
Calpain-Mediated Mitochondrial Damage: An Emerging Mechanism Contributing to Cardiac Disease

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Regulation of Calpain System
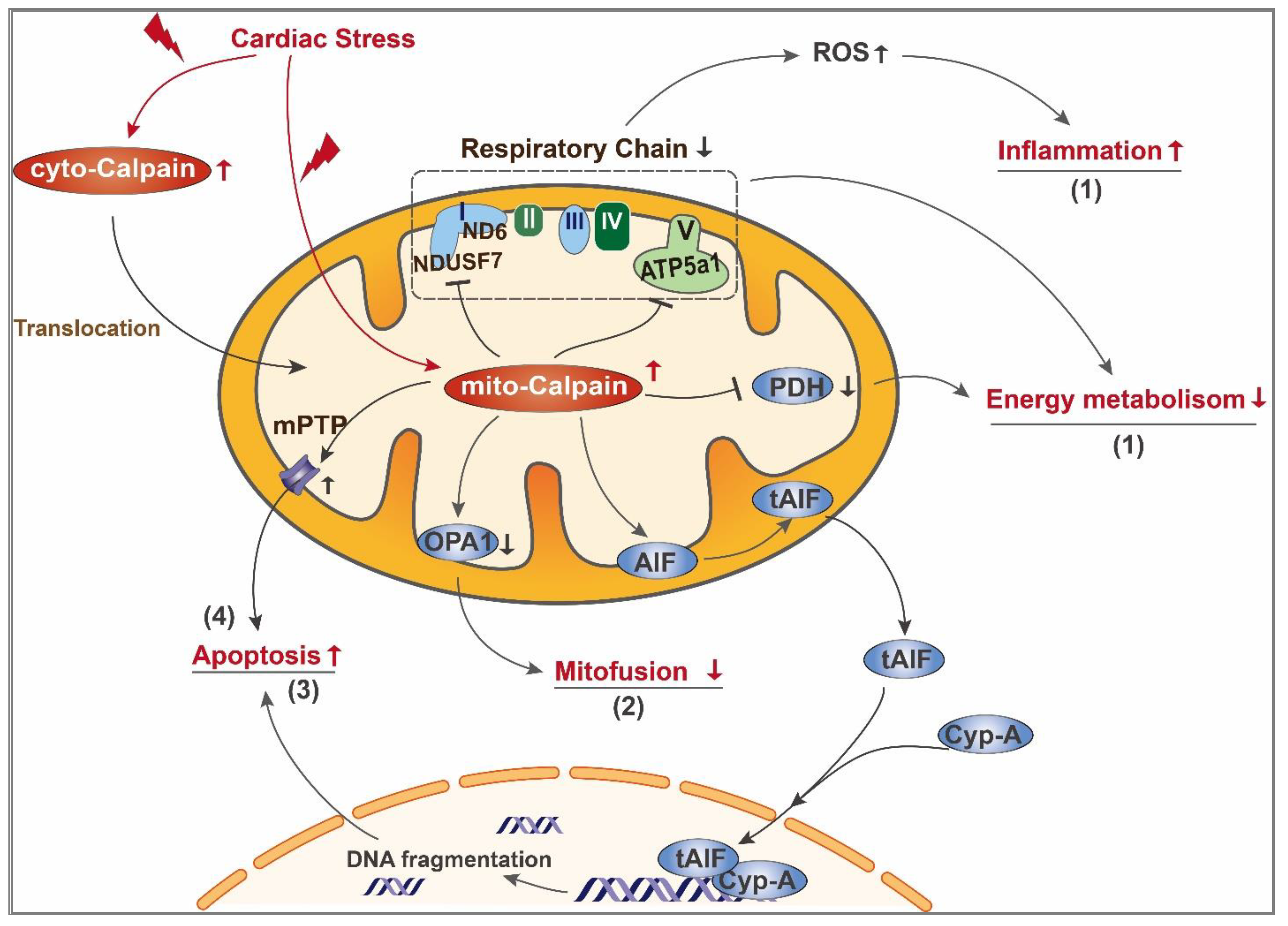
3. Role of Calpain in Mediating Mitochondrial Damage during Cardiac Disease
3.1. Cyto-Calpain-Mediated Mitochondrial Damage
3.1.1. Impairment of Mitochondrial Quality Control
3.1.2. Initiating Mitochondria-Dependent Apoptosis
3.1.3. Indirectly Influencing Mitochondrial Biogenesis
3.1.4. Others
3.2. Mito-Calpain-Mediated Mitochondrial Damage
3.2.1. Impairment of Mitochondrial Energy Metabolism
3.2.2. Imbalance of Mitochondrial Fission and Fusion
3.2.3. Mitochondrial Apoptosis
4. Precisely Targeted Inhibition of Mito-Calpain as a Potential Therapeutic Strategy for Cardiac Disease
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cardiovascular Diseases (CVDs). Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 3 April 2021).
- Protasoni, M.; Zeviani, M. Mitochondrial Structure and Bioenergetics in Normal and Disease Conditions. Int. J. Mol. Sci. 2021, 22, 586. [Google Scholar] [CrossRef]
- Melber, A.; Haynes, C.M. UPRmt Regulation and Output: A Stress Response Mediated by Mitochondrial-Nuclear Communication. Cell Res. 2018, 28, 281–295. [Google Scholar] [CrossRef]
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac Metabolism in Heart Failure-Implications beyond ATP Production. Circ. Res. 2013, 113, 709–724. [Google Scholar] [CrossRef]
- Hom, J.; Sheu, S.-S. Morphological Dynamics of Mitochondria—A Special Emphasis on Cardiac Muscle Cells. J. Mol. Cell. Cardiol. 2009, 46, 811–820. [Google Scholar] [CrossRef]
- Schilling, J.D. The Mitochondria in Diabetic Heart Failure: From Pathogenesis to Therapeutic Promise. Antioxid Redox Signal. 2015, 22, 1515–1526. [Google Scholar] [CrossRef] [PubMed]
- Montaigne, D.; Marechal, X.; Coisne, A.; Debry, N.; Modine, T.; Fayad, G.; Potelle, C.; El Arid, J.-M.; Mouton, S.; Sebti, Y.; et al. Myocardial Contractile Dysfunction Is Associated With Impaired Mitochondrial Function and Dynamics in Type 2 Diabetic but Not in Obese Patients. Circulation 2014, 130, 554–564. [Google Scholar] [CrossRef]
- Del Re, D.P.; Amgalan, D.; Linkermann, A.; Liu, Q.; Kitsis, R.N. Fundamental Mechanisms of Regulated Cell Death and Implications for Heart Disease. Physiol. Rev. 2019, 99, 1765–1817. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Tian, R. Mitochondrial Dysfunction in Pathophysiology of Heart Failure. J. Clin. Investig. 2018, 128, 3716–3726. [Google Scholar] [CrossRef]
- Chen, Q.; Thompson, J.; Hu, Y.; Dean, J.; Lesnefsky, E.J. Inhibition of the Ubiquitous Calpains Protects Complex I Activity and Enables Improved Mitophagy in the Heart Following Ischemia-Reperfusion. Am. J. Physiol. Cell Physiol. 2019, 317, C910–C921. [Google Scholar] [CrossRef] [PubMed]
- Neuhof, C.; Neuhof, H. Calpain System and Its Involvement in Myocardial Ischemia and Reperfusion Injury. World J. Cardiol. 2014, 6, 638–652. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, B.; Huang, C.-K.; Guo, A.; Wu, J.; Zhang, X.; Chen, R.; Chen, C.; Kutschke, W.; Weiss, R.M.; et al. Targeting Calpain for Heart Failure Therapy: Implications From Multiple Murine Models. JACC Basic Transl. Sci. 2018, 3, 503–517. [Google Scholar] [CrossRef] [PubMed]
- Yi, C.; Wu, W.; Zheng, D.; Peng, G.; Huang, H.; Shen, Z.; Teng, X. Targeted Inhibition of Endothelial Calpain Delays Wound Healing by Reducing Inflammation and Angiogenesis. Cell Death Dis. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Carragher, N.O.; Frame, M.C. Calpain: A Role in Cell Transformation and Migration. Int. J. Biochem. Cell Biol. 2002, 34, 1539–1543. [Google Scholar] [CrossRef]
- Davis, M.A.; Fairgrieve, M.R.; Den Hartigh, A.; Yakovenko, O.; Duvvuri, B.; Lood, C.; Thomas, W.E.; Fink, S.L.; Gale, M. Calpain Drives Pyroptotic Vimentin Cleavage, Intermediate Filament Loss, and Cell Rupture That Mediates Immunostimulation. Proc. Natl. Acad. Sci. USA 2019, 116, 5061–5070. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kain, V.; Sitasawad, S.L. High Glucose-Induced Ca2+ Overload and Oxidative Stress Contribute to Apoptosis of Cardiac Cells through Mitochondrial Dependent and Independent Pathways. Biochim. Biophys. Acta 2012, 1820, 907–920. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Zhang, T.; Sun, L.; Luo, Y.; Liu, D.-H.; Xie, S.-T.; Song, X.-Y.; Wang, G.-F.; Chen, X.-L.; Zhou, B.-C.; et al. Calpain, Atg5 and Bak Play Important Roles in the Crosstalk between Apoptosis and Autophagy Induced by Influx of Extracellular Calcium. Apoptosis 2013, 18, 435–451. [Google Scholar] [CrossRef]
- Yu, Y.; Shi, H.; Yu, Y.; Liu, M.; Li, M.; Liu, X.; Wang, Y.; Chen, R. Inhibition of Calpain Alleviates Coxsackievirus B3-Induced Myocarditis through Suppressing the Canonical NLRP3 Inflammasome/Caspase-1-Mediated and Noncanonical Caspase-11-Mediated Pyroptosis Pathways. Am. J. Transl. Res. 2020, 12, 1954–1964. [Google Scholar]
- Galvez, A.S.; Diwan, A.; Odley, A.M.; Hahn, H.S.; Osinska, H.; Melendez, J.G.; Robbins, J.; Lynch, R.A.; Marreez, Y.; Dorn, G.W. Cardiomyocyte Degeneration with Calpain Deficiency Reveals a Critical Role in Protein Homeostasis. Circ. Res. 2007, 100, 1071–1078. [Google Scholar] [CrossRef]
- Taneike, M.; Mizote, I.; Morita, T.; Watanabe, T.; Hikoso, S.; Yamaguchi, O.; Takeda, T.; Oka, T.; Tamai, T.; Oyabu, J.; et al. Calpain Protects the Heart from Hemodynamic Stress. J. Biol. Chem. 2011, 286, 32170–32177. [Google Scholar] [CrossRef]
- Zheng, X.; Zhou, A.-X.; Rouhi, P.; Uramoto, H.; Borén, J.; Cao, Y.; Pereira, T.; Akyürek, L.M.; Poellinger, L. Hypoxia-Induced and Calpain-Dependent Cleavage of Filamin A Regulates the Hypoxic Response. Proc. Natl. Acad. Sci. USA 2014, 111, 2560–2565. [Google Scholar] [CrossRef]
- Lee, S.H.; Wolf, P.L.; Escudero, R.; Deutsch, R.; Jamieson, S.W.; Thistlethwaite, P.A. Early Expression of Angiogenesis Factors in Acute Myocardial Ischemia and Infarction. N. Engl. J. Med. 2000, 342, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Chen, P.; Zhong, J.; Cheng, Y.; Chen, H.; He, Y.; Chen, C. HIF-1α in Myocardial Ischemia-Reperfusion Injury. Mol. Med. Rep. 2021, 23, 352. [Google Scholar] [CrossRef]
- Mo, X.-G.; Chen, Q.-W.; Li, X.-S.; Zheng, M.-M.; Ke, D.-Z.; Deng, W.; Li, G.-Q.; Jiang, J.; Wu, Z.-Q.; Wang, L.; et al. Suppression of NHE1 by Small Interfering RNA Inhibits HIF-1α-Induced Angiogenesis in Vitro via Modulation of Calpain Activity. Microvasc. Res. 2011, 81, 160–168. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, N.M.; Wang, Y.; Youn, J.Y.; Cai, H. Endothelial Cell Calpain as a Critical Modulator of Angiogenesis. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1326–1335. [Google Scholar] [CrossRef]
- Han, Q.; Liu, Q.; Zhang, H.; Lu, M.; Wang, H.; Tang, F.; Zhang, Y. Simvastatin Improves Cardiac Hypertrophy in Diabetic Rats by Attenuation of Oxidative Stress and Inflammation Induced by Calpain-1-Mediated Activation of Nuclear Factor-ΚB (NF-ΚB). Med. Sci. Monit. 2019, 25, 1232–1241. [Google Scholar] [CrossRef]
- Dong, L.-Y.; Yao, L.-P.; Zhao, J.; Jin, K.-K.; Qiu, X.-X. Captopril Inhibits Calpain-mediated Apoptosis of Myocardial Cells in Diabetic Rats and Improves Cardiac Function. Mol. Med. Rep. 2018, 18, 2300–2306. [Google Scholar] [CrossRef]
- Chen, K.; He, L.; Li, Y.; Li, X.; Qiu, C.; Pei, H.; Yang, D. Inhibition of GPR35 Preserves Mitochondrial Function After Myocardial Infarction by Targeting Calpain 1/2. J. Cardiovasc. Pharm. 2020, 75, 556–563. [Google Scholar] [CrossRef]
- Lu, H.-T.; Feng, R.-Q.; Tang, J.-K.; Zhou, J.-J.; Gao, F.; Ren, J. CaMKII/Calpain Interaction Mediates Ischemia/Reperfusion Injury in Isolated Rat Hearts. Cell Death Dis. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Freitas, A.C.S.; Figueiredo, M.J.; Campos, E.C.; Soave, D.F.; Ramos, S.G.; Tanowitz, H.B.; Celes, M.R.N. Activation of Both the Calpain and Ubiquitin-Proteasome Systems Contributes to Septic Cardiomyopathy through Dystrophin Loss/Disruption and MTOR Inhibition. PLoS ONE 2016, 11, e0166839. [Google Scholar] [CrossRef]
- Portbury, A.L.; Willis, M.S.; Patterson, C. Tearin’ Up My Heart: Proteolysis in the Cardiac Sarcomere. J. Biol. Chem. 2011, 286, 9929–9934. [Google Scholar] [CrossRef]
- Patterson, C.; Portbury, A.; Schisler, J.C.; Willis, M.S. Tear Me down: Role of Calpain in the Development of Cardiac Ventricular Hypertrophy. Circ. Res. 2011, 109, 453–462. [Google Scholar] [CrossRef]
- Wang, S.; Wang, H.; Su, X.; Liu, B.; Wang, L.; Yan, H.; Mao, S.; Huang, H.; Huang, C.; Cheng, M.; et al. β-Adrenergic Activation May Promote Myosin Light Chain Kinase Degradation through Calpain in Pressure Overload-Induced Cardiac Hypertrophy: β-Adrenergic Activation Results in MLCK Degradation. Biomed. Pharmacother. 2020, 129, 110438. [Google Scholar] [CrossRef]
- Guo, A.; Wang, Y.; Chen, B.; Wang, Y.; Yuan, J.; Zhang, L.; Hall, D.; Wu, J.; Shi, Y.; Zhu, Q.; et al. E-C Coupling Structural Protein Junctophilin-2 Encodes a Stress-Adaptive Transcription Regulator. Science 2018, 362, 1359–1360. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, S.K.; Quick, A.P.; Samson-Couterie, B.; Hulsurkar, M.; Elzenaar, I.; van Oort, R.J.; Wehrens, X.H.T. Nuclear Localization of a Novel Calpain-2 Mediated Junctophilin-2 C-Terminal Cleavage Peptide Promotes Cardiomyocyte Remodeling. Basic Res. Cardiol. 2020, 115, 49. [Google Scholar] [CrossRef]
- Ni, R.; Zheng, D.; Xiong, S.; Hill, D.J.; Sun, T.; Gardiner, R.B.; Fan, G.-C.; Lu, Y.; Abel, E.D.; Greer, P.A.; et al. Mitochondrial Calpain-1 Disrupts ATP Synthase and Induces Superoxide Generation in Type 1 Diabetic Hearts: A Novel Mechanism Contributing to Diabetic Cardiomyopathy. Diabetes 2016, 65, 255–268. [Google Scholar] [CrossRef]
- Ni, R.; Zheng, D.; Wang, Q.; Yu, Y.; Chen, R.; Sun, T.; Wang, W.; Fan, G.-C.; Greer, P.A.; Gardiner, R.B.; et al. Deletion of Capn4 Protects the Heart Against Endotoxemic Injury by Preventing ATP Synthase Disruption and Inhibiting Mitochondrial Superoxide Generation. Circ. Heart Fail. 2015, 8, 988–996. [Google Scholar] [CrossRef]
- Liang, L.; Li, H.; Cao, T.; Qu, L.; Zhang, L.; Fan, G.-C.; Greer, P.A.; Li, J.; Jones, D.L.; Peng, T. Calpain Activation Mediates Microgravity-Induced Myocardial Abnormalities in Mice via P38 and ERK1/2 MAPK Pathways. J. Biol. Chem. 2020, 295, 16840–16851. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhang, L.; Ni, R.; Cao, T.; Zheng, D.; Xiong, S.; Greer, P.A.; Fan, G.-C.; Peng, T. Disruption of Calpain Reduces Lipotoxicity-Induced Cardiac Injury by Preventing Endoplasmic Reticulum Stress. Biochim. Biophys. Acta 2016, 1862, 2023–2033. [Google Scholar] [CrossRef]
- Cao, T.; Fan, S.; Zheng, D.; Wang, G.; Yu, Y.; Chen, R.; Song, L.-S.; Fan, G.-C.; Zhang, Z.; Peng, T. Increased Calpain-1 in Mitochondria Induces Dilated Heart Failure in Mice: Role of Mitochondrial Superoxide Anion. Basic Res. Cardiol. 2019, 114, 17. [Google Scholar] [CrossRef] [PubMed]
- Teng, X.; Ji, C.; Zhong, H.; Zheng, D.; Ni, R.; Hill, D.J.; Xiong, S.; Fan, G.-C.; Greer, P.A.; Shen, Z.; et al. Selective Deletion of Endothelial Cell Calpain in Mice Reduces Diabetic Cardiomyopathy by Improving Angiogenesis. Diabetologia 2019, 62, 860–872. [Google Scholar] [CrossRef]
- Zheng, D.; Cao, T.; Zhang, L.-L.; Fan, G.-C.; Qiu, J.; Peng, T.-Q. Targeted Inhibition of Calpain in Mitochondria Alleviates Oxidative Stress-Induced Myocardial Injury. Acta Pharmacol. Sin. 2021, 42, 909–920. [Google Scholar] [CrossRef]
- Yue, R.-C.; Lu, S.-Z.; Luo, Y.; Wang, T.; Liang, H.; Zeng, J.; Liu, J.; Hu, H.-X. Calpain Silencing Alleviates Myocardial Ischemia-Reperfusion Injury through the NLRP3/ASC/Caspase-1 Axis in Mice. Life Sci. 2019, 233, 116631. [Google Scholar] [CrossRef]
- Li, X.; Luo, R.; Chen, R.; Song, L.; Zhang, S.; Hua, W.; Chen, H. Cleavage of IκBα by Calpain Induces Myocardial NF-ΚB Activation, TNF-α Expression, and Cardiac Dysfunction in Septic Mice. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H833–H843. [Google Scholar] [CrossRef]
- Xiao, T.-T.; Wang, Y.-Y.; Zhang, Y.; Bai, C.-H.; Shen, X.-C. Similar to Spironolactone, Oxymatrine Is Protective in Aldosterone-Induced Cardiomyocyte Injury via Inhibition of Calpain and Apoptosis-Inducing Factor Signaling. PLoS ONE 2014, 9, e88856. [Google Scholar] [CrossRef]
- Li, W.; Yang, J.; Lyu, Q.; Wu, G.; Lin, S.; Yang, Q.; Hu, J. Taurine Attenuates Isoproterenol-Induced H9c2 Cardiomyocytes Hypertrophy by Improving Antioxidative Ability and Inhibiting Calpain-1-Mediated Apoptosis. Mol. Cell. Biochem. 2020, 469, 119–132. [Google Scholar] [CrossRef]
- Letavernier, E.; Perez, J.; Bellocq, A.; Mesnard, L.; de Castro Keller, A.; Haymann, J.-P.; Baud, L. Targeting the Calpain/Calpastatin System as a New Strategy to Prevent Cardiovascular Remodeling in Angiotensin II-Induced Hypertension. Circ. Res. 2008, 102, 720–728. [Google Scholar] [CrossRef]
- Miyazaki, T.; Akasu, R.; Miyazaki, A. Calpain Proteolytic Systems Counteract Endothelial Cell Adaptation to Inflammatory Environments. Inflamm. Regen. 2020, 40, 5. [Google Scholar] [CrossRef]
- Chen, Q.; Lesnefsky, E.J. Heart Mitochondria and Calpain 1: Location, Function, and Targets. Biochim. Biophys. Acta 2015, 1852, 2372–2378. [Google Scholar] [CrossRef]
- Arrington, D.D.; Van Vleet, T.R.; Schnellmann, R.G. Calpain 10: A Mitochondrial Calpain and Its Role in Calcium-Induced Mitochondrial Dysfunction. Am. J. Physiol. Cell Physiol. 2006, 291, C1159–C1171. [Google Scholar] [CrossRef]
- Kumchantuek, T.; Nakata, H.; Sakulsak, N.; Yamamoto, M.; Iseki, S. Expression and Localization of Calpain 3 in the Submandibular Gland of Mice. Arch. Oral Biol. 2016, 70, 9–15. [Google Scholar] [CrossRef]
- Hata, S.; Abe, M.; Suzuki, H.; Kitamura, F.; Toyama-Sorimachi, N.; Abe, K.; Sakimura, K.; Sorimachi, H. Calpain 8/NCL-2 and Calpain 9/NCL-4 Constitute an Active Protease Complex, G-Calpain, Involved in Gastric Mucosal Defense. PLoS Genet. 2010, 6, e1001040. [Google Scholar] [CrossRef] [PubMed]
- Pandurangan, M.; Hwang, I.; Orhirbat, C.; Jieun, Y.; Cho, S.-H. The Calpain System and Diabetes. Pathophysiology 2014, 21, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Wei, M.; Wang, Q.; Li, J.; Wang, H.; Liu, W.; Lacefield, J.C.; Greer, P.A.; Karmazyn, M.; Fan, G.-C.; et al. Deficiency of Capn4 Gene Inhibits Nuclear Factor-ΚB (NF-ΚB) Protein Signaling/Inflammation and Reduces Remodeling after Myocardial Infarction. J. Biol. Chem. 2012, 287, 27480–27489. [Google Scholar] [CrossRef] [PubMed]
- Hanna, R.A.; Garcia-Diaz, B.E.; Davies, P.L. Calpastatin Simultaneously Binds Four Calpains with Different Kinetic Constants. FEBS Lett. 2007, 581, 2894–2898. [Google Scholar] [CrossRef] [PubMed]
- Wanichawan, P.; Hafver, T.L.; Hodne, K.; Aronsen, J.M.; Lunde, I.G.; Dalhus, B.; Lunde, M.; Kvaløy, H.; Louch, W.E.; Tønnessen, T.; et al. Molecular Basis of Calpain Cleavage and Inactivation of the Sodium-Calcium Exchanger 1 in Heart Failure. J. Biol. Chem. 2014, 289, 33984–33998. [Google Scholar] [CrossRef]
- Sandmann, S.; Yu, M.; Unger, T. Transcriptional and Translational Regulation of Calpain in the Rat Heart after Myocardial Infarction – Effects of AT1 and AT2 Receptor Antagonists and ACE Inhibitor. Br. J. Pharmacol. 2001, 132, 767–777. [Google Scholar] [CrossRef]
- Loonat, A.A.; Martin, E.D.; Sarafraz-Shekary, N.; Tilgner, K.; Hertz, N.T.; Levin, R.; Shokat, K.M.; Burlingame, A.L.; Arabacilar, P.; Uddin, S.; et al. P38γ MAPK Contributes to Left Ventricular Remodeling after Pathologic Stress and Disinhibits Calpain through Phosphorylation of Calpastatin. FASEB J. 2019, 33, 13131–13144. [Google Scholar] [CrossRef]
- Glading, A.; Chang, P.; Lauffenburger, D.A.; Wells, A. Epidermal Growth Factor Receptor Activation of Calpain Is Required for Fibroblast Motility and Occurs via an ERK/MAP Kinase Signaling Pathway *. J. Biol. Chem. 2000, 275, 2390–2398. [Google Scholar] [CrossRef]
- Leloup, L.; Daury, L.; Mazères, G.; Cottin, P.; Brustis, J.-J. Involvement of the ERK/MAP Kinase Signalling Pathway in Milli-Calpain Activation and Myogenic Cell Migration. Int. J. Biochem. Cell Biol. 2007, 39, 1177–1189. [Google Scholar] [CrossRef]
- Leloup, L.; Shao, H.; Bae, Y.H.; Deasy, B.; Stolz, D.; Roy, P.; Wells, A. M-Calpain Activation Is Regulated by Its Membrane Localization and by Its Binding to Phosphatidylinositol 4,5-Bisphosphate. J. Biol. Chem. 2010, 285, 33549–33566. [Google Scholar] [CrossRef]
- Randriamboavonjy, V.; Kyselova, A.; Fleming, I. Redox Regulation of Calpains: Consequences on Vascular Function. Antioxid. Redox Signal. 2019, 30, 1011–1026. [Google Scholar] [CrossRef]
- Rios, F.J.; Zou, Z.-G.; Harvey, A.P.; Harvey, K.Y.; Nosalski, R.; Anyfanti, P.; Camargo, L.L.; Lacchini, S.; Ryazanov, A.G.; Ryazanova, L.; et al. Chanzyme TRPM7 Protects against Cardiovascular Inflammation and Fibrosis. Cardiovasc. Res. 2020, 116, 721–735. [Google Scholar] [CrossRef]
- Hernando, V.; Inserte, J.; Sartório, C.L.; Parra, V.M.; Poncelas-Nozal, M.; Garcia-Dorado, D. Calpain Translocation and Activation as Pharmacological Targets during Myocardial Ischemia/Reperfusion. J. Mol. Cell. Cardiol. 2010, 49, 271–279. [Google Scholar] [CrossRef]
- Bi, S.-H.; Jin, Z.-X.; Zhang, J.-Y.; Chen, T.; Zhang, S.-L.; Yang, Y.; Duan, W.-X.; Yi, D.-H.; Zhou, J.-J.; Ren, J. Calpain Inhibitor MDL 28170 Protects against the Ca2+ Paradox in Rat Hearts. Clin. Exp. Pharmacol. Physiol. 2012, 39, 385–392. [Google Scholar] [CrossRef]
- Chang, H.; Sheng, J.-J.; Zhang, L.; Yue, Z.-J.; Jiao, B.; Li, J.-S.; Yu, Z.-B. ROS-Induced Nuclear Translocation of Calpain-2 Facilitates Cardiomyocyte Apoptosis in Tail-Suspended Rats. J. Cell. Biochem. 2015, 116, 2258–2269. [Google Scholar] [CrossRef]
- Sheng, J.-J.; Chang, H.; Yu, Z.-B. Nuclear Translocation of Calpain-2 Mediates Apoptosis of Hypertrophied Cardiomyocytes in Transverse Aortic Constriction Rat. J. Cell. Physiol. 2015, 230, 2743–2754. [Google Scholar] [CrossRef]
- Tombo, N.; Imam Aliagan, A.D.; Feng, Y.; Singh, H.; Bopassa, J.C. Cardiac Ischemia/Reperfusion Stress Reduces Inner Mitochondrial Membrane Protein (Mitofilin) Levels during Early Reperfusion. Free Radic. Biol. Med. 2020, 158, 181–194. [Google Scholar] [CrossRef]
- Shintani-Ishida, K.; Yoshida, K.-I. Mitochondrial M-Calpain Opens the Mitochondrial Permeability Transition Pore in Ischemia-Reperfusion. Int. J. Cardiol. 2015, 197, 26–32. [Google Scholar] [CrossRef]
- Badugu, R.; Garcia, M.; Bondada, V.; Joshi, A.; Geddes, J.W. N Terminus of Calpain 1 Is a Mitochondrial Targeting Sequence. J. Biol. Chem. 2008, 283, 3409–3417. [Google Scholar] [CrossRef]
- Ozaki, T.; Tomita, H.; Tamai, M.; Ishiguro, S. -i. Characteristics of Mitochondrial Calpains. J. Biochem. 2007, 142, 365–376. [Google Scholar] [CrossRef]
- Luo, T.; Yue, R.; Hu, H.; Zhou, Z.; Yiu, K.H.; Zhang, S.; Xu, L.; Li, K.; Yu, Z. PD150606 Protects against Ischemia/Reperfusion Injury by Preventing μ-Calpain-Induced Mitochondrial Apoptosis. Arch. Biochem. Biophys. 2015, 586, 1–9. [Google Scholar] [CrossRef]
- Thompson, J.; Maceyka, M.; Chen, Q. Targeting ER Stress and Calpain Activation to Reverse Age-Dependent Mitochondrial Damage in the Heart. Mech. Ageing Dev. 2020, 192, 111380. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Takahashi, M.; Koshimizu, M.; Tanonaka, K.; Oikawa, R.; Toyo-oka, T.; Takeo, S. Decrease in Sarcoglycans and Dystrophin in Failing Heart Following Acute Myocardial Infarction. Cardiovasc. Res. 2003, 59, 419–427. [Google Scholar] [CrossRef]
- Meng, Y.; Sun, T.; Wu, C.; Dong, C.; Xiong, S. Calpain Regulates CVB3 Induced Viral Myocarditis by Promoting Autophagic Flux upon Infection. Microbes Infect. 2020, 22, 46–54. [Google Scholar] [CrossRef]
- Ke, L.; Qi, X.Y.; Dijkhuis, A.-J.; Chartier, D.; Nattel, S.; Henning, R.H.; Kampinga, H.H.; Brundel, B.J.J.M. Calpain Mediates Cardiac Troponin Degradation and Contractile Dysfunction in Atrial Fibrillation. J. Mol. Cell. Cardiol. 2008, 45, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Pickles, S.; Vigié, P.; Youle, R.J. The Art of Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Baechler, B.L.; Bloemberg, D.; Quadrilatero, J. Mitophagy Regulates Mitochondrial Network Signaling, Oxidative Stress, and Apoptosis during Myoblast Differentiation. Autophagy 2019, 15, 1606–1619. [Google Scholar] [CrossRef]
- Russo, R.; Berliocchi, L.; Adornetto, A.; Varano, G.P.; Cavaliere, F.; Nucci, C.; Rotiroti, D.; Morrone, L.A.; Bagetta, G.; Corasaniti, M.T. Calpain-Mediated Cleavage of Beclin-1 and Autophagy Deregulation Following Retinal Ischemic Injury in Vivo. Cell Death Dis. 2011, 2, e144. [Google Scholar] [CrossRef]
- Kim, J.-S.; Wang, J.-H.; Biel, T.G.; Kim, D.-S.; Flores-Toro, J.A.; Vijayvargiya, R.; Zendejas, I.; Behrns, K.E. Carbamazepine suppresses calpain-mediated autophagy impairment after ischemia/reperfusion in mouse livers. Toxicol. Appl. Pharmacol. 2013, 273, 600–610. [Google Scholar] [CrossRef]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The Autophagosome: Origins Unknown, Biogenesis Complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Gelmetti, V.; De Rosa, P.; Torosantucci, L.; Marini, E.S.; Romagnoli, A.; Di Rienzo, M.; Arena, G.; Vignone, D.; Fimia, G.M.; Valente, E.M. PINK1 and BECN1 Relocalize at Mitochondria-Associated Membranes during Mitophagy and Promote ER-Mitochondria Tethering and Autophagosome Formation. Autophagy 2017, 13, 654–669. [Google Scholar] [CrossRef]
- Zhao, Q.; Guo, Z.; Deng, W.; Fu, S.; Zhang, C.; Chen, M.; Ju, W.; Wang, D.; He, X. Calpain 2-Mediated Autophagy Defect Increases Susceptibility of Fatty Livers to Ischemia–Reperfusion Injury. Cell Death Dis. 2016, 7, e2186. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.Q.; Zada, S.; Lai, T.H.; Pham, T.M.; Hwang, J.S.; Ahmed, M.; Kim, D.R. Calpain-Dependent Beclin1 Cleavage Stimulates Senescence-Associated Cell Death in HT22 Hippocampal Cells under the Oxidative Stress Conditions. Neurosci. Lett. 2019, 701, 106–111. [Google Scholar] [CrossRef]
- Zhu, X.; Messer, J.S.; Wang, Y.; Lin, F.; Cham, C.M.; Chang, J.; Billiar, T.R.; Lotze, M.T.; Boone, D.L.; Chang, E.B. Cytosolic HMGB1 Controls the Cellular Autophagy/Apoptosis Checkpoint during Inflammation. J. Clin. Investig. 2015, 125, 1098–1110. [Google Scholar] [CrossRef] [PubMed]
- Guan, L.; Che, Z.; Meng, X.; Yu, Y.; Li, M.; Yu, Z.; Shi, H.; Yang, D.; Yu, M. MCU Up-Regulation Contributes to Myocardial Ischemia-Reperfusion Injury through Calpain/OPA-1-Mediated Mitochondrial Fusion/Mitophagy Inhibition. J. Cell. Mol. Med. 2019, 23, 7830–7843. [Google Scholar] [CrossRef]
- Tong, M.; Zablocki, D.; Sadoshima, J. The Role of Drp1 in Mitophagy and Cell Death in the Heart. J. Mol. Cell. Cardiol. 2020, 142, 138–145. [Google Scholar] [CrossRef]
- Ikeda, Y.; Shirakabe, A.; Maejima, Y.; Zhai, P.; Sciarretta, S.; Toli, J.; Nomura, M.; Mihara, K.; Egashira, K.; Ohishi, M.; et al. Endogenous Drp1 Mediates Mitochondrial Autophagy and Protects the Heart Against Energy Stress. Circ. Res. 2015, 116, 264–278. [Google Scholar] [CrossRef]
- Sharp, W.W.; Fang, Y.H.; Han, M.; Zhang, H.J.; Hong, Z.; Banathy, A.; Morrow, E.; Ryan, J.J.; Archer, S.L. Dynamin-Related Protein 1 (Drp1)-Mediated Diastolic Dysfunction in Myocardial Ischemia-Reperfusion Injury: Therapeutic Benefits of Drp1 Inhibition to Reduce Mitochondrial Fission. FASEB J. 2014, 28, 316–326. [Google Scholar] [CrossRef]
- Hu, Q.; Zhang, H.; Gutierrez Cortes, N.; Wu, D.; Wang, P.; Zhang, J.; Mattison, J.A.; Smith, E.; Bettcher, L.F.; Wang, M.; et al. Increased Drp1 Acetylation by Lipid Overload Induces Cardiomyocyte Death and Heart Dysfunction. Circ. Res. 2020, 126, 456–470. [Google Scholar] [CrossRef]
- Cereghetti, G.M.; Stangherlin, A.; de Brito, O.M.; Chang, C.R.; Blackstone, C.; Bernardi, P.; Scorrano, L. Dephosphorylation by Calcineurin Regulates Translocation of Drp1 to Mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 15803–15808. [Google Scholar] [CrossRef]
- Heineke, J.; Ritter, O. Cardiomyocyte Calcineurin Signaling in Subcellular Domains: From the Sarcolemma to the Nucleus and Beyond. J. Mol. Cell. Cardiol. 2012, 52, 62–73. [Google Scholar] [CrossRef]
- Gahl, R.F.; Dwivedi, P.; Tjandra, N. Bcl-2 Proteins Bid and Bax Form a Network to Permeabilize the Mitochondria at the Onset of Apoptosis. Cell Death Dis. 2016, 7, e2424. [Google Scholar] [CrossRef]
- Ott, M.; Norberg, E.; Zhivotovsky, B.; Orrenius, S. Mitochondrial Targeting of TBid/Bax: A Role for the TOM Complex? Cell Death Differ. 2009, 16, 1075–1082. [Google Scholar] [CrossRef]
- Hou, D.; Che, Z.; Chen, P.; Zhang, W.; Chu, Y.; Yang, D.; Liu, J. Suppression of AURKA Alleviates P27 Inhibition on Bax Cleavage and Induces More Intensive Apoptosis in Gastric Cancer. Cell Death Dis. 2018, 9, 781. [Google Scholar] [CrossRef]
- Chen, M.; He, H.; Zhan, S.; Krajewski, S.; Reed, J.C.; Gottlieb, R.A. Bid Is Cleaved by Calpain to an Active Fragment in Vitro and during Myocardial Ischemia/Reperfusion *. J. Biol. Chem. 2001, 276, 30724–30728. [Google Scholar] [CrossRef]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of Mitochondrial Biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Houten, S.M.; Auwerx, J. PGC-1α: Turbocharging Mitochondria. Cell 2004, 119, 5–7. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, H. Translational Regulation of Mitochondrial Biogenesis. Biochem. Soc. Trans. 2016, 44, 1717–1724. [Google Scholar] [CrossRef] [PubMed]
- Lehman, J.J.; Barger, P.M.; Kovacs, A.; Saffitz, J.E.; Medeiros, D.M.; Kelly, D.P. Peroxisome Proliferator–Activated Receptor γ Coactivator-1 Promotes Cardiac Mitochondrial Biogenesis. J. Clin. Investig. 2000, 106, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Di, W.; Lv, J.; Jiang, S.; Lu, C.; Yang, Z.; Ma, Z.; Hu, W.; Yang, Y.; Xu, B. PGC-1: The Energetic Regulator in Cardiac Metabolism. Curr. Issues Mol. Biol. 2018, 29–46. [Google Scholar] [CrossRef]
- Sano, M.; Tokudome, S.; Shimizu, N.; Yoshikawa, N.; Ogawa, C.; Shirakawa, K.; Endo, J.; Katayama, T.; Yuasa, S.; Ieda, M.; et al. Intramolecular Control of Protein Stability, Subnuclear Compartmentalization, and Coactivator Function of Peroxisome Proliferator-Activated Receptor γ Coactivator 1α *. J. Biol. Chem. 2007, 282, 25970–25980. [Google Scholar] [CrossRef] [PubMed]
- Rasbach, K.A.; Green, P.T.; Schnellmann, R.G. Oxidants and Ca+2 Induce PGC-1α Degradation through Calpain. Arch. Biochem. Biophys. 2008, 478, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Chen, W.; Linsel-Nitschke, P.; Martinez, L.O.; Agerholm-Larsen, B.; Silver, D.L.; Tall, A.R. A PEST Sequence in ABCA1 Regulates Degradation by Calpain Protease and Stabilization of ABCA1 by ApoA-I. J. Clin. Investig. 2003, 111, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Bernaudo, S.; Khazai, S.; Honarparvar, E.; Kopteva, A.; Peng, C. Epidermal Growth Factor Promotes Cyclin G2 Degradation via Calpain-Mediated Proteolysis in Gynaecological Cancer Cells. PLoS ONE 2017, 12, e0179906. [Google Scholar] [CrossRef]
- Liu, W.; Ruiz-Velasco, A.; Wang, S.; Khan, S.; Zi, M.; Jungmann, A.; Dolores Camacho-Muñoz, M.; Guo, J.; Du, G.; Xie, L.; et al. Metabolic Stress-Induced Cardiomyopathy Is Caused by Mitochondrial Dysfunction Due to Attenuated Erk5 Signaling. Nat. Commun. 2017, 8, 494. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Zeng, H.; Ni, L.; Qi, L.; Xu, Y.; Xia, L.; Yu, Y.; Liu, B.; Yang, H.; Hao, H.; et al. HIF-1α Preconditioning Potentiates Antioxidant Activity in Ischemic Injury: The Role of Sequential Administration of Dihydrotanshinone I and Protocatechuic Aldehyde in Cardioprotection. Antioxid. Redox Signal. 2019, 31, 227–242. [Google Scholar] [CrossRef]
- Nanayakkara, G.; Alasmari, A.; Mouli, S.; Eldoumani, H.; Quindry, J.; McGinnis, G.; Fu, X.; Berlin, A.; Peters, B.; Zhong, J.; et al. Cardioprotective HIF-1α-Frataxin Signaling against Ischemia-Reperfusion Injury. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H867–H879. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, S. ROS and Redox Signaling in Myocardial Ischemia-Reperfusion Injury and Cardioprotection. Free Radic. Biol. Med. 2018, 117, 76–89. [Google Scholar] [CrossRef]
- Zhou, J.; Köhl, R.; Herr, B.; Frank, R.; Brüne, B. Calpain Mediates a von Hippel-Lindau Protein-Independent Destruction of Hypoxia-Inducible Factor-1alpha. Mol. Biol. Cell 2006, 17, 1549–1558. [Google Scholar] [CrossRef]
- Gong, X.; Yu, Z.; Huang, Z.; Xie, L.; Zhou, N.; Wang, J.; Liang, Y.; Qin, S.; Nie, Z.; Wei, L.; et al. Protective Effects of Cardiac Resynchronization Therapy in a Canine Model with Experimental Heart Failure by Improving Mitochondrial Function: A Mitochondrial Proteomics Study. J. Interv. Card. Electrophysiol. 2020, 61, 123–135. [Google Scholar] [CrossRef]
- Ozaki, T.; Yamashita, T.; Ishiguro, S. Mitochondrial M-Calpain Plays a Role in the Release of Truncated Apoptosis-Inducing Factor from the Mitochondria. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2009, 1793, 1848–1859. [Google Scholar] [CrossRef] [PubMed]
- Wirth, C.; Brandt, U.; Hunte, C.; Zickermann, V. Structure and Function of Mitochondrial Complex I. Biochim. Biophys. Acta (BBA) Bioenerg. 2016, 1857, 902–914. [Google Scholar] [CrossRef]
- Mohsin, A.A.; Thompson, J.; Hu, Y.; Hollander, J.; Lesnefsky, E.J.; Chen, Q. Endoplasmic Reticulum Stress-Induced Complex I Defect: Central Role of Calcium Overload. Arch. Biochem. Biophys. 2020, 683, 108299. [Google Scholar] [CrossRef]
- Koentges, C.; Cimolai, M.C.; Pfeil, K.; Wolf, D.; Marchini, T.; Tarkhnishvili, A.; Hoffmann, M.M.; Odening, K.E.; Diehl, P.; von zur Mühlen, C.; et al. Impaired SIRT3 Activity Mediates Cardiac Dysfunction in Endotoxemia by Calpain-Dependent Disruption of ATP Synthesis. J. Mol. Cell. Cardiol. 2019, 133, 138–147. [Google Scholar] [CrossRef]
- Hori, S.; Hiramuki, Y.; Nishimura, D.; Sato, F.; Sehara-Fujisawa, A. PDH-Mediated Metabolic Flow Is Critical for Skeletal Muscle Stem Cell Differentiation and Myotube Formation during Regeneration in Mice. FASEB J. 2019, 33, 8094–8109. [Google Scholar] [CrossRef]
- Thompson, J.; Hu, Y.; Lesnefsky, E.J.; Chen, Q. Activation of Mitochondrial Calpain and Increased Cardiac Injury: Beyond AIF Release. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H376–H384. [Google Scholar] [CrossRef]
- Tangmansakulchai, K.; Abubakar, Z.; Kitiyanant, N.; Suwanjang, W.; Leepiyasakulchai, C.; Govitrapong, P.; Chetsawang, B. Calpastatin Overexpression Reduces Oxidative Stress-Induced Mitochondrial Impairment and Cell Death in Human Neuroblastoma SH-SY5Y Cells by Decreasing Calpain and Calcineurin Activation, Induction of Mitochondrial Fission and Destruction of Mitochondrial Fusion. Mitochondrion 2016, 30, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.-P.; Zheng, M. Mitochondrial Dynamics and Inter-Mitochondrial Communication in the Heart. Arch. Biochem. Biophys. 2019, 663, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Mihara, K.; Chen, Y.; Scorrano, L.; Dorn, G.W. Mitochondrial Fission and Fusion Factors Reciprocally Orchestrate Mitophagic Culling in Mouse Hearts and Cultured Fibroblasts. Cell Metab. 2015, 21, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Haileselassie, B.; Mukherjee, R.; Joshi, A.U.; Napier, B.A.; Massis, L.M.; Ostberg, N.P.; Queliconi, B.B.; Monack, D.; Bernstein, D.; Mochly-Rosen, D. Drp1/Fis1 Interaction Mediates Mitochondrial Dysfunction in Septic Cardiomyopathy. J. Mol. Cell. Cardiol. 2019, 130, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Wai, T.; García-Prieto, J.; Baker, M.J.; Merkwirth, C.; Benit, P.; Rustin, P.; Rupérez, F.J.; Barbas, C.; Ibañez, B.; Langer, T. Imbalanced OPA1 Processing and Mitochondrial Fragmentation Cause Heart Failure in Mice. Science 2015, 350, 1162–1163. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Resendiz, S.; Prunier, F.; Girao, H.; Dorn, G.; Hausenloy, D.J. Targeting Mitochondrial Fusion and Fission Proteins for Cardioprotection. J. Cell. Mol. Med. 2020, 24, 6571–6585. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, F.; Li, L.; Tang, F.; Siedlak, S.L.; Fujioka, H.; Liu, Y.; Su, B.; Pi, Y.; Wang, X. MFN2 Couples Glutamate Excitotoxicity and Mitochondrial Dysfunction in Motor Neurons♦. J. Biol. Chem. 2015, 290, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.; Patel, J.; Khacho, M. Linking Mitochondrial Dynamics, Cristae Remodeling and Supercomplex Formation: How Mitochondrial Structure Can Regulate Bioenergetics. Mitochondrion 2019, 49, 259–268. [Google Scholar] [CrossRef]
- Jang, S.; Javadov, S. OPA1 Regulates Respiratory Supercomplexes Assembly: The Role of Mitochondrial Swelling. Mitochondrion 2020, 51, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Jahani-Asl, A.; Pilon-Larose, K.; Xu, W.; MacLaurin, J.G.; Park, D.S.; McBride, H.M.; Slack, R.S. The Mitochondrial Inner Membrane GTPase, Optic Atrophy 1 (Opa1), Restores Mitochondrial Morphology and Promotes Neuronal Survival Following Excitotoxicity. J. Biol. Chem. 2011, 286, 4772–4782. [Google Scholar] [CrossRef] [PubMed]
- Bano, D.; Prehn, J.H.M. Apoptosis-Inducing Factor (AIF) in Physiology and Disease: The Tale of a Repented Natural Born Killer. EBioMedicine 2018, 30, 29–37. [Google Scholar] [CrossRef]
- Chelko, S.P.; Keceli, G.; Carpi, A.; Doti, N.; Agrimi, J.; Asimaki, A.; Beti, C.B.; Miyamoto, M.; Amat-Codina, N.; Bedja, D.; et al. Exercise Triggers CAPN1-Mediated AIF Truncation, Inducing Myocyte Cell Death in Arrhythmogenic Cardiomyopathy. Sci. Transl. Med. 2021, 13, eabf0891. [Google Scholar] [CrossRef]
- Choudhury, S.; Bae, S.; Ke, Q.; Lee, J.Y.; Kim, J.; Kang, P.M. Mitochondria to Nucleus Translocation of AIF in Mice Lacking Hsp70 during Ischemia/Reperfusion. Basic Res. Cardiol. 2011, 106, 397. [Google Scholar] [CrossRef]
- Farina, B.; Di Sorbo, G.; Chambery, A.; Caporale, A.; Leoni, G.; Russo, R.; Mascanzoni, F.; Raimondo, D.; Fattorusso, R.; Ruvo, M.; et al. Structural and Biochemical Insights of CypA and AIF Interaction. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Moreira, A.C.; Branco, A.F.; Sampaio, S.F.; Cunha-Oliveira, T.; Martins, T.R.; Holy, J.; Oliveira, P.J.; Sardão, V.A. Mitochondrial Apoptosis-Inducing Factor Is Involved in Doxorubicin-Induced Toxicity on H9c2 Cardiomyoblasts. Biochim. Biophys. Acta 2014, 1842, 2468–2478. [Google Scholar] [CrossRef]
- Chen, Q.; Paillard, M.; Gomez, L.; Ross, T.; Hu, Y.; Xu, A.; Lesnefsky, E.J. Activation of Mitochondrial μ-Calpain Increases AIF Cleavage in Cardiac Mitochondria during Ischemia-Reperfusion. Biochem. Biophys. Res. Commun. 2011, 415, 533–538. [Google Scholar] [CrossRef]
- Briston, T.; Selwood, D.L.; Szabadkai, G.; Duchen, M.R. Mitochondrial Permeability Transition: A Molecular Lesion with Multiple Drug Targets. Trends Pharmacol. Sci. 2019, 40, 50–70. [Google Scholar] [CrossRef]
- Ong, S.-B.; Lee, W.H.; Shao, N.-Y.; Ismail, N.I.; Katwadi, K.; Lim, M.-M.; Kwek, X.-Y.; Michel, N.A.; Li, J.; Newson, J.; et al. Calpain Inhibition Restores Autophagy and Prevents Mitochondrial Fragmentation in a Human IPSC Model of Diabetic Endotheliopathy. Stem Cell Rep. 2019, 12, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, Y.; Feng, Q.; Arnold, M.; Peng, T. Calpain Activation Contributes to Hyperglycaemia-Induced Apoptosis in Cardiomyocytes. Cardiovasc. Res. 2009, 84, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Poncelas, M.; Inserte, J.; Aluja, D.; Hernando, V.; Vilardosa, U.; Garcia-Dorado, D. Delayed, Oral Pharmacological Inhibition of Calpains Attenuates Adverse Post-Infarction Remodelling. Cardiovasc. Res. 2017, 113, 950–961. [Google Scholar] [CrossRef] [PubMed]
- Da Silva-Ferrada, E.; Ribeiro-Rodrigues, T.M.; Rodríguez, M.S.; Girão, H. Proteostasis and SUMO in the Heart. Int. J. Biochem. Cell Biol. 2016, 79, 443–450. [Google Scholar] [CrossRef]
- Zheng, D.; Su, Z.; Zhang, Y.; Ni, R.; Fan, G.-C.; Robbins, J.; Song, L.-S.; Li, J.; Peng, T. Calpain-2 Promotes MKP-1 Expression Protecting Cardiomyocytes in Both in Vitro and in Vivo Mouse Models of Doxorubicin-Induced Cardiotoxicity. Arch. Toxicol. 2019, 93, 1051–1065. [Google Scholar] [CrossRef]
- Liu, Z.; Ji, J.; Zheng, D.; Su, L.; Peng, T. Calpain-2 Protects against Heat Stress-Induced Cardiomyocyte Apoptosis and Heart Dysfunction by Blocking P38 Mitogen-Activated Protein Kinase Activation. J. Cell. Physiol. 2019, 234, 10761–10770. [Google Scholar] [CrossRef]
- AbbVie. A Multiple-Dose Study to Evaluate the Safety, Tolerability and Pharmacokinetics of ABT-957 in Subjects with Mild-to-Moderate Alzheimer’s Disease on Stable Doses of Acetylcholinesterase Inhibitors. 2016. Available online: clinicaltrials.gov (accessed on 22 April 2021).
- Lon, H.-K.; Mendonca, N.; Goss, S.; Othman, A.A.; Locke, C.; Jin, Z.; Rendenbach-Mueller, B. Pharmacokinetics, Safety, Tolerability, and Pharmacodynamics of Alicapistat, a Selective Inhibitor of Human Calpains 1 and 2 for the Treatment of Alzheimer Disease: An Overview of Phase 1 Studies. Clin. Pharmacol. Drug Dev. 2019, 8, 290–303. [Google Scholar] [CrossRef]
- Blade Therapeutics. Phase 2, Randomized, Double-Blind, Placebo-Controlled Study to Evaluate the Safety and Antiviral Activity of BLD-2660 in Hospitalized Subjects with Recently Diagnosed COVID-19 Compared to Standard of Care Treatment. 2021. Available online: clinicaltrials.gov (accessed on 16 May 2021).



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, M.; Wang, G.; Peng, T. Calpain-Mediated Mitochondrial Damage: An Emerging Mechanism Contributing to Cardiac Disease. Cells 2021, 10, 2024. https://doi.org/10.3390/cells10082024
Zhang M, Wang G, Peng T. Calpain-Mediated Mitochondrial Damage: An Emerging Mechanism Contributing to Cardiac Disease. Cells. 2021; 10(8):2024. https://doi.org/10.3390/cells10082024
Chicago/Turabian StyleZhang, Mengxiao, Grace Wang, and Tianqing Peng. 2021. "Calpain-Mediated Mitochondrial Damage: An Emerging Mechanism Contributing to Cardiac Disease" Cells 10, no. 8: 2024. https://doi.org/10.3390/cells10082024
APA StyleZhang, M., Wang, G., & Peng, T. (2021). Calpain-Mediated Mitochondrial Damage: An Emerging Mechanism Contributing to Cardiac Disease. Cells, 10(8), 2024. https://doi.org/10.3390/cells10082024