
Transcriptional Landscape and Splicing Efficiency in Arabidopsis Mitochondria


Abstract
1. Introduction
2. Results and Discussion
2.1. The Mitochondrial Genome of Arabidopsis Is Entirely Transcribed with Highly Variable Levels of Read Depth
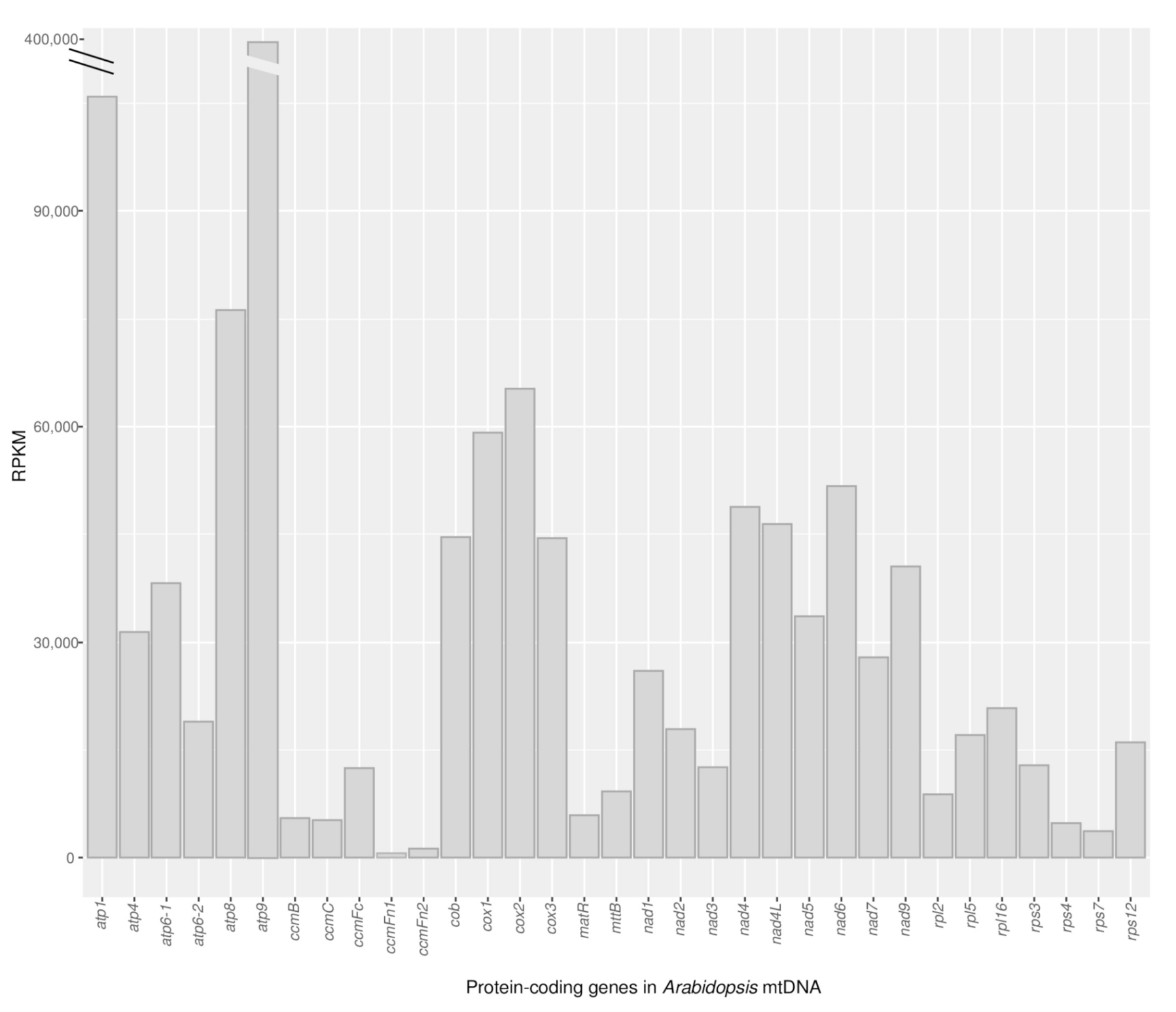
2.2. Protein-Coding Genes Show Elevated RNA Read Depth
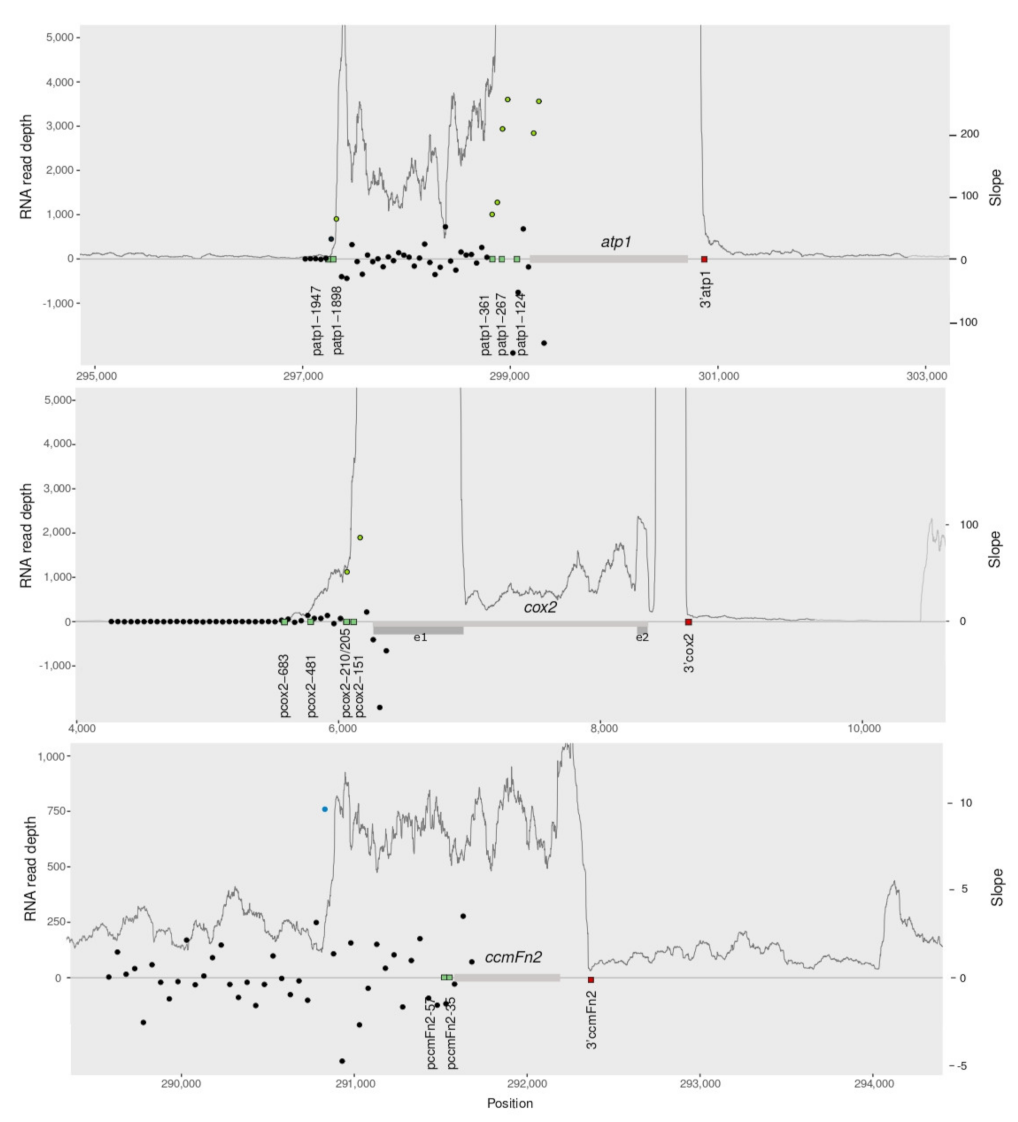
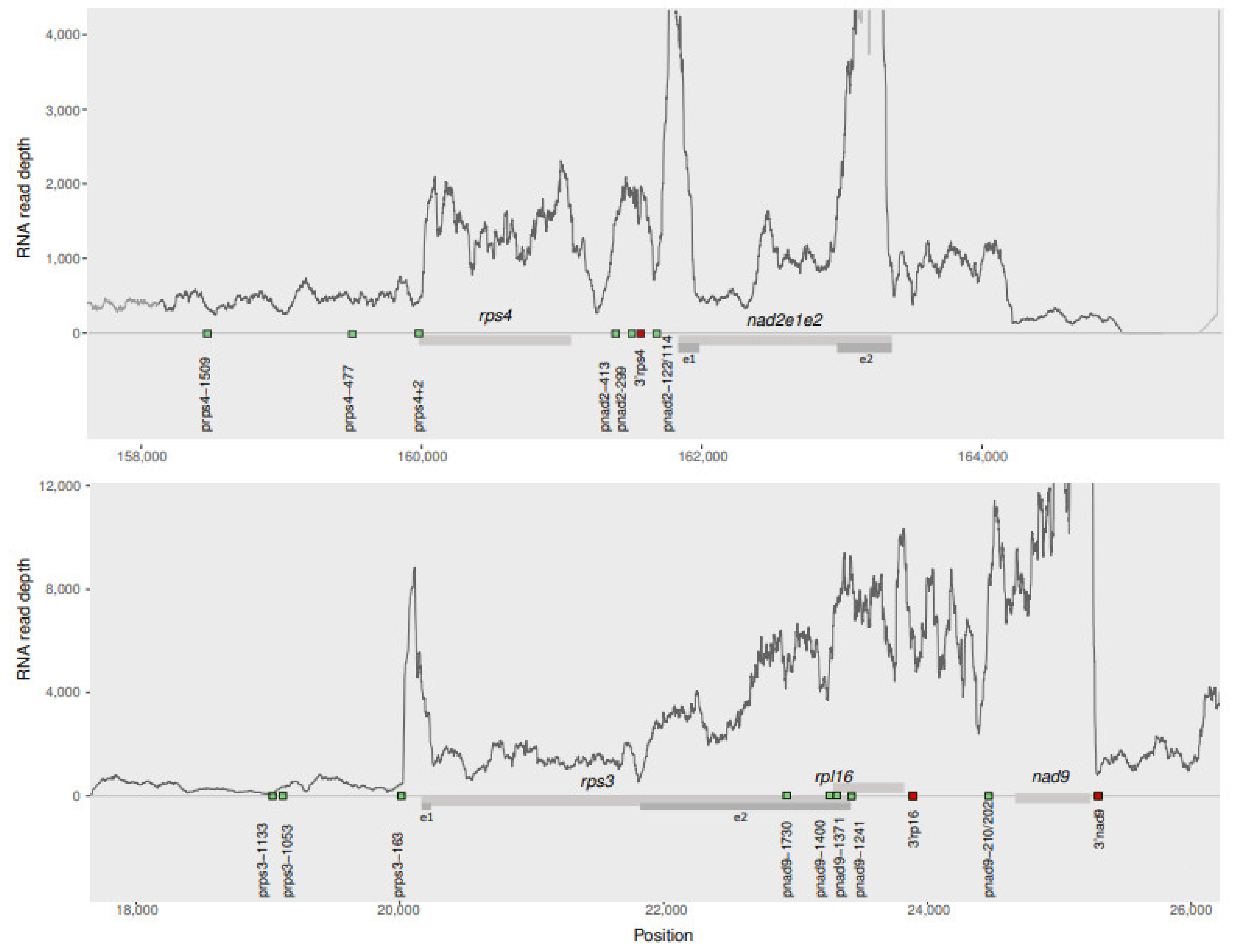
2.3. The RNAseq Transcriptional Landscape Is Useful to Infer the Region of Transcription Start
2.4. RNAseq Mapping Predicts Co-Transcription except When Genes Are Too Closely Located
2.5. Splicing Efficiency of Cis and Trans-Splicing Introns and Those with Different Splicing Mechanisms
3. Materials and Methods
3.1. RNA Data Sets
3.2. RNA Read Alignments
3.3. Initiation and Termination of Transcription Analysis
3.4. Co-Transcription Analysis
3.5. Splicing Efficiency
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gray, M.W.; Burger, G.; Lang, B.F. The origin and early evolution of mitochondria. Genome Biol. 2001, 2, 1–5. [Google Scholar] [CrossRef]
- Putintseva, Y.A.; Bondar, E.I.; Simonov, E.P.; Sharov, V.V.; Oreshkova, N.V.; Kuzmin, D.A.; Konstantinov, Y.M.; Shmakov, V.N.; Belkov, V.I.; Sadovsky, M.G.; et al. Siberian larch (Larix sibirica Ledeb.) mitochondrial genome assembled using both short and long nucleotide sequence reads is currently the largest known mitogenome. BMC Genom. 2020, 21, 1–12. [Google Scholar] [CrossRef]
- Unseld, M.; Marienfeld, J.R.; Brandt, P.; Brennicke, A. The mitochondrial genome of Arabidopsis thaliana contains 57 genes in 366,924 nucleotides. Nat. Genet. 1997, 15, 57–61. [Google Scholar] [CrossRef]
- Kubo, T.; Nishizawa, S.; Sugawara, A.; Itchoda, N.; Estiati, A.; Mikami, T. The complete nucleotide sequence of the mitochondrial genome of sugar beet (Beta vulgaris L.) reveals a novel gene for tRNACys(GCA). Nucleic Acids Res. 2000, 28, 2571–2576. [Google Scholar] [CrossRef]
- Notsu, Y.; Masood, S.; Nishikawa, T.; Kubo, N.; Akiduki, G.; Nakazono, M.; Hirai, A.; Kadowaki, K. The complete sequence of the rice (Oryza sativa L.) mitochondrial genome: Frequent DNA sequence acquisition and loss during the evolution of flowering plants. Mol. Genet. Genom. 2002, 268, 434–445. [Google Scholar] [CrossRef]
- Handa, H. The complete nucleotide sequence and RNA editing content of the mitochondrial genome of rapeseed (Brassica napus L.): Comparative analysis of the mitochondrial genomes of rapeseed and Arabidopsis thaliana. Nucleic Acids Res. 2003, 31, 5907–5916. [Google Scholar] [CrossRef] [PubMed]
- Clifton, S.W.; Minx, P.; Fauron, C.M.-R.; Gibson, M.; Allen, J.O.; Sun, H.; Thompson, M.; Barbazuk, W.; Kanuganti, S.; Tayloe, C.; et al. Sequence and Comparative Analysis of the Maize NB Mitochondrial Genome. Plant Physiol. 2004, 136, 3486–3503. [Google Scholar] [CrossRef]
- Satoh, M.; Kubo, T.; Nishizawa, S.; Estiati, A.; Itchoda, N.; Mikami, T. The cytoplasmic male-sterile type and normal type mitochondrial genomes of sugar beet share the same complement of genes of known function but differ in the content of expressed ORFs. Mol. Genet. Genom. 2004, 272, 247–256. [Google Scholar] [CrossRef]
- Ogihara, Y.; Yamazaki, Y.; Murai, K.; Kanno, A.; Terachi, T.; Shiina, T.; Miyashita, N.; Nasuda, S.; Nakamura, C.; Mori, N.; et al. Structural dynamics of cereal mitochondrial genomes as revealed by complete nucleotide sequencing of the wheat mitochondrial genome. Nucleic Acids Res. 2005, 33, 6235–6250. [Google Scholar] [CrossRef]
- Sugiyama, Y.; Watase, Y.; Nagase, M.; Makita, N.; Yagura, S.; Hirai, A.; Sugiura, M. The complete nucleotide sequence and multipartite organization of the tobacco mitochondrial genome: Comparative analysis of mitochondrial genomes in higher plants. Mol. Genet. Genom. 2004, 272, 603–615. [Google Scholar] [CrossRef]
- Gandini, C.L.; Garcia, L.E.; Abbona, C.; Sanchez-Puerta, M.V. The complete organelle genomes of Physochlaina orientalis: Insights into short sequence repeats across seed plant mitochondrial genomes. Mol. Phylogenetics Evol. 2019, 137, 274–284. [Google Scholar] [CrossRef]
- Kubo, T.; Newton, K.J. Angiosperm mitochondrial genomes and mutations. Mitochondrion 2008, 8, 5–14. [Google Scholar] [CrossRef]
- Kühn, K.; Gualberto, J.M. Chapter Nine-Recombination in the Stability, Repair and Evolution of the Mitochondrial Genome. In Advances in Botanical Research; Maréchal-Drouard, L., Ed.; Academic Press: Cambridge, MA, USA, 2012; Volume 63, pp. 215–252. ISBN 0065-2296. [Google Scholar]
- Holec, S.; Lange, H.; Kühn, K.; Alioua, M.; Börner, T.; Gagliardi, D. Relaxed Transcription in Arabidopsis Mitochondria Is Counterbalanced by RNA Stability Control Mediated by Polyadenylation and Polynucleotide Phosphorylase. Mol. Cell. Biol. 2006, 26, 2869–2876. [Google Scholar] [CrossRef] [PubMed]
- Binder, S.; And, K.S.; Stoll, B. Maturation of 5’ ends of plant mitochondrial RNAs. Physiol. Plant. 2016, 157, 280–288. [Google Scholar] [CrossRef]
- Brown, G.G.; Francs-Small, C.C.D.; Ostersetzer-Biran, O. Group II intron splicing factors in plant mitochondria. Front. Plant Sci. 2014, 5, 35. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Zmudjak, M.; Francs-Small, C.C.D.; Malik, S.; Shaya, F.; Keren, I.; Belausov, E.; Many, Y.; Brown, G.G.; Small, I.; et al. nMAT4, a maturase factor required fornad1pre-mRNA processing and maturation, is essential for holocomplex I biogenesis in Arabidopsis mitochondria. Plant J. 2014, 78, 253–268. [Google Scholar] [CrossRef]
- Gualberto, J.M.; Le Ret, M.; Beator, B.; Kühn, K. The RAD52-like protein ODB1 is required for the efficient excision of two mitochondrial introns spliced via first-step hydrolysis. Nucleic Acids Res. 2015, 43, 6500–6510. [Google Scholar] [CrossRef] [PubMed]
- Shevtsov, S.; Nevo-Dinur, K.; Faigon, L.; Sultan, L.D.; Zmudjak, M.; Markovits, M.; Ostersetzer-Biran, O. Correction: Control of organelle gene expression by the mitochondrial transcription termination factor mTERF22 in Arabidopsis thaliana plants. PLoS ONE 2019, 14, e0214558. [Google Scholar] [CrossRef]
- Giegé, P.; Hoffmann, M.; Binder, S.; Brennicke, A. RNA degradation buffers asymmetries of transcription in Arabidopsis mitochondria. EMBO Rep. 2000, 1, 164–170. [Google Scholar] [CrossRef]
- Brandt, P.; Sünkel, S.; Unseld, M.; Brennicke, A.; Knoop, V. The nad4L gene is encoded between exon c of nad5 and orf25 in the Arabidopsis mitochondrial genome. Mol. Genet. Genom. 1992, 236, 33–38. [Google Scholar] [CrossRef]
- Lippok, B.; Brennicke, A.; Unseld, M. The rps4-Gene Is Encoded Upstream of the nad2-Gene in Arabidopsis Mitochondria. Biol. Chem. Hoppe-Seyler 1996, 377, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Varré, J.-S.; D’Agostino, N.; Touzet, P.; Gallina, S.; Tamburino, R.; Cantarella, C.; Ubrig, E.; Cardi, T.; Drouard, L.; Gualberto, J.M.; et al. Complete Sequence, Multichromosomal Architecture and Transcriptome Analysis of the Solanum tuberosum Mitochondrial Genome. Int. J. Mol. Sci. 2019, 20, 4788. [Google Scholar] [CrossRef]
- Tsujimura, M.; Kaneko, T.; Sakamoto, T.; Kimura, S.; Shigyo, M.; Yamagishi, H.; Terachi, T. Multichromosomal structure of the onion mitochondrial genome and a transcript analysis. Mitochondrion 2019, 46, 179–186. [Google Scholar] [CrossRef]
- Kwasniak-Owczarek, M.; Kazmierczak, U.; Tomal, A.; Mackiewicz, P.; Janska, H. Deficiency of mitoribosomal S10 protein affects translation and splicing in Arabidopsis mitochondria. Nucleic Acids Res. 2019, 47, 11790–11806. [Google Scholar] [CrossRef]
- Sun, Y.; Xie, M.; Xu, Z.; Chan, K.C.; Zhong, J.Y.; Fan, K.; Wong-Bajracharya, J.; Lam, H.-M.; Lim, B.L. Differential RNA Editing and Intron Splicing in Soybean Mitochondria During Nodulation. Int. J. Mol. Sci. 2020, 21, 9378. [Google Scholar] [CrossRef]
- Lima, M.S.; Smith, D.R. Pervasive, Genome-Wide Transcription in the Organelle Genomes of Diverse Plastid-Bearing Protists. G3 Genes Genomes Genet. 2017, 7, 3789–3796. [Google Scholar] [CrossRef][Green Version]
- Fujii, S.; Toda, T.; Kikuchi, S.; Suzuki, R.; Yokoyama, K.; Tsuchida, H.; Yano, K.; Toriyama, K. Transcriptome map of plant mitochondria reveals islands of unexpected transcribed regions. BMC Genom. 2011, 12, 279. [Google Scholar] [CrossRef] [PubMed]
- Grimes, B.T.; Sisay, A.K.; Carroll, H.D.; Cahoon, A.B. Deep sequencing of the tobacco mitochondrial transcriptome reveals expressed ORFs and numerous editing sites outside coding regions. BMC Genom. 2014, 15, 31. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Stone, J.D.; Štorchová, H.; Sloan, D.B. High transcript abundance, RNA editing, and small RNAs in intergenic regions within the massive mitochondrial genome of the angiosperm Silene noctiflora. BMC Genom. 2015, 16, 938. [Google Scholar] [CrossRef] [PubMed]
- Silva, S.R.; Alvarenga, D.O.; Aranguren, Y.; Penha, H.; Fernandes, C.C.; Pinheiro, D.G.; Oliveira, M.T.; Michael, T.P.; Miranda, V.F.O.; Varani, A.M. The mitochondrial genome of the terrestrial carnivorous plant Utricularia reniformis (Lentibulariaceae): Structure, comparative analysis and evolutionary landmarks. PLoS ONE 2017, 12, e0180484. [Google Scholar] [CrossRef]
- Evans, D.L.; Hlongwane, T.T.; Joshi, S.V.; Pachón, D.M.R. The sugarcane mitochondrial genome: Assembly, phylogenetics and transcriptomics. PeerJ 2019, 7, e7558. [Google Scholar] [CrossRef]
- Garcia, L.E.; Edera, A.A.; Palmer, J.D.; Sato, H.; Sanchez-Puerta, M.V. Horizontal gene transfers dominate the functional mitochondrial gene space of a holoparasitic plant. New Phytol. 2021, 229, 1701–1714. [Google Scholar] [CrossRef]
- Grewe, F.; Edger, P.P.; Keren, I.; Sultan, L.; Pires, J.C.; Ostersetzer-Biran, O.; Mower, J.P. Comparative analysis of 11 Brassicales mitochondrial genomes and the mitochondrial transcriptome of Brassica oleracea. Mitochondrion 2014, 19, 135–143. [Google Scholar] [CrossRef]
- Shtratnikova, V.Y.; Schelkunov, M.I.; Penin, A.A.; Logacheva, M.D. Mitochondrial genome of the nonphotosynthetic mycoheterotrophic plant Hypopitys monotropa, its structure, gene expression and RNA editing. PeerJ 2020, 8, e9309. [Google Scholar] [CrossRef]
- Forner, J.; Weber, B.; Thuss, S.; Wildum, S.; Binder, S. Mapping of mitochondrial mRNA termini in Arabidopsis thaliana: T-elements contribute to 5′ and 3′ end formation. Nucleic Acids Res. 2007, 35, 3676–3692. [Google Scholar] [CrossRef]
- Zhao, P.; Wang, F.; Li, N.; Shi, D.-Q.; Yang, W.-C. Pentatricopeptide repeat protein MID1 modulates nad2 intron 1 splicing and Arabidopsis development. Sci. Rep. 2020, 10, 2008–2016. [Google Scholar] [CrossRef]
- Zmudjak, M.; Shevtsov, S.; Sultan, L.D.; Keren, I.; Ostersetzer-Biran, O. Analysis of the Roles of the Arabidopsis nMAT2 and PMH2 Proteins Provided with New Insights into the Regulation of Group II Intron Splicing in Land-Plant Mitochondria. Int. J. Mol. Sci. 2017, 18, 2428. [Google Scholar] [CrossRef]
- de Longevialle, A.F.; Meyer, E.; Andrés, C.; Taylor, N.L.; Lurin, C.; Millar, A.H.; Small, I.D. The Pentatricopeptide Repeat Gene OTP43 Is Required for trans-Splicing of the Mitochondrial nad1 Intron 1 in Arabidopsis thaliana. Plant Cell 2007, 19, 3256–3265. [Google Scholar] [CrossRef] [PubMed]
- Arrieta-Montiel, M.P.; Shedge, V.; Davila, J.; Christensen, A.C.; Mackenzie, S.A. Diversity of the Arabidopsis Mitochondrial Genome Occurs via Nuclear-Controlled Recombination Activity. Genetics 2009, 183, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Hedtke, B.; Börner, T.; Weihe, A. Mitochondrial and Chloroplast Phage-Type RNA Polymerases in Arabidopsis. Science 1997, 277, 809–811. [Google Scholar] [CrossRef] [PubMed]
- Kühn, K.; Richter, U.; Meyer, E.; Delannoy, E.; de Longevialle, A.F.; O’Toole, N.; Börner, T.; Millar, A.H.; Small, I.; Whelan, J. Phage-Type RNA Polymerase RPOTmp Performs Gene-Specific Transcription in Mitochondria of Arabidopsis thaliana. Plant Cell 2009, 21, 2762–2779. [Google Scholar] [CrossRef]
- Giegé, P.; Brennicke, A. RNA editing in Arabidopsis mitochondria effects 441 C to U changes in ORFs. Proc. Natl. Acad. Sci. USA 1999, 96, 15324–15329. [Google Scholar] [CrossRef]
- Kühn, K.; Weihe, A.; Börner, T. Multiple promoters are a common feature of mitochondrial genes in Arabidopsis. Nucleic Acids Res. 2005, 33, 337–346. [Google Scholar] [CrossRef]
- Wu, Z.; Hu, K.; Yan, M.; Song, L.; Wen, J.; Ma, C.; Shen, J.; Fu, T.; Yi, B.; Tu, J. Mitochondrial genome and transcriptome analysis of five alloplasmic male-sterile lines in Brassica juncea. BMC Genom. 2019, 20, 348. [Google Scholar] [CrossRef]
- Sloan, D.B.; Wu, Z.; Sharbrough, J. Correction of Persistent Errors in Arabidopsis Reference Mitochondrial Genomes. Plant Cell 2018, 30, 525–527. [Google Scholar] [CrossRef]
- Kubo, T.; Mikami, T. Organization and variation of angiosperm mitochondrial genome. Physiol. Plant. 2007, 129, 6–13. [Google Scholar] [CrossRef]
- Forner, J.; Hölzle, A.; Jonietz, C.; Thuss, S.; Schwarzländer, M.; Weber, B.; Meyer, R.C.; Binder, S. Mitochondrial mRNA Polymorphisms in Different Arabidopsis Accessions. Plant Physiol. 2008, 148, 1106–1116. [Google Scholar] [CrossRef]
- Qiu, Y.; Filipenko, S.J.; Darracq, A.; Adams, K.L. Expression of a transferred nuclear gene in a mitochondrial genome. Curr. Plant Biol. 2014, 1, 68–72. [Google Scholar] [CrossRef]
- Planchard, N.; Bertin, P.; Quadrado, M.; Dargel-Graffin, C.; Hatin, I.; Namy, O.; Mireau, H. The translational landscape of Arabidopsis mitochondria. Nucleic Acids Res. 2018, 46, 6218–6228. [Google Scholar] [CrossRef]
- Marienfeld, J.; Unseld, M.; Brandt, P.; Brennicke, A. Genomic Recombination of the Mitochondrial atp6 Gene in Arabidopsis thaliana at the Protein Processing Site Creates Two Different Presequences. DNA Res. 1996, 3, 287–290. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Binder, S.; Brennicke, A. Gene expression in plant mitochondria: Transcriptional and post–transcriptional control. Philos. Trans. R. Soc. B Biol. Sci. 2003, 358, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Raczynska, K.D.; Le Ret, M.; Rurek, M.; Bonnard, G.; Augustyniak, H.; Gualberto, J.M. Plant mitochondrial genes can be expressed from mRNAs lacking stop codons. FEBS Lett. 2006, 580, 5641–5646. [Google Scholar] [CrossRef]
- Gagliardi, D.; Gualberto, J.M. Gene Expression in Higher Plant Mitochondria. In Plant Mitochondria: From Genome to Function; Day, D.A., Millar, A.H., Whelan, J., Eds.; Springer: Dordrecht, The Netherlands, 2004; pp. 55–81. ISBN 978-1-4020-2400-9. [Google Scholar]
- Newton, K.J.; Walbot, V. Maize mitochondria synthesize organ-specific polypeptides. Proc. Natl. Acad. Sci. USA 1985, 82, 6879–6883. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, D.; Binder, S. Expression of the Plant Mitochondrial Genome. In Annual Plant Reviews Online; American Cancer Society: Atlanta, GA, USA, 2018; pp. 50–96. ISBN 978-1-119-31299-4. [Google Scholar]
- Bonen, L. Cis- and trans-splicing of group II introns in plant mitochondria. Mitochondrion 2008, 8, 26–34. [Google Scholar] [CrossRef]
- Schmitz-Linneweber, C.; Lampe, M.-K.; Sultan, L.D.; Ostersetzer-Biran, O. Organellar maturases: A window into the evolution of the spliceosome. Biochim. et Biophys. Acta (BBA)-Bioenerg. 2015, 1847, 798–808. [Google Scholar] [CrossRef]
- Lambowitz, A.M.; Zimmerly, S. Group II Introns: Mobile Ribozymes that Invade DNA. Cold Spring Harb. Perspect. Biol. 2010, 3, a003616. [Google Scholar] [CrossRef]
- Podar, M.; Chu, V.T.; Pyle, A.M.; Perlman, P.S. Group II intron splicing in vivo by first-step hydrolysis. Nat. Cell Biol. 1998, 391, 915–918. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kuhn, J.; Tengler, U.; Binder, S. Transcript Lifetime Is Balanced between Stabilizing Stem-Loop Structures and Degradation-Promoting Polyadenylation in Plant Mitochondria. Mol. Cell. Biol. 2001, 21, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, T.; Matsuura, T.; Ushiyama, S.; Narusaka, M.; Kurihara, Y.; Yasuda, M.; Ohtani, M.; Seki, M.; Demura, T.; Nakashita, H.; et al. A poly(A)-specific ribonuclease directly regulates the poly(A) status of mitochondrial mRNA in Arabidopsis. Nat. Commun. 2013, 4, 2247. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suit of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Xia, X. RNA-Seq approach for accurate characterization of splicing efficiency of yeast introns. Methods 2020, 176, 25–33. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Cis or Trans-Splicing a | Splicing Mechanism b | RNA Read Depth | I3′E/EI5′ | Splicing Efficiency (SE) | |||
|---|---|---|---|---|---|---|---|
| EI5′ c | I3′E d | EE e | |||||
| ccmFci | Cis-splicing | Branching pathway | 192 | 356 | 1149 | 1.85 | 0.81 |
| cox2i | Cis-splicing | Branching pathway | 129 | 282 | 12,433 | 2.19 | 0.98 |
| nad1i1 | Trans-splicing | Hydrolytic pathway | 180 | 1197 | 944 | 6.65 | 0.58 |
| nad1i2 | Cis-splicing | Hydrolytic pathway | 151 | 174 | 2322 | 1.15 | 0.93 |
| nad1i3 | Trans-splicing | Both pathways were reported | 187 | 945 | 3210 | 5.05 | 0.85 |
| nad1i4 | Cis-splicing | Branching pathway | 240 | 1280 | 1982 | 5.33 | 0.72 |
| nad2i1 | Cis-splicing | Hydrolytic pathway | 141 | 437 | 919 | 3.10 | 0.76 |
| nad2i2 | Trans-splicing | Branching pathway | 54 | 476 | 1958 | 8.81 | 0.88 |
| nad2i3 | Cis-splicing | Branching pathway | 59 | 581 | 1471 | 9.85 | 0.82 |
| nad2i4 | Cis-splicing | Branching pathway | 389 | 460 | 2688 | 1.18 | 0.86 |
| nad4i1 | Cis-splicing | Both pathways were reported | 480 | 633 | 2498 | 1.32 | 0.82 |
| nad4i2 | Cis-splicing | Hydrolytic pathway | 144 | 125 | 6924 | 0.87 | 0.98 |
| nad4i3 | Cis-splicing | Branching pathway | 163 | 468 | 8402 | 2.87 | 0.96 |
| nad5i1 | Cis-splicing | Branching pathway | 278 | 817 | 1452 | 2.94 | 0.73 |
| nad5i4 | Cis-splicing | Branching pathway | 189 | 1031 | 17,008 | 5.46 | 0.97 |
| nad7i1 | Cis-splicing | Hydrolytic pathway | 75 | 135 | 639 | 1.80 | 0.86 |
| nad7i2 | Cis-splicing | Branching pathway | 177 | 1379 | 1169 | 7.79 | 0.60 |
| nad7i3 | Cis-splicing | Branching pathway | 104 | 332 | 4001 | 3.19 | 0.95 |
| nad7i4 | Cis-intron | Branching pathway | 209 | 304 | 2685 | 1.45 | 0.91 |
| rpl2i | Cis-splicing | Both pathways were reported | 122 | 685 | 465 | 5.61 | 0.54 |
| rps3i | Cis-splicing | Branching pathway | 496 | 211 | 734 | 0.43 | 0.67 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia, L.E.; Sanchez-Puerta, M.V. Transcriptional Landscape and Splicing Efficiency in Arabidopsis Mitochondria. Cells 2021, 10, 2054. https://doi.org/10.3390/cells10082054
Garcia LE, Sanchez-Puerta MV. Transcriptional Landscape and Splicing Efficiency in Arabidopsis Mitochondria. Cells. 2021; 10(8):2054. https://doi.org/10.3390/cells10082054
Chicago/Turabian StyleGarcia, Laura E., and M. Virginia Sanchez-Puerta. 2021. "Transcriptional Landscape and Splicing Efficiency in Arabidopsis Mitochondria" Cells 10, no. 8: 2054. https://doi.org/10.3390/cells10082054
APA StyleGarcia, L. E., & Sanchez-Puerta, M. V. (2021). Transcriptional Landscape and Splicing Efficiency in Arabidopsis Mitochondria. Cells, 10(8), 2054. https://doi.org/10.3390/cells10082054