
Interaction of Fibromodulin and Myostatin to Regulate Skeletal Muscle Aging: An Opposite Regulation in Muscle Aging, Diabetes, and Intracellular Lipid Accumulation

 ,
,  ,
,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Mouse Experiment
2.2. Cell Culture
2.3. Metabolite Analysis
2.4. MTT Assay
2.5. Galactosidase Staining
2.6. Oil Red O Staining
2.7. Gene Knockdown
2.8. Giemsa Staining and Fusion Indices
2.9. RNA Extraction, cDNA Synthesis, and Real-Time PCR
2.10. Western Blot
2.11. Immunohistochemistry
2.12. ELISA Analysis
2.13. Statistical Analysis
3. Results
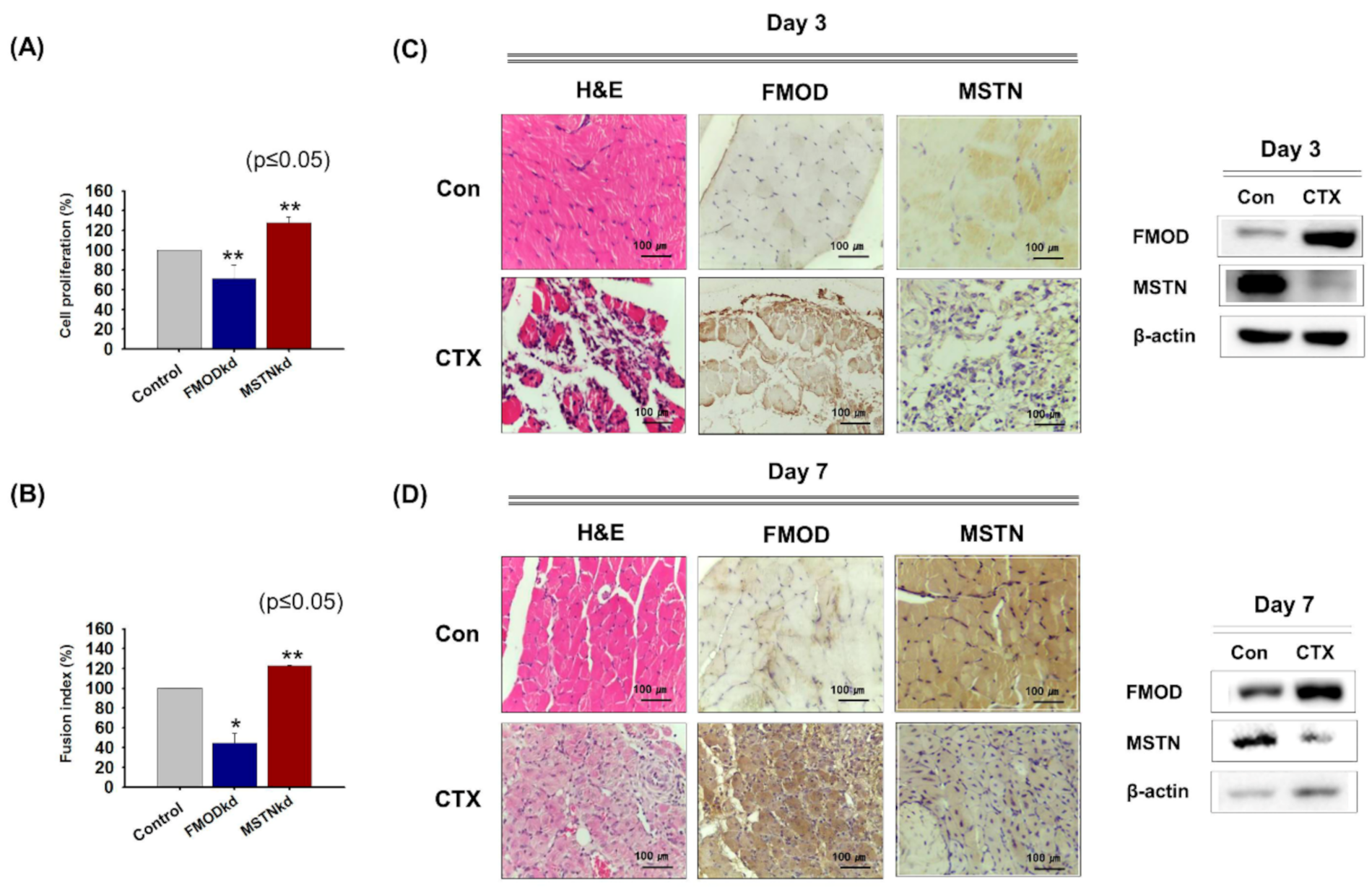
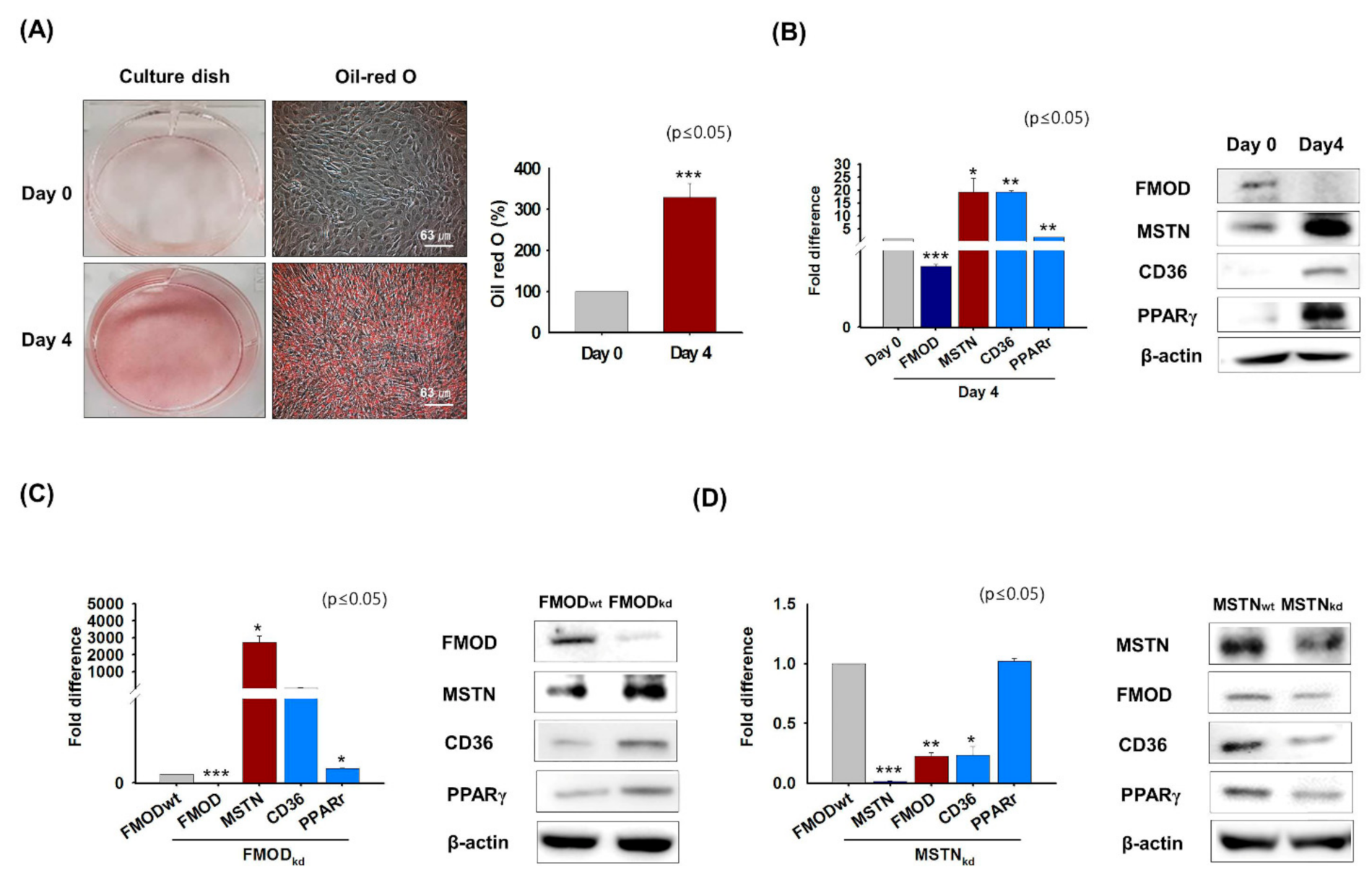
3.1. FMOD and MSTN Gene Expressions during Myoblast Proliferation, Differentiation, and Muscle Regeneration
3.2. Gene Expressions in MSTN Knockout Mice
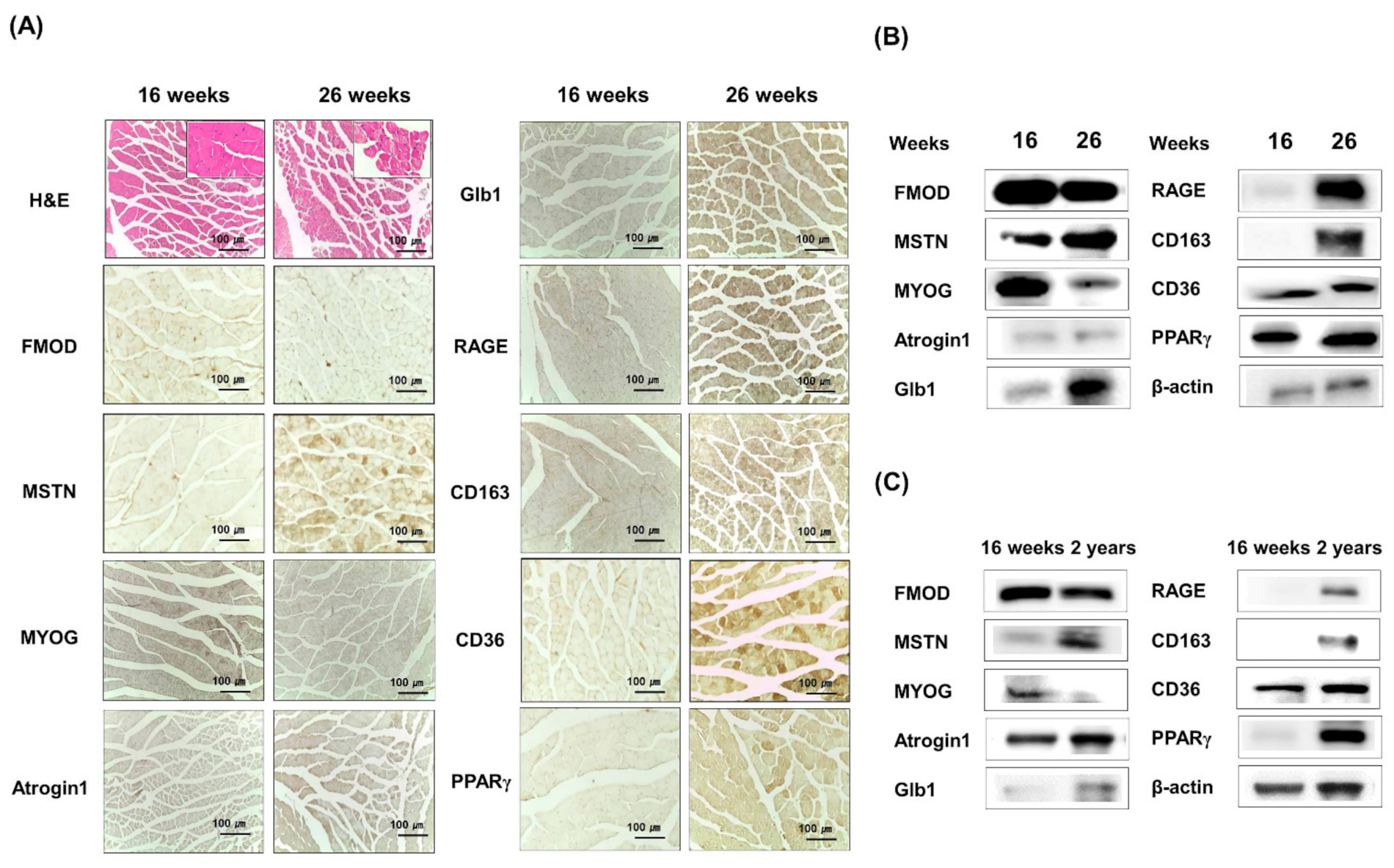
3.3. Age-Dependent FMOD and MSTN Expressions in Muscles
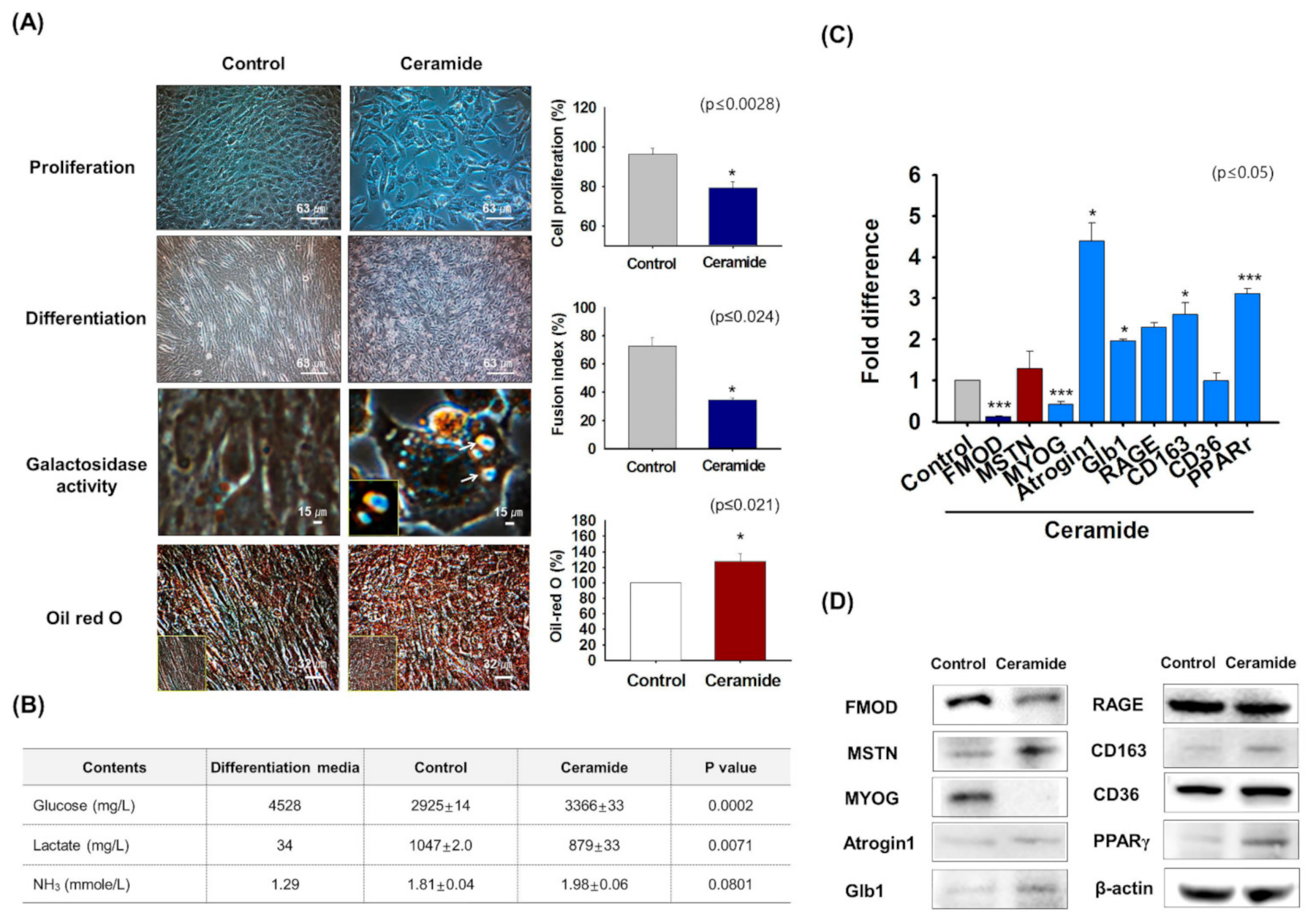
3.4. FMOD and MSTN Gene Expression in Ceramide Treated Cells
3.5. Effect of Adipogenic Conditions on FMOD and MSTN Gene Expressions in Myoblasts
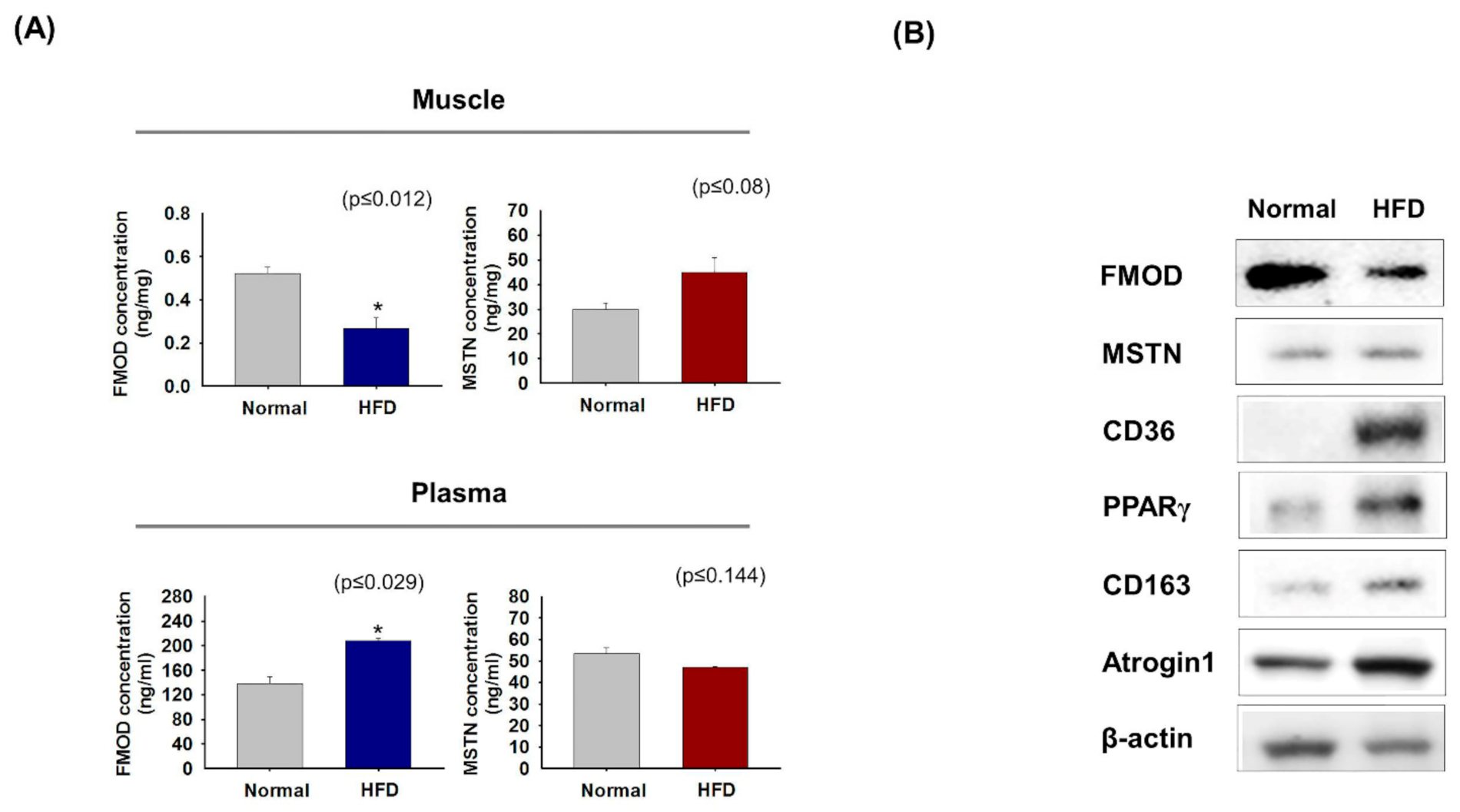
3.6. FMOD and MSTN Gene Expressions in High-Fat Diet Mice
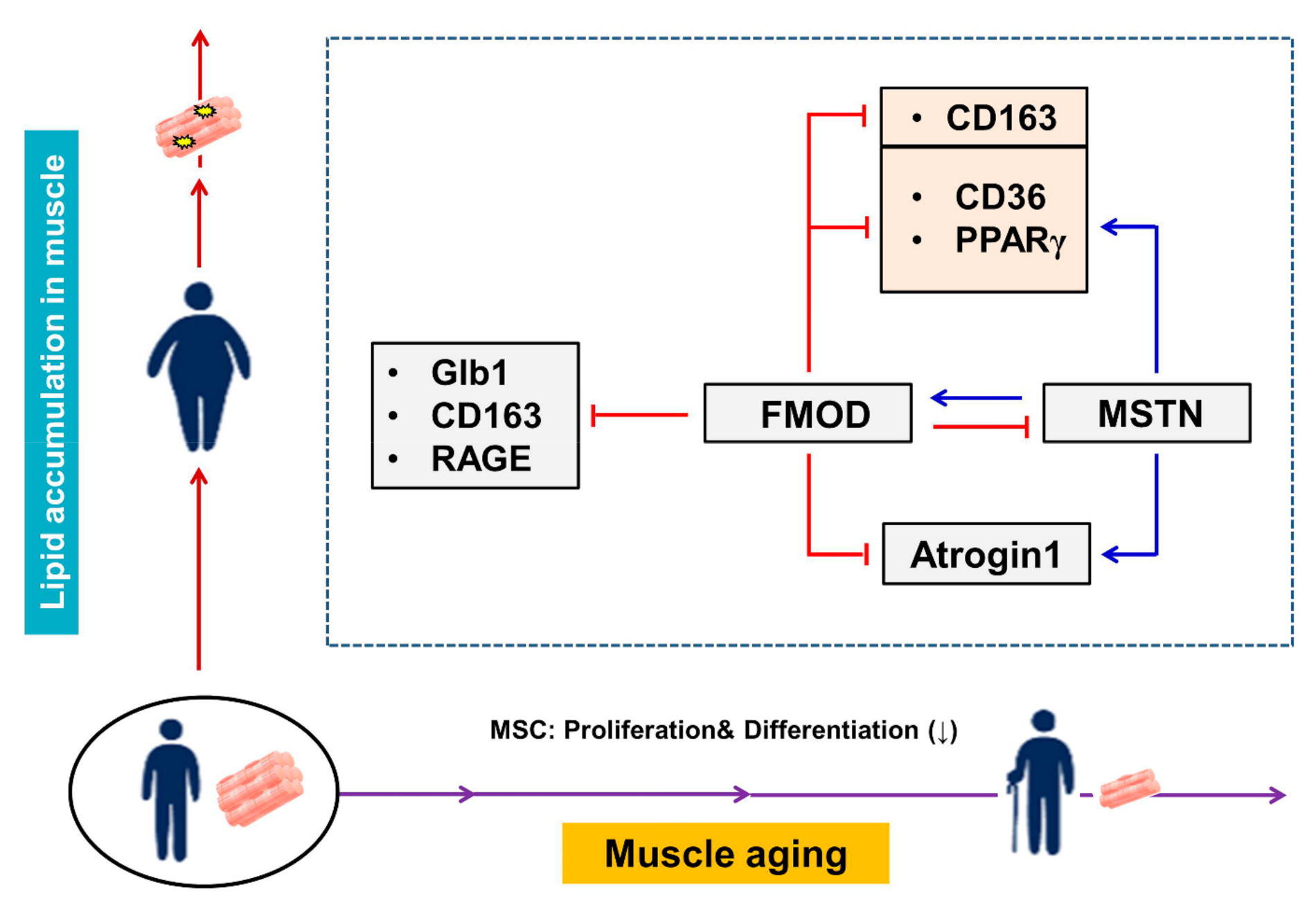
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| SM | Skeletal muscle |
| MSCs | Muscle stem cells |
| MYOG | Myogenin |
| MSTN | Myostatin |
| TGF-β | Transforming growth factor-β |
| FMOD | Fibromodulin |
| ECM | Extracellular matrix |
| CTX | Cardiotoxin |
| PPARγ | Peroxisome proliferator-activated receptor gamma |
| Glb1 | Galactosidase Beta 1 |
| ECM | Extracellular matrix |
| Gas muscle | Gastrocnemius muscle |
| HFD | High-fat diet |
| ACVRIIB | Type IIB activin receptor |
| MRF4 | Muscle regulatory factor 4 |
| IMCL | Intramyocellular lipid |
| AGE | Advanced glycation end product |
| FST | Follistatin |
| FLRG | FST-like related gene |
| GASP-1 | Growth and differentiation factor-associated serum protein-1 |
| MYH | Myosin heavy chain |
| RAGE | Receptor for advanced glycation end-products |
| GASP-2 | Growth and differentiation factor-associated serum protein-2 |
References
- Baig, M.H.; Rahman, S.; Rabbani, G.; Imran, M.; Ahmad, K.; Choi, I. Multi-Spectroscopic Characterization of Human Serum Albumin Binding with Cyclobenzaprine Hydrochloride: Insights from Biophysical and In Silico Approaches. Int. J. Mol. Sci. 2019, 20, 662. [Google Scholar] [CrossRef]
- Dumont, N.A.; Bentzinger, C.F.; Sincennes, M.C.; Rudnicki, M.A. Satellite Cells and Skeletal Muscle Regeneration. Compr. Physiol. 2015, 5, 1027–1059. [Google Scholar] [CrossRef]
- Ahmad, K.; Lee, E.J.; Moon, J.S.; Park, S.Y.; Choi, I. Multifaceted Interweaving Between Extracellular Matrix, Insulin Resistance, and Skeletal Muscle. Cells 2018, 7, 148. [Google Scholar] [CrossRef]
- Lee, E.J.; Jan, A.T.; Baig, M.H.; Ahmad, K.; Malik, A.; Rabbani, G.; Kim, T.; Lee, I.K.; Lee, Y.H.; Park, S.Y.; et al. Fibromodulin and regulation of the intricate balance between myoblast differentiation to myocytes or adipocyte-like cells. FASEB J. 2018, 32, 768–781. [Google Scholar] [CrossRef] [PubMed]
- Asakura, A.; Komaki, M.; Rudnicki, M. Muscle satellite cells are multipotential stem cells that exhibit myogenic, osteogenic, and adipogenic differentiation. Differentiation 2001, 68, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Zammit, P.; Beauchamp, J. The skeletal muscle satellite cell: Stem cell or son of stem cell? Differentiation 2001, 68, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Day, K.; Paterson, B.; Yablonka-Reuveni, Z. A distinct profile of myogenic regulatory factor detection within Pax7+ cells at S phase supports a unique role of Myf5 during posthatch chicken myogenesis. Dev. Dyn 2009, 238, 1001–1009. [Google Scholar] [CrossRef]
- Yin, H.; Price, F.; Rudnicki, M.A. Satellite cells and the muscle stem cell niche. Physiol. Rev. 2013, 93, 23–67. [Google Scholar] [CrossRef] [PubMed]
- Bonaldo, P.; Sandri, M. Cellular and molecular mechanisms of muscle atrophy. Dis. Model. Mech. 2013, 6, 25–39. [Google Scholar] [CrossRef]
- Lee, S.J.; McPherron, A.C. Regulation of myostatin activity and muscle growth. Proc. Natl. Acad. Sci. USA 2001, 98, 9306–9311. [Google Scholar] [CrossRef]
- Dong, J.; Dong, Y.; Dong, Y.; Chen, F.; Mitch, W.E.; Zhang, L. Inhibition of myostatin in mice improves insulin sensitivity via irisin-mediated cross talk between muscle and adipose tissues. Int. J. Obes. 2016, 40, 434–442. [Google Scholar] [CrossRef]
- Joulia-Ekaza, D.; Cabello, G. Myostatin regulation of muscle development: Molecular basis, natural mutations, physiopathological aspects. Exp. Cell. Res. 2006, 312, 2401–2414. [Google Scholar] [CrossRef]
- McFarlane, C.; Hennebry, A.; Thomas, M.; Plummer, E.; Ling, N.; Sharma, M.; Kambadur, R. Myostatin signals through Pax7 to regulate satellite cell self-renewal. Exp. Cell. Res. 2008, 314, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Han, H.Q.; Zhou, X.; Mitch, W.E.; Goldberg, A.L. Myostatin/activin pathway antagonism: Molecular basis and therapeutic potential. Int. J. Biochem. Cell. Biol. 2013, 45, 2333–2347. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Wall, R.J.; Yang, J. Transgenic expression of myostatin propeptide prevents diet-induced obesity and insulin resistance. Biochem. Biophys. Res. Commun. 2005, 337, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Lebrasseur, N.K. Building muscle, browning fat and preventing obesity by inhibiting myostatin. Diabetologia 2012, 55, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Jou, W.; Chanturiya, T.; Portas, J.; Gavrilova, O.; McPherron, A.C. Myostatin inhibition in muscle, but not adipose tissue, decreases fat mass and improves insulin sensitivity. PLoS ONE 2009, 4, e4937. [Google Scholar] [CrossRef]
- McPherron, A.C.; Lee, S.J. Double muscling in cattle due to mutations in the myostatin gene. Proc. Natl. Acad. Sci. USA 1997, 94, 12457–12461. [Google Scholar] [CrossRef]
- Singh, R.; Braga, M.; Pervin, S. Regulation of brown adipocyte metabolism by myostatin/follistatin signaling. Front. Cell. Dev. Biol. 2014, 2, 60. [Google Scholar] [CrossRef]
- Li, N.; Yang, Q.; Walker, R.G.; Thompson, T.B.; Du, M.; Rodgers, B.D. Myostatin Attenuation In Vivo Reduces Adiposity, but Activates Adipogenesis. Endocrinology 2016, 157, 282–291. [Google Scholar] [CrossRef]
- Lin, J.; Arnold, H.B.; Della-Fera, M.A.; Azain, M.J.; Hartzell, D.L.; Baile, C.A. Myostatin knockout in mice increases myogenesis and decreases adipogenesis. Biochem. Biophys. Res. Commun. 2002, 291, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell. Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, K.; Lee, E.J.; Shaikh, S.; Kumar, A.; Rao, K.M.; Park, S.Y.; Jin, J.O.; Han, S.S.; Choi, I. Targeting integrins for cancer management using nanotherapeutic approaches: Recent advances and challenges. Semin. Cancer Biol. 2019, 69, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Zaidel-Bar, R.; Itzkovitz, S.; Ma’ayan, A.; Iyengar, R.; Geiger, B. Functional atlas of the integrin adhesome. Nat. Cell. Biol. 2007, 9, 858–867. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.S.; Kang, L.; Wasserman, D.H. The extracellular matrix and insulin resistance. Trends Endocrinol. Metab. 2015, 26, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Jan, A.T.; Baig, M.H.; Ashraf, J.M.; Nahm, S.S.; Kim, Y.W.; Park, S.Y.; Choi, I. Fibromodulin: A master regulator of myostatin controlling progression of satellite cells through a myogenic program. FASEB J. 2016, 30, 2708–2719. [Google Scholar] [CrossRef]
- Kim, T.; Ahmad, K.; Shaikh, S.; Jan, A.T.; Seo, M.G.; Lee, E.J.; Choi, I. Dermatopontin in Skeletal Muscle Extracellular Matrix Regulates Myogenesis. Cells 2019, 8, 332. [Google Scholar] [CrossRef]
- Hildebrand, A.; Romaris, M.; Rasmussen, L.M.; Heinegard, D.; Twardzik, D.R.; Border, W.A.; Ruoslahti, E. Interaction of the small interstitial proteoglycans biglycan, decorin and fibromodulin with transforming growth factor beta. Biochem. J. 1994, 302, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Onnerfjord, P.; Heathfield, T.F.; Heinegard, D. Identification of tyrosine sulfation in extracellular leucine-rich repeat proteins using mass spectrometry. J. Biol. Chem. 2004, 279, 26–33. [Google Scholar] [CrossRef]
- Pourhanifeh, M.H.; Mohammadi, R.; Noruzi, S.; Hosseini, S.A.; Fanoudi, S.; Mohamadi, Y.; Hashemzehi, M.; Asemi, Z.; Mirzaei, H.R.; Salarinia, R.; et al. The role of fibromodulin in cancer pathogenesis: Implications for diagnosis and therapy. Cancer Cell Int. 2019, 19, 157. [Google Scholar] [CrossRef]
- Chowdhury, S.; Maria, L.; Cruz, A.; Manara, D.; Dieste-Blanco, O.; Stora, T.; Goncalves, A.P. Uranium Carbide Fibers with Nano-Grains as Starting Materials for ISOL Targets. Nanomaterials 2020, 10, 2458. [Google Scholar] [CrossRef] [PubMed]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef]
- Cong, H.; Sun, L.; Liu, C.; Tien, P. Inhibition of atrogin-1/MAFbx expression by adenovirus-delivered small hairpin RNAs attenuates muscle atrophy in fasting mice. Hum. Gene. Ther. 2011, 22, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Faitg, J.; Reynaud, O.; Leduc-Gaudet, J.P.; Gouspillou, G. Skeletal muscle aging and mitochondrial dysfunction: An update. Med. Sci. 2017, 33, 955–962. [Google Scholar] [CrossRef]
- Park, D.C.; Yeo, S.G. Aging. Korean J. Audiol. 2013, 17, 39–44. [Google Scholar] [CrossRef][Green Version]
- Saito, M.; Okamatsu-Ogura, Y.; Matsushita, M.; Watanabe, K.; Yoneshiro, T.; Nio-Kobayashi, J.; Iwanaga, T.; Miyagawa, M.; Kameya, T.; Nakada, K.; et al. High incidence of metabolically active brown adipose tissue in healthy adult humans: Effects of cold exposure and adiposity. Diabetes 2009, 58, 1526–1531. [Google Scholar] [CrossRef]
- Rivas, D.A.; Morris, E.P.; Fielding, R.A. Lipogenic regulators are elevated with age and chronic overload in rat skeletal muscle. Acta. Physiol. 2011, 202, 691–701. [Google Scholar] [CrossRef] [PubMed]
- Rivas, D.A.; Morris, E.P.; Haran, P.H.; Pasha, E.P.; Morais Mda, S.; Dolnikowski, G.G.; Phillips, E.M.; Fielding, R.A. Increased ceramide content and NFkappaB signaling may contribute to the attenuation of anabolic signaling after resistance exercise in aged males. J. Appl. Physiol. 2012, 113, 1727–1736. [Google Scholar] [CrossRef] [PubMed]
- Goodpaster, B.H.; Carlson, C.L.; Visser, M.; Kelley, D.E.; Scherzinger, A.; Harris, T.B.; Stamm, E.; Newman, A.B. Attenuation of skeletal muscle and strength in the elderly: The Health ABC Study. J. Appl. Physiol. 2001, 90, 2157–2165. [Google Scholar] [CrossRef]
- Gueugneau, M.; Coudy-Gandilhon, C.; Theron, L.; Meunier, B.; Barboiron, C.; Combaret, L.; Taillandier, D.; Polge, C.; Attaix, D.; Picard, B.; et al. Skeletal muscle lipid content and oxidative activity in relation to muscle fiber type in aging and metabolic syndrome. J. Gerontol. A Biol. Sci. Med. Sci. 2015, 70, 566–576. [Google Scholar] [CrossRef]
- Stephens, F.B.; Chee, C.; Wall, B.T.; Murton, A.J.; Shannon, C.E.; van Loon, L.J.; Tsintzas, K. Lipid-induced insulin resistance is associated with an impaired skeletal muscle protein synthetic response to amino acid ingestion in healthy young men. Diabetes 2015, 64, 1615–1620. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Lee, H.J.; Kamli, M.R.; Pokharel, S.; Bhat, A.R.; Lee, Y.H.; Choi, B.H.; Chun, T.; Kang, S.W.; Lee, Y.S.; et al. Depot-specific gene expression profiles during differentiation and transdifferentiation of bovine muscle satellite cells, and differentiation of preadipocytes. Genomics 2012, 100, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Baig, M.H.; Jan, A.T.; Rabbani, G.; Ahmad, K.; Ashraf, J.M.; Kim, T.; Min, H.S.; Lee, Y.H.; Cho, W.K.; Ma, J.Y.; et al. Methylglyoxal and Advanced Glycation End products: Insight of the regulatory machinery affecting the myogenic program and of its modulation by natural compounds. Sci. Rep. 2017, 7, 5916. [Google Scholar] [CrossRef]
- Lee, E.J.; Nam, J.H.; Choi, I. Fibromodulin modulates myoblast differentiation by controlling calcium channel. Biochem. Biophys. Res. Commun. 2018, 503, 580–585. [Google Scholar] [CrossRef]
- McPherron, A.C.; Lawler, A.M.; Lee, S.J. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature 1997, 387, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, Y.; Hattori, M.; Harada, K.; Shirase, R.; Bando, M.; Okano, G. Age-related changes in intramyocellular lipid in humans by in vivo H-MR spectroscopy. Gerontology 2007, 53, 218–223. [Google Scholar] [CrossRef]
- Lee, S.J. Regulation of muscle mass by myostatin. Annu. Rev. Cell Dev. Biol. 2004, 20, 61–86. [Google Scholar] [CrossRef]
- Rodgers, B.D.; Garikipati, D.K. Clinical, agricultural, and evolutionary biology of myostatin: A comparative review. Endocr. Rev. 2008, 29, 513–534. [Google Scholar] [CrossRef]
- Liu, C.; Li, W.; Zhang, X.; Zhang, N.; He, S.; Huang, J.; Ge, Y.; Liu, M. The critical role of myostatin in differentiation of sheep myoblasts. Biochem. Biophys. Res. Commun. 2012, 422, 381–386. [Google Scholar] [CrossRef]
- Chen, J.L.; Colgan, T.D.; Walton, K.L.; Gregorevic, P.; Harrison, C.A. The TGF-beta Signalling Network in Muscle Development, Adaptation and Disease. Adv. Exp. Med. Biol. 2016, 900, 97–131. [Google Scholar] [CrossRef]
- Brack, A.S.; Bildsoe, H.; Hughes, S.M. Evidence that satellite cell decrement contributes to preferential decline in nuclear number from large fibres during murine age-related muscle atrophy. J. Cell Sci. 2005, 118, 4813–4821. [Google Scholar] [CrossRef] [PubMed]
- Argiles, J.M.; Orpi, M.; Busquets, S.; Lopez-Soriano, F.J. Myostatin: More than just a regulator of muscle mass. Drug Discov. Today 2012, 17, 702–709. [Google Scholar] [CrossRef]
- Jespersen, J.; Kjaer, M.; Schjerling, P. The possible role of myostatin in skeletal muscle atrophy and cachexia. Scand. J. Med. Sci. Sports 2006, 16, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Doherty, T.J. Invited review: Aging and sarcopenia. J. Appl. Physiol. 2003, 95, 1717–1727. [Google Scholar] [CrossRef]
- Zhang, C.; McFarlane, C.; Lokireddy, S.; Masuda, S.; Ge, X.; Gluckman, P.D.; Sharma, M.; Kambadur, R. Inhibition of myostatin protects against diet-induced obesity by enhancing fatty acid oxidation and promoting a brown adipose phenotype in mice. Diabetologia 2012, 55, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Mosler, S.; Relizani, K.; Mouisel, E.; Amthor, H.; Diel, P. Combinatory effects of siRNA-induced myostatin inhibition and exercise on skeletal muscle homeostasis and body composition. Physiol. Rep. 2014, 2, e00262. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Cao, Y.; Qiu, B.; Zhou, Z.; Deng, R.; Chen, Z.; Li, R.; Li, X.; Wei, Q.; Xia, X.; et al. Establishment and phenotypic analysis of an Mstn knockout rat. Biochem. Biophys. Res. Commun. 2016, 477, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.; Kishioka, Y.; Wakamatsu, J.; Hattori, A.; Hennebry, A.; Berry, C.J.; Sharma, M.; Kambadur, R.; Nishimura, T. Decorin binds myostatin and modulates its activity to muscle cells. Biochem. Biophys. Res. Commun. 2006, 340, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Cash, J.N.; Rejon, C.A.; McPherron, A.C.; Bernard, D.J.; Thompson, T.B. The structure of myostatin:follistatin 288: Insights into receptor utilization and heparin binding. EMBO J. 2009, 28, 2662–2676. [Google Scholar] [CrossRef]
- Hill, J.J.; Davies, M.V.; Pearson, A.A.; Wang, J.H.; Hewick, R.M.; Wolfman, N.M.; Qiu, Y. The myostatin propeptide and the follistatin-related gene are inhibitory binding proteins of myostatin in normal serum. J. Biol. Chem. 2002, 277, 40735–40741. [Google Scholar] [CrossRef]
- Lee, Y.S.; Lee, S.J. Regulation of GDF-11 and myostatin activity by GASP-1 and GASP-2. Proc. Natl. Acad. Sci. USA 2013, 110, E3713–E3722. [Google Scholar] [CrossRef] [PubMed]
- Amthor, H.; Macharia, R.; Navarrete, R.; Schuelke, M.; Brown, S.C.; Otto, A.; Voit, T.; Muntoni, F.; Vrbova, G.; Partridge, T.; et al. Lack of myostatin results in excessive muscle growth but impaired force generation. Proc. Natl. Acad. Sci. USA 2007, 104, 1835–1840. [Google Scholar] [CrossRef] [PubMed]
- Baumann, A.P.; Ibebunjo, C.; Grasser, W.A.; Paralkar, V.M. Myostatin expression in age and denervation-induced skeletal muscle atrophy. J. Musculoskelet Neuronal Interact. 2003, 3, 8–16. [Google Scholar]
- Adams, J.M., 2nd; Pratipanawatr, T.; Berria, R.; Wang, E.; DeFronzo, R.A.; Sullards, M.C.; Mandarino, L.J. Ceramide content is increased in skeletal muscle from obese insulin-resistant humans. Diabetes 2004, 53, 25–31. [Google Scholar] [CrossRef]
- Jadhav, K.S.; Dungan, C.M.; Williamson, D.L. Metformin limits ceramide-induced senescence in C2C12 myoblasts. Mech. Ageing Dev. 2013, 134, 548–559. [Google Scholar] [CrossRef]
- Mahfouz, R.; Khoury, R.; Blachnio-Zabielska, A.; Turban, S.; Loiseau, N.; Lipina, C.; Stretton, C.; Bourron, O.; Ferre, P.; Foufelle, F.; et al. Characterising the inhibitory actions of ceramide upon insulin signaling in different skeletal muscle cell models: A mechanistic insight. PLoS ONE 2014, 9, e101865. [Google Scholar] [CrossRef] [PubMed]
- Miramontes, E.; Kempisty, B.; Petitte, J.; Dasarathy, S.; Kulus, M.; Wieczorkiewicz, M.; Mozdziak, P. Myogenic Response to Increasing Concentrations of Ammonia Differs between Mammalian, Avian, and Fish Species: Cell Differentiation and Genetic Study. Genes 2020, 11, 840. [Google Scholar] [CrossRef] [PubMed]
- Nagy, L.; Tontonoz, P.; Alvarez, J.G.; Chen, H.; Evans, R.M. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell 1998, 93, 229–240. [Google Scholar] [CrossRef]
- Hajri, T.; Han, X.X.; Bonen, A.; Abumrad, N.A. Defective fatty acid uptake modulates insulin responsiveness and metabolic responses to diet in CD36-null mice. J. Clin. Investig. 2002, 109, 1381–1389. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, E.J.; Ahmad, S.S.; Lim, J.H.; Ahmad, K.; Shaikh, S.; Lee, Y.-S.; Park, S.J.; Jin, J.O.; Lee, Y.-H.; Choi, I. Interaction of Fibromodulin and Myostatin to Regulate Skeletal Muscle Aging: An Opposite Regulation in Muscle Aging, Diabetes, and Intracellular Lipid Accumulation. Cells 2021, 10, 2083. https://doi.org/10.3390/cells10082083
Lee EJ, Ahmad SS, Lim JH, Ahmad K, Shaikh S, Lee Y-S, Park SJ, Jin JO, Lee Y-H, Choi I. Interaction of Fibromodulin and Myostatin to Regulate Skeletal Muscle Aging: An Opposite Regulation in Muscle Aging, Diabetes, and Intracellular Lipid Accumulation. Cells. 2021; 10(8):2083. https://doi.org/10.3390/cells10082083
Chicago/Turabian StyleLee, Eun Ju, Syed Sayeed Ahmad, Jeong Ho Lim, Khurshid Ahmad, Sibhghatulla Shaikh, Yun-Sil Lee, Sang Joon Park, Jun O. Jin, Yong-Ho Lee, and Inho Choi. 2021. "Interaction of Fibromodulin and Myostatin to Regulate Skeletal Muscle Aging: An Opposite Regulation in Muscle Aging, Diabetes, and Intracellular Lipid Accumulation" Cells 10, no. 8: 2083. https://doi.org/10.3390/cells10082083
APA StyleLee, E. J., Ahmad, S. S., Lim, J. H., Ahmad, K., Shaikh, S., Lee, Y.-S., Park, S. J., Jin, J. O., Lee, Y.-H., & Choi, I. (2021). Interaction of Fibromodulin and Myostatin to Regulate Skeletal Muscle Aging: An Opposite Regulation in Muscle Aging, Diabetes, and Intracellular Lipid Accumulation. Cells, 10(8), 2083. https://doi.org/10.3390/cells10082083