
Organ Crosstalk and the Modulation of Insulin Signaling


Abstract
1. Introduction
2. Secreted Modulators of Early Insulin Signaling
2.1. Inhibitory Modulators
2.2. Stimulatory Modulators
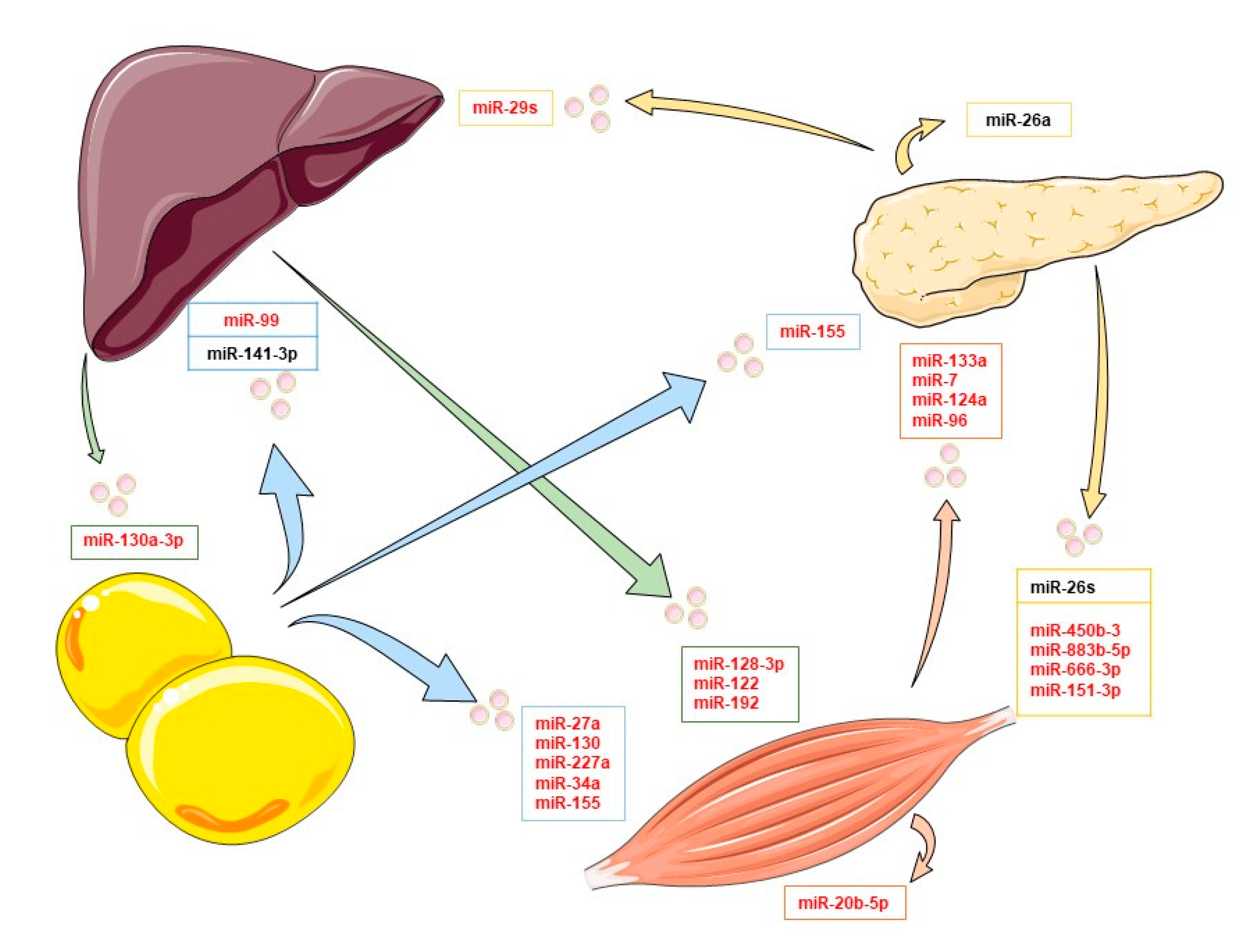
3. Extracellular Vesicles and Insulin Signaling
3.1. Adipose Tissue Exosomal miRNAs
3.2. Muscle Exosomal miRNAs
3.3. Pancreatic Exosomal miRNAs
3.4. Liver miRNA Exosomes
3.5. Exosomal miRNA and Classical Modulators in Metabolic Diseases
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Priest, C.; Tontonoz, P. Inter-organ cross-talk in metabolic syndrome. Nat. Metab. 2019, 1, 1177–1188. [Google Scholar] [CrossRef]
- Eckel, J. Myokines in metabolic homeostasis and diabetes. Diabetology 2019, 62, 1523–1528. [Google Scholar] [CrossRef] [PubMed]
- Eckel, J. Intestinal microbiota and host metabolism—A complex relationship. Acta Physiol. 2021, 232, e13638. [Google Scholar] [CrossRef] [PubMed]
- Dahlman, I.; Elsen, M.; Tennagels, N.; Korn, M.; Brockmann, B.; Sell, H.; Eckel, J.; Arner, P. Functional annotation of the human fat cell secretome. Arch. Physiol. Biochem. 2012, 118, 84–91. [Google Scholar] [CrossRef]
- Raschke, S.; Eckardt, K.; Holven, K.B.; Jensen, J.; Eckel, J. Identification and Validation of Novel Contraction-Regulated Myokines Released from Primary Human Skeletal Muscle Cells. PLoS ONE 2013, 8, e62008. [Google Scholar] [CrossRef] [PubMed]
- Romacho, T.; Elsen, M.; Röhrborn, D.; Eckel, J. Adipose tissue and its role in organ crosstalk. Acta Physiol. 2014, 210, 733–753. [Google Scholar] [CrossRef]
- Hotamisligil, G.; Shargill, N.; Spiegelman, B. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Peraldi, P.; Budavari, A.; Ellis, R.; White, M.F.; Spiegelman, B.M. IRS-1-Mediated Inhibition of Insulin Receptor Tyrosine Kinase Activity in TNF-alpha- and Obesity-Induced Insulin Resistance. Science 1996, 271, 665–670. [Google Scholar] [CrossRef]
- Jensen-Cody, S.O.; Potthoff, M.J. Hepatokines and metabolism: Deciphering communication from the liver. Mol. Metab. 2020, 44, 101138. [Google Scholar] [CrossRef]
- Whitham, M.; Parker, B.L.; Friedrichsen, M.; Hingst, J.R.; Hjorth, M.; Hughes, W.; Egan, C.L.; Cron, L.; Watt, K.; Kuchel, R.P.; et al. Extracellular Vesicles Provide a Means for Tissue Crosstalk during Exercise. Cell Metab. 2018, 27, 237–251.e4. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.; Wang, J. Exosomes and Their Noncoding RNA Cargo Are Emerging as New Modulators for Diabetes Mellitus. Cells 2019, 8, 853. [Google Scholar] [CrossRef]
- Thomou, T.; Mori, M.; Dreyfuss, J.; Konishi, M.; Sakaguchi, M.; Wolfrum, C.; Rao, T.N.; Winnay, J.N.; Garcia-Martin, R.; Grinspoon, S.K.; et al. Adipose-derived circulating miRNAs regulate gene expression in other tissues. Nat. Cell Biol. 2017, 542, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Hirsova, P.; Ibrahim, S.H.; Verma, V.; Morton, L.A.; Shah, V.H.; LaRusso, N.F.; Gores, G.J.; Malhi, H. Extracellular vesicles in liver pathobiology: Small particles with big impact. Hepatology 2016, 64, 2219–2233. [Google Scholar] [CrossRef] [PubMed]
- Batista, T.M.; Haider, N.; Kahn, C.R. Defining the underlying defect in insulin action in type 2 diabetes. Diabetology 2021, 64, 994–1006. [Google Scholar] [CrossRef]
- Szendroedi, J.; Yoshimura, T.; Phielix, E.; Koliaki, C.; Marcucci, M.; Zhang, D.; Jelenik, T.; Müller, J.; Herder, C.; Nowotny, P.; et al. Role of diacylglycerol activation of PKCθ in lipid-induced muscle insulin resistance in humans. Proc. Natl. Acad. Sci. USA 2014, 111, 9597–9602. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.; Shulman, G. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef]
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes. Front. Physiol. 2020, 10, 1607. [Google Scholar] [CrossRef] [PubMed]
- Stefan, N.; Häring, H.-U. The role of hepatokines in metabolism. Nat. Rev. Endocrinol. 2013, 9, 144–152. [Google Scholar] [CrossRef]
- Srinivas, P.R.; Wagner, A.S.; Reddy, L.V.; Deutsch, D.D.; Leon, M.A.; Goustin, A.-S.; Grunberger, G. Serum alpha 2-HS-glycoprotein is an inhibitor of the human insulin receptor at the tyrosine kinase level. Mol. Endocrinol. 1993, 7, 1445–1455. [Google Scholar] [CrossRef] [PubMed]
- Haukeland, J.W.; Dahl, T.B.; Yndestad, A.; Gladhaug, I.P.; Løberg, E.M.; Haaland, T.; Konopski, Z.; Wium, C.; Aasheim, E.T.; Johansen, O.E.; et al. Fetuin A in nonalcoholic fatty liver disease: In vivo and in vitro studies. Eur. J. Endocrinol. 2012, 166, 503–510. [Google Scholar] [CrossRef]
- Reinehr, T.; Roth, C.L. Fetuin-A and Its Relation to Metabolic Syndrome and Fatty Liver Disease in Obese Children Before and After Weight Loss. J. Clin. Endocrinol. Metab. 2008, 93, 4479–4485. [Google Scholar] [CrossRef]
- Pan, X.; Wen, S.W.; Bestman, P.L.; Kaminga, A.C.; Acheampong, K.; Liu, A. Fetuin-A in Metabolic syndrome: A systematic review and meta-analysis. PLoS ONE 2020, 15, e0229776. [Google Scholar] [CrossRef] [PubMed]
- Bourebaba, L.; Marycz, K. Pathophysiological Implication of Fetuin-A Glycoprotein in the Development of Metabolic Disorders: A Concise Review. J. Clin. Med. 2019, 8, 2033. [Google Scholar] [CrossRef] [PubMed]
- Meex, R.C.; Hoy, A.; Morris, A.; Brown, R.D.; Lo, J.C.; Burke, M.; Goode, R.; Kingwell, B.A.; Kraakman, M.; Febbraio, M.A.; et al. Fetuin B Is a Secreted Hepatocyte Factor Linking Steatosis to Impaired Glucose Metabolism. Cell Metab. 2015, 22, 1078–1089. [Google Scholar] [CrossRef]
- Peter, A.; Kovarova, M.; Staiger, H.; Machann, J.; Schick, F.; Königsrainer, A.; Königsrainer, I.; Schleicher, E.; Fritsche, A.; Häring, H.-U.; et al. The hepatokines fetuin-A and fetuin-B are upregulated in the state of hepatic steatosis and may differently impact on glucose homeostasis in humans. Am. J. Physiol. Metab. 2018, 314, E266–E273. [Google Scholar] [CrossRef]
- Benissan-Messan, D.Z.; Zhu, H.; Zhong, W.; Tan, T.; Ma, J.; Lee, P.H.U. Multi-Cellular Functions of MG53 in Muscle Calcium Signaling and Regeneration. Front. Physiol. 2020, 11, 1410. [Google Scholar] [CrossRef]
- Song, R.; Peng, W.; Zhang, Y.; Lv, F.; Wu, H.-K.; Guo, J.; Cao, Y.; Pi, Y.; Zhang, X.; Jin, L.; et al. Central role of E3 ubiquitin ligase MG53 in insulin resistance and metabolic disorders. Nat. Cell Biol. 2013, 494, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-K.; Zhang, Y.; Cao, C.-M.; Hu, X.; Fang, M.; Yao, Y.; Jin, L.; Chen, G.; Jiang, P.; Zhang, S.; et al. Glucose-Sensitive Myokine/Cardiokine MG53 Regulates Systemic Insulin Response and Metabolic Homeostasis. Circulation 2019, 139, 901–914. [Google Scholar] [CrossRef]
- Bian, Z.; Wang, Q.; Zhou, X.; Tan, T.; Park, K.H.; Kramer, H.F.; McDougal, A.; Laping, N.J.; Kumar, S.; Adesanya, T.M.A.; et al. Sustained elevation of MG53 in the bloodstream increases tissue regenerative capacity without compromising metabolic function. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Philouze, C.; Turban, S.; Cremers, B.; Caliez, A.; Lamarche, G.; Bernard, C.; Provost, N.; Delerive, P. MG53 is not a critical regulator of insulin signaling pathway in skeletal muscle. PLoS ONE 2021, 16, e0245179. [Google Scholar] [CrossRef]
- Whitson, B.A.; Tan, T.; Gong, N.; Zhu, H.; Ma, J. Muscle multiorgan crosstalk with MG53 as a myokine for tissue repair and regeneration. Curr. Opin. Pharmacol. 2021, 59, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.; Dong, L.Q. Adiponectin signaling and function in insulin target tissues. J. Mol. Cell Biol. 2016, 8, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Iwabu, M.; Okada-Iwabu, M.; Kadowaki, T. Adiponectin receptors: A review of their structure, function and how they work. Best Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 15–23. [Google Scholar] [CrossRef]
- Tanabe, H.; Fujii, Y.; Okada-Iwabu, M.; Iwabu, M.; Nakamura, Y.; Hosaka, T.; Motoyama, K.; Ikeda, M.; Wakiyama, M.; Terada, T.; et al. Crystal structures of the human adiponectin receptors. Nat. Cell Biol. 2015, 520, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Kikani, C.; Riojas, R.A.; Langlais, P.; Wang, L.; Ramos, F.J.; Fang, Q.; Christ-Roberts, C.Y.; Hong, J.Y.; Kim, R.-Y.; et al. APPL1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nat. Cell Biol. 2006, 8, 516–523. [Google Scholar] [CrossRef]
- Ryu, J.; Galan, A.K.; Xin, X.; Dong, F.; Abdul-Ghani, M.A.; Zhou, L.; Wang, C.; Li, C.; Holmes, B.M.; Sloane, L.B.; et al. APPL1 Potentiates Insulin Sensitivity by Facilitating the Binding of IRS1/2 to the Insulin Receptor. Cell Rep. 2014, 7, 1227–1238. [Google Scholar] [CrossRef]
- Kita, S.; Maeda, N.; Shimomura, I. Interorgan communication by exosomes, adipose tissue, and adiponectin in metabolic syndrome. J. Clin. Investig. 2019, 129, 4041–4049. [Google Scholar] [CrossRef]
- Denzel, M.; Scimia, M.-C.; Zumstein, P.M.; Walsh, K.; Ruiz-Lozano, P.; Ranscht, B. T-cadherin is critical for adiponectin-mediated cardioprotection in mice. J. Clin. Investig. 2010, 120, 4342–4352. [Google Scholar] [CrossRef]
- Obata, Y.; Kita, S.; Koyama, Y.; Fukuda, S.; Takeda, H.; Takahashi, M.; Fujishima, Y.; Nagao, H.; Masuda, S.; Tanaka, Y.; et al. Adiponectin/T-cadherin system enhances exosome biogenesis and decreases cellular ceramides by exosomal release. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Castaño, C.; Novials, A.; Párrizas, M. Exosomes and diabetes. Diabetes/Metab. Res. Rev. 2018, 35, e3107. [Google Scholar] [CrossRef]
- Hooten, N.N.; Evans, M.K. Extracellular vesicles as signaling mediators in type 2 diabetes mellitus. Am. J. Physiol. Physiol. 2020, 318, C1189–C1199. [Google Scholar] [CrossRef]
- Kalra, H.; Drummen, G.P.C.; Mathivanan, S. Focus on Extracellular Vesicles: Introducing the Next Small Big Thing. Int. J. Mol. Sci. 2016, 17, 170. [Google Scholar] [CrossRef]
- Pardo, F.; Villalobos-Labra, R.; Sobrevia, B.; Toledo, F.; Sobrevia, L. Extracellular vesicles in obesity and diabetes mellitus. Mol. Asp. Med. 2017, 60, 81–91. [Google Scholar] [CrossRef]
- Dorairaj, V.; Sulaiman, S.A.; Abu, N.; Murad, N.A.A. Extracellular Vesicles in the Development of the Non-Alcoholic Fatty Liver Disease: An Update. Biomology 2020, 10, 1494. [Google Scholar] [CrossRef]
- Kumar, A.; Sundaram, K.; Mu, J.; Dryden, G.W.; Sriwastva, M.K.; Lei, C.; Zhang, L.; Qiu, X.; Xu, F.; Yan, J.; et al. High-fat diet-induced upregulation of exosomal phosphatidylcholine contributes to insulin resistance. Nat. Commun. 2021, 12, 1–21. [Google Scholar] [CrossRef]
- Sáez, T.; Toledo, F.; Sobrevia, L. Impaired signalling pathways mediated by extracellular vesicles in diabesity. Mol. Asp. Med. 2019, 66, 13–20. [Google Scholar] [CrossRef]
- Lakhter, A.J.; Sims, E.K. Minireview: Emerging Roles for Extracellular Vesicles in Diabetes and Related Metabolic Disorders. Mol. Endocrinol. 2015, 29, 1535–1548. [Google Scholar] [CrossRef] [PubMed]
- Katayama, M.; Wiklander, O.P.; Fritz, T.; Caidahl, K.; Andaloussi, S.E.; Zierath, J.R.; Krook, A. Circulating Exosomal miR-20b-5p is Elevated in Type 2 Diabetes and Could Impair Insulin Action in Human Skeletal Muscle. Diabetes 2018, db180470. [Google Scholar] [CrossRef] [PubMed]
- Eissa, S.; Matboli, M.; Bekhet, M.M. Clinical verification of a novel urinary microRNA panal: 133b, -342 and -30 as biomarkers for diabetic nephropathy identified by bioinformatics analysis. Biomed. Pharmacother. 2016, 83, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Jia, Y.; Cuihua, X.; Hu, F.; Xue, M.; Xue, Y. Urinary Exosomal MicroRNA Profiling in Incipient Type 2 Diabetic Kidney Disease. J. Diabetes Res. 2017, 2017, 1–10. [Google Scholar] [CrossRef]
- Xu, H.; Du, X.; Xu, J.; Zhang, Y.; Tian, Y.; Liu, G.; Wang, X.; Ma, M.; Du, W.; Liu, Y.; et al. Pancreatic β cell microRNA-26a alleviates type 2 diabetes by improving peripheral insulin sensitivity and preserving β cell function. PLoS Biol. 2020, 18, e3000603. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Du, H.; Wei, S.; Feng, L.; Li, J.; Yao, F.; Zhang, M.; Hatch, G.M.; Chen, L. Adipocyte-Derived Exosomal MiR-27a Induces Insulin Resistance in Skeletal Muscle Through Repression of PPARγ. Theranostics 2018, 8, 2171–2188. [Google Scholar] [CrossRef]
- Huang, Z.; Xu, A. Adipose Extracellular Vesicles in Intercellular and Inter-Organ Crosstalk in Metabolic Health and Diseases. Front. Immunol. 2021, 12, 463. [Google Scholar] [CrossRef]
- Lee, M.-W.; Lee, M.; Oh, K.-J. Adipose Tissue-Derived Signatures for Obesity and Type 2 Diabetes: Adipokines, Batokines and MicroRNAs. J. Clin. Med. 2019, 8, 854. [Google Scholar] [CrossRef] [PubMed]
- Emanuelli, B.; Vienberg, S.G.; Smyth, G.; Cheng, C.; Stanford, K.I.; Arumugam, M.; Michael, M.D.; Adams, A.C.; Kharitonenkov, A.; Kahn, C.R. Interplay between FGF21 and insulin action in the liver regulates metabolism. J. Clin. Investig. 2014, 124, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.-M.; Lin, X.; Xu, F.; Shan, S.-K.; Guo, B.; Li, F.-X.; Zheng, M.-H.; Wang, Y.; Xu, Q.-S.; Yuan, L.-Q. Exosomes and Obesity-Related Insulin Resistance. Front. Cell Dev. Biol. 2021, 9, 588. [Google Scholar] [CrossRef] [PubMed]
- Rome, S.; Forterre, A.; Mizgier, M.L.; Bouzakri, K. Skeletal Muscle-Released Extracellular Vesicles: State of the Art. Front. Physiol. 2019, 10, 929. [Google Scholar] [CrossRef] [PubMed]
- Bonen, A. PGC-1α-induced improvements in skeletal muscle metabolism and insulin sensitivityThis paper is one of a selection of papers published in this Special Issue, entitled 14th International Biochemistry of Exercise Conference—Muscles as Molecular and Metabolic Machines, and has undergone the Journal’s usual peer review process. Appl. Physiol. Nutr. Metab. 2009, 34, 307–314. [Google Scholar] [CrossRef]
- Xiao, Y.; Zheng, L.; Zou, X.; Wang, J.; Zhong, J.; Zhong, T. Extracellular vesicles in type 2 diabetes mellitus: Key roles in pathogenesis, complications, and therapy. J. Extracell. Vesicles 2019, 8, 1625677. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, O.-K. Potential Roles of Adipocyte Extracellular Vesicle–Derived miRNAs in Obesity-Mediated Insulin Resistance. Adv. Nutr. 2020, 12, 566–574. [Google Scholar] [CrossRef]
- Yao, F.; Yu, Y.; Feng, L.; Li, J.; Zhang, M.; Lan, X.; Yan, X.; Liu, Y.; Guan, F.; Zhang, M.; et al. Adipogenic miR-27a in adipose tissue upregulates macrophage activation via inhibiting PPARγ of insulin resistance induced by high-fat diet-associated obesity. Exp. Cell Res. 2017, 355, 105–112. [Google Scholar] [CrossRef]
- Kukreti, H.; Amuthavalli, K. MicroRNA-34a causes ceramide accumulation and effects insulin signaling pathway by targeting ceramide kinase (CERK) in aging skeletal muscle. J. Cell. Biochem. 2020, 121, 3070–3089. [Google Scholar] [CrossRef]
- Nigi, L.; Grieco, G.E.; Ventriglia, G.; Brusco, N.; Mancarella, F.; Formichi, C.; Dotta, F.; Sebastiani, G. MicroRNAs as Regulators of Insulin Signaling: Research Updates and Potential Therapeutic Perspectives in Type 2 Diabetes. Int. J. Mol. Sci. 2018, 19, 3705. [Google Scholar] [CrossRef]
- Leonardini, A.; Laviola, L.; Perrini, S.; Natalicchio, A.; Giorgino, F. Cross-Talk between PPARγand Insulin Signaling and Modulation of Insulin Sensitivity. PPAR Res. 2009, 2009, 1–12. [Google Scholar] [CrossRef]
- Chen, Y.; Siegel, F.; Kipschull, S.; Haas, B.; Fröhlich, H.; Meister, G.; Pfeifer, A. miR-155 regulates differentiation of brown and beige adipocytes via a bistable circuit. Nat. Commun. 2013, 4, 1769. [Google Scholar] [CrossRef] [PubMed]
- Ables, G.P. Update on Ppar and Nonalcoholic Fatty Liver Disease. PPAR Res. 2012, 2012, 1–5. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Akbar, N.; Azzimato, V.; Choudhury, R.P.; Aouadi, M. Extracellular vesicles in metabolic disease. Diabetology 2019, 62, 2179–2187. [Google Scholar] [CrossRef]
- Guay, C.; Regazzi, R. Exosomes as new players in metabolic organ cross-talk. Diabetes Obes. Metab. 2017, 19, 137–146. [Google Scholar] [CrossRef]
- Barlow, J.P.; Solomon, T.P. Do skeletal muscle-secreted factors influence the function of pancreatic β-cells? Am. J. Physiol. Metab. 2018, 314, E297–E307. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, K.; Chen, J.; Guo, J.; Yin, Y.; Cai, X.; Guo, X.; Wang, G.; Yang, R.; Zhu, L.; et al. In Vitro Evidence Suggests That miR-133a-mediated Regulation of Uncoupling Protein 2 (UCP2) Is an Indispensable Step in Myogenic Differentiation. J. Biol. Chem. 2009, 284, 5362–5369. [Google Scholar] [CrossRef]
- Nieto, M.; Hevia, P.; Garcia, E.; Klein, D.; Alvarez-Cubela, S.; Bravo-Egana, V.; Rosero, S.; Molano, R.; Vargas, N.; Ricordi, C.; et al. Antisense miR-7 Impairs Insulin Expression in Developing Pancreas and in Cultured Pancreatic Buds. Cell Transplant. 2012, 21, 1761–1774. [Google Scholar] [CrossRef] [PubMed]
- Lovis, P.; Gattesco, S.; Regazzi, R. Regulation of the expression of components of the exocytotic machinery of insulin-secreting cells by microRNAs. Biol. Chem. 2008, 389, 305–312. [Google Scholar] [CrossRef]
- Baroukh, N.; Ravier, M.; Loder, M.K.; Hill, E.V.; Bounacer, A.; Scharfmann, R.; Rutter, G.; Van Obberghen, E. MicroRNA-124a Regulates Foxa2 Expression and Intracellular Signaling in Pancreatic β-Cell Lines. J. Biol. Chem. 2007, 282, 19575–19588. [Google Scholar] [CrossRef]
- Foudi, N.; Legeay, S. Effects of physical activity on cell-to-cell communication during type 2 diabetes: A focus on miRNA signaling. Fundam. Clin. Pharmacol. 2021. [Google Scholar] [CrossRef]
- Calderari, S.; Diawara, M.R.; Garaud, A.; Gauguier, D. Biological roles of microRNAs in the control of insulin secretion and action. Physiol. Genom. 2017, 49, 1–10. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, Z.; Wang, Y.; Tweardy, D.J.; Mitch, W.E. Stat3 activation induces insulin resistance via a muscle-specific E3 ubiquitin ligase Fbxo40. Am. J. Physiol. Metab. 2020, 318, E625–E635. [Google Scholar] [CrossRef] [PubMed]
- Castaño, C.; Mirasierra, M.; Vallejo, M.; Novials, A.; Párrizas, M. Delivery of muscle-derived exosomal miRNAs induced by HIIT improves insulin sensitivity through down-regulation of hepatic FoxO1 in mice. Proc. Natl. Acad. Sci. USA 2020, 117, 30335–30343. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Li, D.; Liu, Y.; Li, J.; Zhang, Y.; Zhang, C.-Y. Islet β cell: An endocrine cell secreting miRNAs. Biochem. Biophys. Res. Commun. 2018, 495, 1648–1654. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Y.; Ye, Y.; Li, D.; Liu, Y.; Lee, E.; Zhang, M.; Dai, X.; Zhang, X.; Wang, S.; et al. Pancreatic β cells control glucose homeostasis via the secretion of exosomal miR-29 family. J. Extracell. Vesicles 2021, 10, e12055. [Google Scholar] [CrossRef]
- Ge, Q.; Xie, X.X.; Xiao, X.; Li, X. Exosome-Like Vesicles as New Mediators and Therapeutic Targets for Treating Insulin Resistance and β-Cell Mass Failure in Type 2 Diabetes Mellitus. J. Diabetes Res. 2019, 2019, 1–7. [Google Scholar] [CrossRef]
- Wu, J.; Dong, T.; Chen, T.; Sun, J.; Luo, J.; He, J.; Wei, L.; Zeng, B.; Zhang, H.; Li, W.; et al. Hepatic exosome-derived miR-130a-3p attenuates glucose intolerance via suppressing PHLPP2 gene in adipocyte. Metabolism 2019, 103, 154006. [Google Scholar] [CrossRef] [PubMed]
- Santovito, D.; De Nardis, V.; Marcantonio, P.; Mandolini, C.; Paganelli, C.; Vitale, E.; Buttitta, F.; Bucci, M.; Mezzetti, A.; Consoli, A.; et al. Plasma Exosome MicroRNA Profiling Unravels a New Potential Modulator of Adiponectin Pathway in Diabetes: Effect of Glycemic Control. J. Clin. Endocrinol. Metab. 2014, 99, E1681–E1685. [Google Scholar] [CrossRef] [PubMed]
- Sabry, D.; Mahmoud, G.; El-Aziz, A.; Mahmoud, K.; Fayez Salama, R. Mirna 326 Derived Exosomes Is a Potential Modulator of Adiponectin in Diabetes Mellitus. Eur. J. Mol. Clin. Med. 2020, 7, 2122–2130. [Google Scholar]


{kind=link}
| Functional Impact | Target | Target Organ | Exosomal miRNA | Effect |
|---|---|---|---|---|
| Insulin receptor and PI3K signaling pathway | IRS1 and PI3K/p-Akt | Liver, Muscle | miR 29s, miR 450b-3, miR-227a, miR-27a, miR 883b-5p, miR 666-3p, miR 151-3p, miR-155 | ↓ |
| MAFB | Pancreas | miR-155 | ↓ | |
| Akt phosphorylation | Liver | miR- 141-3p | ↑ | |
| AKTIP/STAT3 | Muscle | miR-20b-5p | ↓ | |
| GLUT-4 translocation and glucose homeostasis | PI3K/p-Akt and GLUT-4 trafficking | Muscle | miR-450b-3, miR-883b-5p, miR-666-3p, miR-151-3p | ↓ |
| CERK | Muscle | miR-34a | ↓ | |
| IRS phosphorylation | Muscle | miR-155 | ↓ | |
| PGC1α | Muscle | miR-130 | ↓ | |
| PPAR-γ | Adipose tissue, Muscle, Liver | miR-130a-3p, miR 128-3p, miR-155, miR 122, miR 192, miR-227a, miR-27a | ↓ | |
| Organokine expression | FGF-21 | Liver | miR-99b | ↓ |
| Adiponectin | Adipose tissue | miR-326 | ↓ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romero, A.; Eckel, J. Organ Crosstalk and the Modulation of Insulin Signaling. Cells 2021, 10, 2082. https://doi.org/10.3390/cells10082082
Romero A, Eckel J. Organ Crosstalk and the Modulation of Insulin Signaling. Cells. 2021; 10(8):2082. https://doi.org/10.3390/cells10082082
Chicago/Turabian StyleRomero, Alejandra, and Juergen Eckel. 2021. "Organ Crosstalk and the Modulation of Insulin Signaling" Cells 10, no. 8: 2082. https://doi.org/10.3390/cells10082082
APA StyleRomero, A., & Eckel, J. (2021). Organ Crosstalk and the Modulation of Insulin Signaling. Cells, 10(8), 2082. https://doi.org/10.3390/cells10082082