [Google Scholar] [CrossRef] [Green Version]
- Yokota, S.; Fahimi, H.D. Degradation of excess peroxisomes in mammalian liver cells by autophagy and other mechanisms. Histochem. Cell Biol. 2009, 131, 455–458. [Google Scholar] [CrossRef]
- Yang, J.S.; Lu, C.C.; Kuo, S.C.; Hsu, Y.M.; Tsai, S.C.; Chen, S.Y.; Chen, Y.T.; Lin, Y.J.; Huang, Y.C.; Chen, C.J.; et al. Autophagy and its link to type II diabetes mellitus. Biomedicine 2017, 7, 8. [Google Scholar] [CrossRef] [Green Version]
- Shigihara, N.; Fukunaka, A.; Hara, A.; Komiya, K.; Honda, A.; Uchida, T.; Abe, H.; Toyofuku, Y.; Tamaki, M.; Ogihara, T.; et al. Human IAPP-induced pancreatic β cell toxicity and its regulation by autophagy. J. Clin. Investig. 2014, 124, 3634–3644. [Google Scholar] [CrossRef]
- Hoshino, A.; Ariyoshi, M.; Okawa, Y.; Kaimoto, S.; Uchihashi, M.; Fukai, K.; Iwai-Kanai, E.; Ikeda, K.; Ueyama, T.; Ogata, T.; et al. Inhibition of p53 preserves Parkin-mediated mitophagy and pancreatic β-cell function in diabetes. Proc. Natl. Acad. Sci. USA 2014, 111, 3116–3121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Völkers, M.; Doroudgar, S.; Nguyen, N.; Konstandin, M.H.; Quijada, P.; Din, S.; Ornelas, L.; Thuerauf, D.J.; Gude, N.; Friedrich, K.; et al. PRAS40 prevents development of diabetic cardiomyopathy and improves hepatic insulin sensitivity in obesity. EMBO Mol. Med. 2014, 6, 57–65. [Google Scholar] [CrossRef]
- Das, A.; Durrant, D.; Koka, S.; Salloum, F.N.; Xi, L.; Kukreja, R.C. Mammalian target of rapamycin (mTOR) inhibition with rapamycin improves cardiac function in type 2 diabetic mice: Potential role of attenuated oxidative stress and altered contractile protein expression. J. Biol. Chem. 2014, 289, 4145–4160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Yang, Q.; Sun, Y.Y.; Xing, Y.F.; Wang, Y.-b.; Lu, X.T.; Bai, W.W.; Liu, X.Q.; Zhao, Y.X. Resveratrol-enhanced autophagic flux ameliorates myocardial oxidative stress injury in diabetic mice. J. Cell. Mol. Med. 2014, 18, 1599–1611. [Google Scholar] [CrossRef]
- Quan, W.; Hur, K.Y.; Lim, Y.; Oh, S.H.; Lee, J.C.; Kim, K.H.; Kim, G.H.; Kim, S.W.; Kim, H.L.; Lee, M.K.; et al. Autophagy deficiency in beta cells leads to compromised unfolded protein response and progression from obesity to diabetes in mice. Diabetologia 2012, 55, 392–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, Q.; Xiao, X.; Prasadan, K.; Chen, C.; Ming, Y.; Fusco, J.; Gangopadhyay, N.N.; Ricks, D.; Gittes, G.K. Autophagy protects pancreatic beta cell mass and function in the setting of a high-fat and high-glucose diet. Sci. Rep. 2017, 7, 16348. [Google Scholar] [CrossRef]
- Kosacka, J.; Kern, M.; Klöting, N.; Paeschke, S.; Rudich, A.; Haim, Y.; Gericke, M.; Serke, H.; Stumvoll, M.; Bechmann, I.; et al. Autophagy in adipose tissue of patients with obesity and type 2 diabetes. Mol. Cell. Endocrinol. 2015, 409, 21–32. [Google Scholar] [CrossRef]
- Ji, J.; Petropavlovskaia, M.; Khatchadourian, A.; Patapas, J.; Makhlin, J.; Rosenberg, L.; Maysinger, D. Type 2 diabetes is associated with suppression of autophagy and lipid accumulation in β-cells. J. Cell. Mol. Med. 2019, 23, 2890–2900. [Google Scholar] [CrossRef] [Green Version]
- Qian, Q.; Zhang, Z.; Orwig, A.; Chen, S.; Ding, W.X.; Xu, Y.; Kunz, R.C.; Lind, N.R.L.; Stamler, J.S.; Yang, L. S-nitrosoglutathione reductase dysfunction contributes to obesity-associated hepatic insulin resistance via regulating autophagy. Diabetes 2018, 67, 193–207. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Huang, W.; Wang, J.; Xu, Z.; He, J.; Lin, X.; Zhou, Z.; Zhang, J. Metformin plays a dual role in min6 pancreatic β cell function through AMPK-dependent autophagy. Int. J. Biol. Sci. 2014, 10, 268–277. [Google Scholar] [CrossRef]
- Marasco, M.R.; Linnemann, A.K. B-cell autophagy in diabetes pathogenesis. Endocrinology 2018, 159, 2127–2141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, F. Autophagy in vascular endothelial cells. Clin. Exp. Pharmacol. Physiol. 2016, 43, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Osonoi, Y.; Mita, T.; Azuma, K.; Nakajima, K.; Masuyama, A.; Goto, H.; Nishida, Y.; Miyatsuka, T.; Fujitani, Y.; Koike, M.; et al. Defective autophagy in vascular smooth muscle cells enhances cell death and atherosclerosis. Autophagy 2018, 14, 1991–2006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nussenzweig, S.C.; Verma, S.; Finkel, T. The role of autophagy in vascular biology. Circ. Res. 2015, 116, 480–488. [Google Scholar] [CrossRef]
- Li, S.; Liu, C.; Gu, L.; Wang, L.; Shang, Y.; Liu, Q.; Wan, J.; Shi, J.; Wang, F.; Xu, Z.; et al. Autophagy protects cardiomyocytes from the myocardial ischaemia-reperfusion injury through the clearance of CLP36. Open Biol. 2016, 6, 160177. [Google Scholar] [CrossRef]
- Shao, B.Z.; Han, B.Z.; Zeng, Y.X.; Su, D.F.; Liu, C. The roles of macrophage autophagy in atherosclerosis. Acta Pharmacol. Sin. 2016, 37, 150–156. [Google Scholar] [CrossRef] [Green Version]
- Hassanpour, M.; Rahbarghazi, R.; Nouri, M.; Aghamohammadzadeh, N.; Safaei, N.; Ahmadi, M. Role of autophagy in atherosclerosis: Foe or friend? J. Inflamm. 2019, 16, 8. [Google Scholar] [CrossRef] [Green Version]
- Menikdiwela, K.R.; Ramalingam, L.; Rasha, F.; Wang, S.; Dufour, J.M.; Kalupahana, N.S.; Sunahara, K.K.S.; Martins, J.O.; Moustaid-Moussa, N. Autophagy in metabolic syndrome: Breaking the wheel by targeting the renin–angiotensin system. Cell Death Dis. 2020, 11, 87. [Google Scholar] [CrossRef]
- Yang, M.; Zhang, Y.; Ren, J. Autophagic regulation of lipid homeostasis in cardiometabolic syndrome. Front. Cardiovasc. Med. 2018, 5, 38. [Google Scholar] [CrossRef] [Green Version]
- Kuma, A.; Hatano, M.; Matsui, M.; Yamamoto, A.; Nakaya, H.; Yoshimori, T.; Ohsumi, Y.; Tokuhisa, T.; Mizushima, N. The role of autophagy during the early neonatal starvation period. Nature 2004, 432, 1032–1036. [Google Scholar] [CrossRef]
- Tocchi, A.; Quarles, E.K.; Basisty, N.; Gitari, L.; Rabinovitch, P.S. Mitochondrial dysfunction in cardiac aging. Biochim. Biophys. Acta Bioenerg. 2015, 1847, 1424–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Jovinge, S.; Frisén, J.; et al. Linked references are available on JSTOR for this article: Renewal in humans evidence for cardiomyocyte. Natl. Inst. Health 2017, 324, 98–102. [Google Scholar] [CrossRef]
- Abdellatif, M.; Sedej, S.; Carmona-Gutierrez, D.; Madeo, F.; Kroemer, G. Autophagy in cardiovascular aging. Circ. Res. 2018, 123, 803–824. [Google Scholar] [CrossRef]
- Gu, J.; Hu, W.; Song, Z.P.; Chen, Y.G.; Zhang, D.D.; Wang, C.Q. Rapamycin inhibits cardiac hypertrophy by promoting autophagy via the MEK/ERK/Beclin-1 pathway. Front. Physiol. 2016, 7, 104. [Google Scholar] [CrossRef]
- Ott, C.; Jung, T.; Brix, S.; John, C.; Betz, I.R.; Foryst-Ludwig, A.; Deubel, S.; Kuebler, W.M.; Grune, T.; Kintscher, U.; et al. Hypertrophy-reduced autophagy causes cardiac dysfunction by directly impacting cardiomyocyte contractility. Cells 2021, 10, 805. [Google Scholar] [CrossRef]
- Kanamori, H.; Takemura, G.; Goto, K.; Maruyama, R.; Ono, K.; Nagao, K.; Tsujimoto, A.; Ogino, A.; Takeyama, T.; Kawaguchi, T.; et al. Autophagy limits acute myocardial infarction induced by permanent coronary artery occlusion. Am. J. Physiol. Hear. Circ. Physiol. 2011, 300, 2261–2271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Liu, H.; Foyil, S.R.; Godar, R.J.; Weinheimer, C.J.; Hill, J.A.; Diwan, A. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia-reperfusion injury. Circulation 2012, 125, 3170–3181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaludercic, N.; Maiuri, M.C.; Kaushik, S.; Fernández, Á.F.; De Bruijn, J.; Castoldi, F.; Chen, Y.; Ito, J.; Mukai, R.; Murakawa, T.; et al. Comprehensive autophagy evaluation in cardiac disease models. Cardiovasc. Res. 2020, 116, 483–504. [Google Scholar] [CrossRef] [Green Version]
- Larocca, T.J.; Henson, G.D.; Thorburn, A.; Sindler, A.L.; Pierce, G.L.; Seals, D.R. Translational evidence that impaired autophagy contributes to arterial ageing. J. Physiol. 2012, 590, 3305–3316. [Google Scholar] [CrossRef]
- Michiels, C.F.; Fransen, P.; De Munck, D.G.; De Meyer, G.R.Y.; Martinet, W. Defective autophagy in vascular smooth muscle cells alters contractility and Ca2+ homeostasis in mice. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H557–H567. [Google Scholar] [CrossRef]
- Torisu, T.; Torisu, K.; Lee, I.H.; Liu, J.; Malide, D.; Combs, C.A.; Wu, X.S.; Rovira, I.I.; Fergusson, M.M.; Weigert, R.; et al. Autophagy regulates endothelial cell processing, maturation and secretion of von Willebrand factor. Nat. Med. 2013, 19, 1281–1287. [Google Scholar] [CrossRef] [Green Version]
- Nah, J.; Zablocki, D.; Sadoshima, J. Autosis: A new target to prevent cell death. JACC Basic Transl. Sci. 2020, 5, 857–869. [Google Scholar] [CrossRef]
- Liu, Y.; Levine, B. Autosis and autophagic cell death: The dark side of autophagy. Cell Death Differ. 2015, 22, 367–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valentim, L.; Laurence, K.M.; Townsend, P.A.; Knight, R.A.; Latchman, D.S.; Stephanou, A. Urocortin inhibits Beclin1-mediated autophagic cell death in cardiac myocytes exposed to ischaemia/reperfusion injury. J. Mol. Cell. Cardiol. 2006, 40, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.P.; Kanasaki, K.; Goodwin, J.E. Loss of mitochondrial control impacts renal health. Front. Pharmacol. 2020, 11, 2133. [Google Scholar] [CrossRef]
- Tang, C.; Han, H.; Yan, M.; Zhu, S.; Liu, J.; Liu, Z.; He, L.; Tan, J.; Liu, Y.; Liu, H.; et al. PINK1-PRKN/PARK2 pathway of mitophagy is activated to protect against renal ischemia-reperfusion injury. Autophagy 2018, 14, 880–897. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.; Takabatake, Y.; Takahashi, A.; Kaimori, J.-y.; Matsui, I.; Namba, T.; Kitamura, H.; Niimura, F.; Matsusaka, T.; Soga, T.; et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J. Am. Soc. Nephrol. 2011, 22, 902–913. [Google Scholar] [CrossRef]
- Sun, J.; Zhu, H.; Wang, X.; Gao, Q.; Li, Z.; Huang, H. CoQ10 ameliorates mitochondrial dysfunction in diabetic nephropathy through mitophagy. J. Endocrinol. 2019, 240, 445–465. [Google Scholar] [CrossRef]
- Sheng, J.; Li, H.; Dai, Q.; Lu, C.; Xu, M.; Zhang, J.; Feng, J. NR4A1 promotes diabetic nephropathy by activating mff-mediated mitochondrial fission and suppressing parkin-mediated mitophagy. Cell. Physiol. Biochem. 2018, 48, 1675–1693. [Google Scholar] [CrossRef]
- Liu, X.; Wang, W.; Song, G.; Wei, X.; Zeng, Y.; Han, P.; Wang, D.; Shao, M.; Wu, J.; Sun, H.; et al. Astragaloside IV ameliorates diabetic nephropathy by modulating the mitochondrial quality control network. PLoS ONE 2017, 12, e0182558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hombrebueno, J.R.; Cairns, L.; Dutton, L.R.; Lyons, T.J.; Brazil, D.P.; Moynagh, P.; Curtis, T.M.; Xu, H. Uncoupled turnover disrupts mitochondrial quality control in diabetic retinopathy. JCI Insight 2019, 4, e129760. [Google Scholar] [CrossRef] [PubMed]
- Tien, T.; Zhang, J.; Muto, T.; Kim, D.; Sarthy, V.P.; Roy, S. High glucose induces mitochondrial dysfunction in retinal müller cells: Implications for diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 2915–2921. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.J.; Cascio, M.A.; Rosca, M.G. Diabetic retinopathy: The role of mitochondria in the neural retina and microvascular disease. Antioxidants 2020, 9, 905. [Google Scholar] [CrossRef] [PubMed]
- Devi, T.S.; Somayajulu, M.; Kowluru, R.A.; Singh, L.P. TXNIP regulates mitophagy in retinal Müller cells under high-glucose conditions: Implications for diabetic retinopathy. Cell Death Dis. 2017, 8, e2777. [Google Scholar] [CrossRef]
- Evans, C.S.; Holzbaur, E.L.F. Degradation of engulfed mitochondria is rate-limiting in optineurin-mediated mitophagy in neurons. Elife 2020, 9, e50260. [Google Scholar] [CrossRef]
- Jin, H.; Zhu, Y.; Li, Y.; Ding, X.; Ma, W.; Han, X.; Wang, B. BDNF-mediated mitophagy alleviates high-glucose-induced brain microvascular endothelial cell injury. Apoptosis 2019, 24, 511–528. [Google Scholar] [CrossRef]
- Onphachanh, X.; Lee, H.J.; Lim, J.R.; Jung, Y.H.; Kim, J.S.; Chae, C.W.; Lee, S.J.; Gabr, A.A.; Han, H.J. Enhancement of high glucose-induced PINK1 expression by melatonin stimulates neuronal cell survival: Involvement of MT2/Akt/NF-κB pathway. J. Pineal Res. 2017, 63, e12427. [Google Scholar] [CrossRef] [Green Version]
- Su, C.J.; Shen, Z.; Cui, R.X.; Huang, Y.; Xu, D.L.; Zhao, F.L.; Pan, J.; Shi, A.M.; Liu, T.; Yu, Y.L. Thioredoxin-interacting protein (TXNIP) regulates parkin/PINK1-mediated mitophagy in dopaminergic neurons under high-glucose conditions: Implications for molecular links between parkinson’s disease and diabetes. Neurosci. Bull. 2020, 36, 346–358. [Google Scholar] [CrossRef]
- Ferreiro, J.L.; Angiolillo, D.J. Diabetes and antiplatelet therapy in acute coronary syndrome. Circulation 2011, 123, 798–813. [Google Scholar] [CrossRef]
- Song, M.; Gong, G.; Burelle, Y.; Gustafsson, A.B.; Kitsis, R.N.; Matkovich, S.J.; Dorn, G.W. Interdependence of parkin-mediated mitophagy and mitochondrial fission in adult mouse hearts. Circ. Res. 2015, 117, 346–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.; Yuan, Y.; Liao, G.; Li, L.; Liu, J.; Chen, Y.; Zhang, J.; Cheng, J.; Lu, Y. Mesenchymal stem cells ameliorate hyperglycemia-induced endothelial injury through modulation of mitophagy. Cell Death Dis. 2018, 9, 837. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Du, J.; Stitham, J.; Atteya, G.; Lee, S.; Xiang, Y.; Wang, D.; Jin, Y.; Leslie, K.L.; Spollett, G.; et al. Inducing mitophagy in diabetic platelets protects against severe oxidative stress. EMBO Mol. Med. 2016, 8, 779–795. [Google Scholar] [CrossRef]
- Kobayashi, S.; Zhao, F.; Zhang, Z.; Kobayashi, T.; Huang, Y.; Shi, B.; Wu, W.; Liang, Q. Mitochondrial fission and mitophagy coordinately restrict high glucose toxicity in cardiomyocytes. Front. Physiol. 2020, 11, 1596. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.R.; Zheng, D.L.; Liu, P.M.; Yang, H.; Li, L.A.; Kuang, S.J.; Lai, Y.Y.; Rao, F.; Xue, Y.M.; Lin, J.J.; et al. High glucose induces Drp1-mediated mitochondrial fission via the Orai1 calcium channel to participate in diabetic cardiomyocyte hypertrophy. Cell Death Dis. 2021, 12, 216. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Zhang, H.; Cortés, N.G.; Wu, D.; Wang, P.; Zhang, J.; Mattison, J.A.; Smith, E.; Bettcher, L.F.; Wang, M.; et al. Increased Drp1 acetylation by lipid overload induces cardiomyocyte death and heart dysfunction. Circ. Res. 2020, 126, 456–470. [Google Scholar] [CrossRef] [PubMed]
- Tong, M.; Saito, T.; Zhai, P.; Oka, S.I.; Mizushima, W.; Nakamura, M.; Ikeda, S.; Shirakabe, A.; Sadoshima, J. Mitophagy is essential for maintaining cardiac function during high fat diet-induced diabetic cardiomyopathy. Circ. Res. 2019, 124, 1360–1371. [Google Scholar] [CrossRef] [PubMed]
- Koncsos, G.; Varga, Z.V.; Baranyai, T.; Boengler, K.; Rohrbach, S.; Li, L.; Schlüter, K.-D.; Schreckenberg, R.; Radovits, T.; Oláh, A.; et al. Diastolic dysfunction in prediabetic male rats: Role of mitochondrial oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H927–H943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, J.; Zhang, D.; Tian, Y.; Xie, Z.; Zou, M.-H. BRD4 inhibition by JQ1 prevents high-fat diet-induced diabetic cardiomyopathy by activating PINK1/Parkin-mediated mitophagy in vivo. J. Mol. Cell. Cardiol. 2020, 149, 1–14. [Google Scholar] [CrossRef]
- Zhang, Y.; Su, W.; Zhang, Q.; Xu, J.; Liu, H.; Luo, J.; Zhan, L.; Xia, Z.; Lei, S. Glycine protects H9c2 cardiomyocytes from high glucose-and hypoxia/reoxygenation-induced injury via inhibiting PKCβ2 activation and improving mitochondrial quality. J. Diabetes Res. 2018, 2018, 9502895. [Google Scholar] [CrossRef]
- Fang, L.; Zhou, Y.; Cao, H.; Wen, P.; Jiang, L.; He, W.; Dai, C.; Yang, J. Autophagy attenuates diabetic glomerular damage through protection of hyperglycemia-induced podocyte injury. PLoS ONE 2013, 8, e60546. [Google Scholar] [CrossRef]
- Fu, D.; Yu, J.Y.; Yang, S.; Wu, M.; Hammad, S.M.; Connell, A.R.; Du, M.; Chen, J.; Lyons, T.J. Survival or death: A dual role for autophagy in stress-induced pericyte loss in diabetic retinopathy. Diabetologia 2016, 59, 2251–2261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.; He, L.; Li, L.; Chen, L. Apelin/APJ system as a therapeutic target in diabetes and its complications. Mol. Genet. Metab. 2016, 119, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Castan-Laurell, I.; El Boustany, R.; Pereira, O.; Potier, L.; Marre, M.; Fumeron, F.; Valet, P.; Gourdy, P.; Velho, G.; Roussel, R. Plasma apelin and risk of type 2 diabetes in a cohort from the community. Diabetes Care 2020, 43, E15–E16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, F.; Wu, D.; Huang, S.F.; Cao, J.G.; Li, H.N.; He, L.; Liu, M.Q.; Li, L.F.; Chen, L.X. The endoplasmic reticulum stress-autophagy pathway is involved in apelin-13-induced cardiomyocyte hypertrophy in vitro. Acta Pharmacol. Sin. 2017, 38, 1589–1600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cipolla, C.M.; Lodhi, I.J. Peroxisomal dysfunction in age-related diseases. Trends Endocrinol. Metab. 2017, 28, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Cheung, N.; Mitchell, P.; Wong, T.Y. Diabetic retinopathy. Lancet 2010, 367, 124–136. [Google Scholar]
- Marcassa, E.; Kallinos, A.; Jardine, J.; Rusilowicz-Jones, E.V.; Martinez, A.; Kuehl, S.; Islinger, M.; Clague, M.J.; Urbé, S. Dual role of USP 30 in controlling basal pexophagy and mitophagy. EMBO Rep. 2018, 19, e45595. [Google Scholar] [CrossRef]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef]
- Wu, S.; Lu, Q.; Wang, Q.; Ding, Y.; Ma, Z.; Mao, X.; Huang, K.; Xie, Z.; Zou, M.H. Binding of FUN14 domain containing 1 with inositol 1,4,5-trisphosphate receptor in mitochondria-associated endoplasmic reticulum membranes maintains mitochondrial dynamics and function in hearts in vivo. Circulation 2017, 136, 2248–2266. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bharath, L.P.; Rockhold, J.D.; Conway, R. Selective Autophagy in Hyperglycemia-Induced Microvascular and Macrovascular Diseases. Cells 2021, 10, 2114. https://doi.org/10.3390/cells10082114
Bharath LP, Rockhold JD, Conway R. Selective Autophagy in Hyperglycemia-Induced Microvascular and Macrovascular Diseases. Cells. 2021; 10(8):2114. https://doi.org/10.3390/cells10082114
Chicago/Turabian StyleBharath, Leena P., Jack Donato Rockhold, and Rachel Conway. 2021. "Selective Autophagy in Hyperglycemia-Induced Microvascular and Macrovascular Diseases" Cells 10, no. 8: 2114. https://doi.org/10.3390/cells10082114
APA StyleBharath, L. P., Rockhold, J. D., & Conway, R. (2021). Selective Autophagy in Hyperglycemia-Induced Microvascular and Macrovascular Diseases. Cells, 10(8), 2114. https://doi.org/10.3390/cells10082114