
ChroMo, an Application for Unsupervised Analysis of Chromosome Movements in Meiosis


Abstract
1. Introduction
2. Materials and Methods
2.1. ChroMo Platform
2.2. Type of Data Required as Input
2.3. Synthetic Time-Series Generation
2.4. Time-Series Behavioural Segmentation and Statistics
- -
- Interaction of cluster factor, its starting and ending time;
- -
- Interaction of time;
- -
- Interaction of cluster factor and its proportion;
- -
- Interaction of cluster factor;
- -
- Intercept-only.
2.5. Global and Per-Segment Motifs Discovery
2.6. Global Covariate and Time-Series Causality Analysis
2.7. Limitations of ChroMo
2.7.1. Which Data Can Be Used?
2.7.2. Parameterization and Interpretation
2.7.3. Causality Graphs
2.8. Strains, Growth Conditions, and Meiosis Induction
2.9. Fluorescence Microscopy, Image Processing, and Analysis
3. Results
3.1. First Steps with ChroMo
3.2. ChroMo Provides More Detailed Information about Fission Yeast Meiotic Prophase
3.3. ChroMo Detects Causal Relations in CM Time-Series
3.4. Tuning up ChroMo Analysis with a Synthetic Dataset
3.5. ChroMo Performs Segmented and Time-Wise Analysis of Spectrum and Velocities
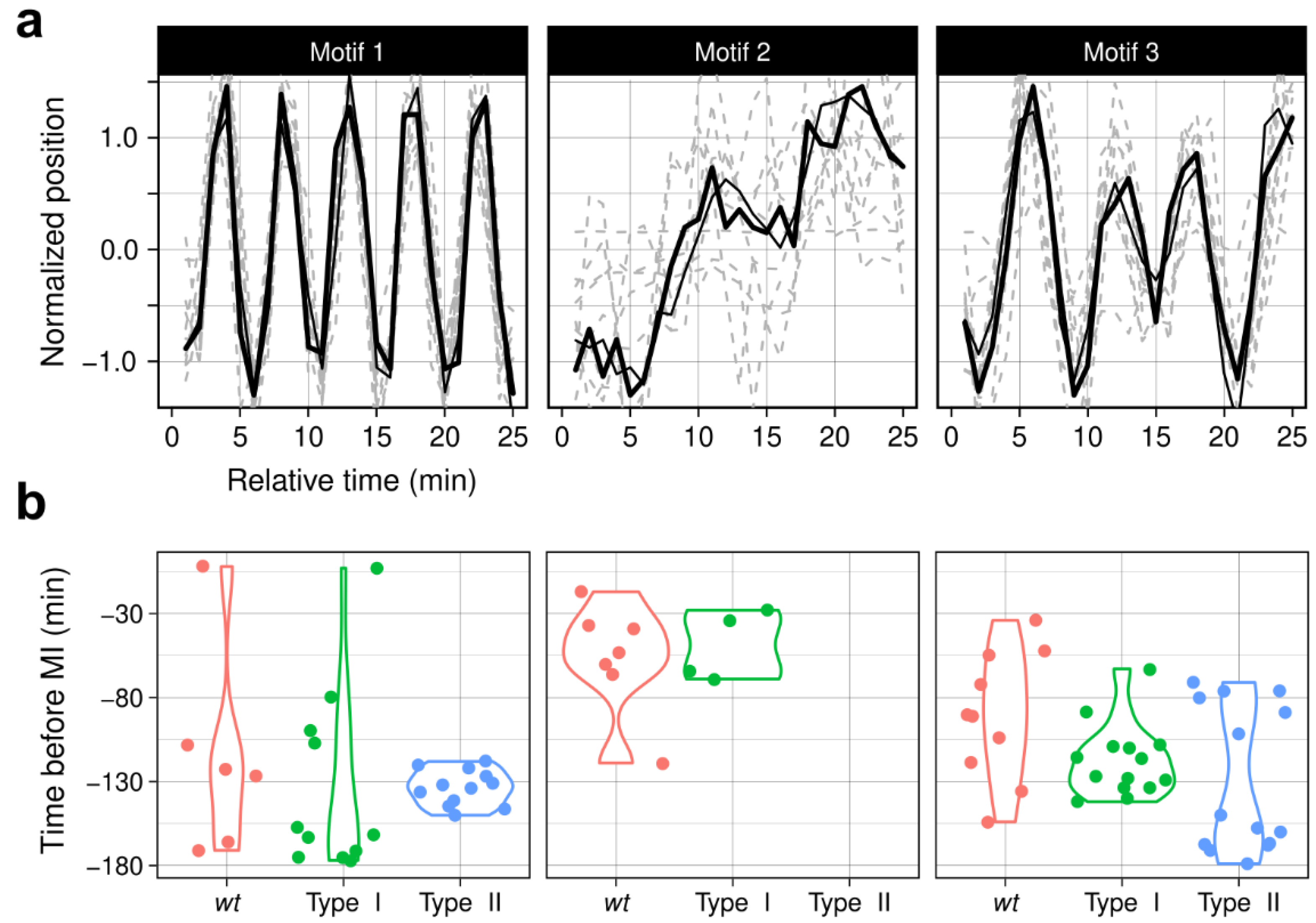
3.6. ChroMo Uses Motifs to Add a Complexity and Detail Layer to Behavioural Segments
3.7. ChroMo Finds Undisclosed Features on Known Strains
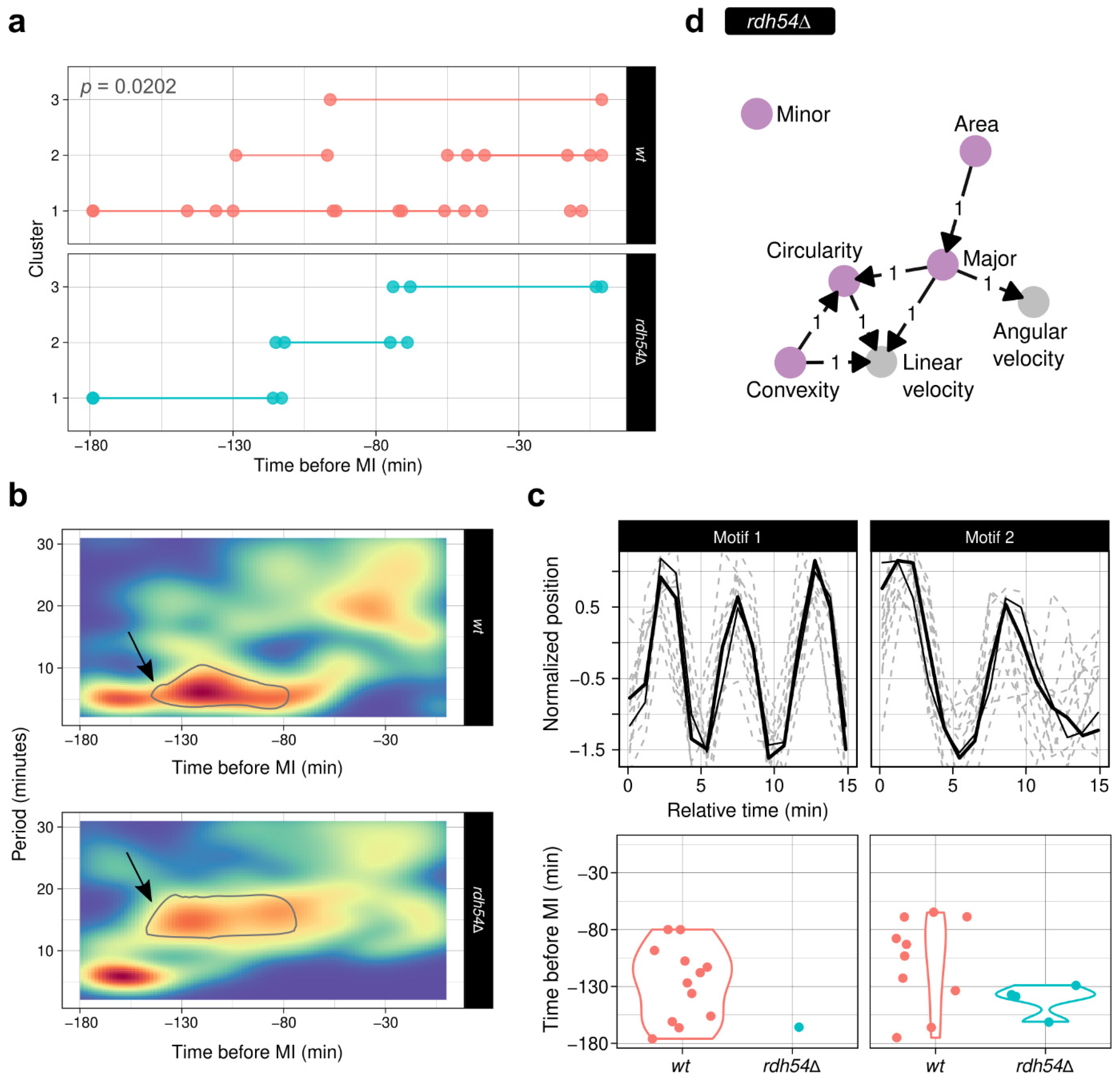
3.7.1. ChroMo Shows That the Oscillatory Movement Patterns Are Conserved in the hrs1Δ Strain
3.7.2. The rdh54 Deletion Leads to Different Oscillatory Movements
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. Strains Used throughout the Study

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Feature | Genotype | Origin | References |
|---|---|---|---|---|
| AFA226 | wt | h90 ade6-M210 his3-D1 leu1-32 Pnda3-mCherry-Atb2:aur1 Sid4-GFP:KanMX6 Hht1-CFP:his3 | Lab stock | – |
| AFA824 | hrs1Δ | h90 ade6-M210 his3-D1 leu1.32 Hht1-CFP:his3 Pnda3-mCherry-Atb2:aur1 | JCF10361 | [70] |
| AFA826 | rdh54Δ | h90 ura4-D18 taz1-YFP:KanMX6 Hht1-CFP:ura4 Sid4-mCherry:NatMX6 mei4-mCherry:NatMX6 rdh54::zeoCV | KT2475 | [31] |
Appendix A.2. Definition of Synthetic Time-Series
References
- Barchi, M.; Cohen, P.; Keeney, S. Special issue on “recent advances in meiotic chromosome structure, recombination and segregation”. Chromosoma 2016, 125, 173–175. [Google Scholar] [CrossRef]
- Petronczki, M.; Siomos, M.F.; Nasmyth, K. Un menage a quatre: The molecular biology of chromosome segregation in meiosis. Cell 2003, 112, 423–440. [Google Scholar] [CrossRef]
- Yanowitz, J. Meiosis: Making a break for it. Curr. Opin. Cell Biol. 2010, 22, 744–751. [Google Scholar] [CrossRef]
- Ohkura, H. Meiosis: An overview of key differences from mitosis. Cold Spring Harb. Perspect. Biol. 2015, 7, a015859. [Google Scholar] [CrossRef]
- Rubin, T.; Macaisne, N.; Huynh, J.R. Mixing and matching chromosomes during female meiosis. Cells 2020, 9, 696. [Google Scholar] [CrossRef] [PubMed]
- Klutstein, M.; Cooper, J.P. The chromosomal courtship dance—Homolog pairing in early meiosis. Curr. Opin. Cell. Biol. 2014, 26, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Link, J.; Jantsch, V. Meiotic chromosomes in motion: A perspective from Mus musculus and Caenorhabditis elegans. Chromosoma 2019, 128, 317–330. [Google Scholar] [CrossRef]
- Koszul, R.; Kim, K.P.; Prentiss, M.; Kleckner, N.; Kameoka, S. Meiotic chromosomes move by linkage to dynamic actin cables with transduction of force through the nuclear envelope. Cell 2008, 133, 1188–1201. [Google Scholar] [CrossRef] [PubMed]
- Christophorou, N.; Rubin, T.; Bonnet, I.; Piolot, T.; Arnaud, M.; Huynh, J.R. Microtubule-driven nuclear rotations promote meiotic chromosome dynamics. Nat. Cell Biol. 2015, 17, 1388–1400. [Google Scholar] [CrossRef] [PubMed]
- Wynne, D.J.; Rog, O.; Carlton, P.M.; Dernburg, A.F. Dynein-dependent processive chromosome motions promote homologous pairing in C. elegans meiosis. J. Cell Biol. 2012, 196, 47–64. [Google Scholar] [CrossRef]
- Baudrimont, A.; Penkner, A.; Woglar, A.; Machacek, T.; Wegrostek, C.; Gloggnitzer, J.; Fridkin, A.; Klein, F.; Gruenbaum, Y.; Pasierbek, P.; et al. Leptotene/zygotene chromosome movement via the SUN/KASH protein bridge in Caenorhabditis elegans. PLoS Genet. 2010, 6, e1001219. [Google Scholar] [CrossRef]
- Morimoto, A.; Shibuya, H.; Zhu, X.; Kim, J.; Ishiguro, K.; Han, M.; Watanabe, Y. A conserved KASH domain protein associates with telomeres, SUN1, and dynactin during mammalian meiosis. J. Cell Biol. 2012, 198, 165–172. [Google Scholar] [CrossRef]
- Sato, A.; Isaac, B.; Phillips, C.M.; Rillo, R.; Carlton, P.M.; Wynne, D.J.; Kasad, R.A.; Dernburg, A.F. Cytoskeletal forces span the nuclear envelope to coordinate meiotic chromosome pairing and synapsis. Cell 2009, 139, 907–919. [Google Scholar] [CrossRef]
- Lee, C.Y.; Bisig, C.G.; Conrad, M.N.; Ditamo, Y.; de Almeida, L.P.; Dresser, M.E.; Pezza, R.J. Telomere-led meiotic chromosome movements: Recent update in structure and function. Nucleus 2020, 11, 111–116. [Google Scholar] [CrossRef]
- Yamamoto, A.; West, R.R.; McIntosh, J.R.; Hiraoka, Y. A cytoplasmic dynein heavy chain is required for oscillatory nuclear movement of meiotic prophase and efficient meiotic recombination in fission yeast. J. Cell Biol. 1999, 145, 1233–1249. [Google Scholar] [CrossRef]
- Chikashige, Y.; Ding, D.Q.; Funabiki, H.; Haraguchi, T.; Mashiko, S.; Yanagida, M.; Hiraoka, Y. Telomere-led premeiotic chromosome movement in fission yeast. Science 1994, 264, 270–273. [Google Scholar] [CrossRef]
- Chikashige, Y.; Ding, D.Q.; Imai, Y.; Yamamoto, M.; Haraguchi, T.; Hiraoka, Y. Meiotic nuclear reorganization: Switching the position of centromeres and telomeres in the fission yeast Schizosaccharomyces pombe. EMBO J. 1997, 16, 193–202. [Google Scholar] [CrossRef]
- Scherthan, H. A bouquet makes ends meet. Nat. Rev. Mol. Cell Biol. 2001, 2, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, H.; Ishiguro, K.; Watanabe, Y. The TRF1-binding protein TERB1 promotes chromosome movement and telomere rigidity in meiosis. Nat. Cell Biol. 2014, 16, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Huang, C.; Chen, Y.; Zhang, Y.; Shi, S.; Wu, L.; Liu, Y.; Liu, C.; Wu, J.; Lei, M. Telomeric TERB1-TRF1 interaction is crucial for male meiosis. Nat. Struct. Mol. Biol. 2017, 24, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Daniel, K.; Trankner, D.; Wojtasz, L.; Shibuya, H.; Watanabe, Y.; Alsheimer, M.; Toth, A. Mouse CCDC79 (TERB1) is a meiosis-specific telomere associated protein. BMC Cell Biol. 2014, 15, 17. [Google Scholar] [CrossRef]
- Shibuya, H.; Hernandez-Hernandez, A.; Morimoto, A.; Negishi, L.; Hoog, C.; Watanabe, Y. MAJIN links telomeric DNA to the nuclear membrane by exchanging telomere cap. Cell 2015, 163, 1252–1266. [Google Scholar] [CrossRef] [PubMed]
- da Cruz, I.; Brochier-Armanet, C.; Benavente, R. The TERB1-TERB2-MAJIN complex of mouse meiotic telomeres dates back to the common ancestor of metazoans. BMC Evol. Biol. 2020, 20, 55. [Google Scholar] [CrossRef]
- Phillips, C.M.; Dernburg, A.F. A family of zinc-finger proteins is required for chromosome-specific pairing and synapsis during meiosis in C. elegans. Dev. Cell 2006, 11, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C.M.; Wong, C.; Bhalla, N.; Carlton, P.M.; Weiser, P.; Meneely, P.M.; Dernburg, A.F. HIM-8 binds to the X chromosome pairing center and mediates chromosome-specific meiotic synapsis. Cell 2005, 123, 1051–1063. [Google Scholar] [CrossRef] [PubMed]
- Scherthan, H.; Wang, H.; Adelfalk, C.; White, E.J.; Cowan, C.; Cande, W.Z.; Kaback, D.B. Chromosome mobility during meiotic prophase in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2007, 104, 16934–16939. [Google Scholar] [CrossRef]
- Conrad, M.N.; Dominguez, A.M.; Dresser, M.E. Ndj1p, a meiotic telomere protein required for normal chromosome synapsis and segregation in yeast. Science 1997, 276, 1252–1255. [Google Scholar] [CrossRef]
- Chikashige, Y.; Tsutsumi, C.; Yamane, M.; Okamasa, K.; Haraguchi, T.; Hiraoka, Y. Meiotic proteins Bqt1 and Bqt2 tether telomeres to form the bouquet arrangement of chromosomes. Cell 2006, 125, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Robinow, C.F. The number of chromosomes in Schizosaccharomyces pombe: Light microscopy of stained preparations. Genetics 1977, 87, 491–497. [Google Scholar] [CrossRef]
- Niwa, O.; Shimanuki, M.; Miki, F. Telomere-led bouquet formation facilitates homologous chromosome pairing and restricts ectopic interaction in fission yeast meiosis. EMBO J. 2000, 19, 3831–3840. [Google Scholar] [CrossRef] [PubMed]
- Moiseeva, V.; Amelina, H.; Collopy, L.C.; Armstrong, C.A.; Pearson, S.R.; Tomita, K. The telomere bouquet facilitates meiotic prophase progression and exit in fission yeast. Cell Discov. 2017, 3, 17041. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.Q.; Yamamoto, A.; Haraguchi, T.; Hiraoka, Y. Dynamics of homologous chromosome pairing during meiotic prophase in fission yeast. Dev. Cell 2004, 6, 329–341. [Google Scholar] [CrossRef]
- Chacon, M.R.; Delivani, P.; Tolic, I.M. Meiotic nuclear oscillations are necessary to avoid excessive chromosome associations. Cell Rep. 2016, 17, 1632–1645. [Google Scholar] [CrossRef]
- Tomita, K.; Cooper, J.P. The telomere bouquet controls the meiotic spindle. Cell 2007, 130, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Labrador, L.; Barroso, C.; Lightfoot, J.; Muller-Reichert, T.; Flibotte, S.; Taylor, J.; Moerman, D.G.; Villeneuve, A.M.; Martinez-Perez, E. Chromosome movements promoted by the mitochondrial protein SPD-3 are required for homology search during Caenorhabditis elegans meiosis. PLoS Genet. 2013, 9, e1003497. [Google Scholar] [CrossRef] [PubMed]
- Rog, O.; Dernburg, A.F. Direct visualization reveals kinetics of meiotic chromosome synapsis. Cell Rep. 2015, 10, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Woglar, A.; Jantsch, V. Chromosome movement in meiosis I prophase of Caenorhabditis elegans. Chromosoma 2014, 123, 15–24. [Google Scholar] [CrossRef][Green Version]
- Conrad, M.N.; Lee, C.Y.; Chao, G.; Shinohara, M.; Kosaka, H.; Shinohara, A.; Conchello, J.A.; Dresser, M.E. Rapid telomere movement in meiotic prophase is promoted by NDJ1, MPS3, and CSM4 and is modulated by recombination. Cell 2008, 133, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Arranz, S.; Gardner, J.M.; Yu, Z.; Patel, N.J.; Heldrich, J.; Santos, B.; Carballo, J.A.; Jaspersen, S.L.; Hochwagen, A.; San-Segundo, P.A. SWR1-independent association of H2A.Z to the LINC complex promotes meiotic chromosome motion. Front. Cell Dev. Biol. 2020, 8, 594092. [Google Scholar] [CrossRef]
- Ananthanarayanan, V.; Schattat, M.; Vogel, S.K.; Krull, A.; Pavin, N.; Tolic-Norrelykke, I.M. Dynein motion switches from diffusive to directed upon cortical anchoring. Cell 2013, 153, 1526–1536. [Google Scholar] [CrossRef]
- Kakui, Y.; Barrington, C.; Barry, D.J.; Gerguri, T.; Fu, X.; Bates, P.A.; Khatri, B.S.; Uhlmann, F. Fission yeast condensin contributes to interphase chromatin organization and prevents transcription-coupled DNA damage. Genome Biol. 2020, 21, 272. [Google Scholar] [CrossRef]
- Caridi, C.P.; Delabaere, L.; Tjong, H.; Hopp, H.; Das, D.; Alber, F.; Chiolo, I. Quantitative methods to investigate the 4D dynamics of heterochromatic repair sites in drosophila cells. Methods Enzym. 2018, 601, 359–389. [Google Scholar] [CrossRef]
- Mine-Hattab, J.; Rothstein, R. Increased chromosome mobility facilitates homology search during recombination. Nat. Cell Biol. 2012, 14, 510–517. [Google Scholar] [CrossRef]
- Thankachan, J.M.; Nuthalapati, S.S.; Tirumala, N.A.; Ananthanarayanan, V. Fission yeast myosin I facilitates PI(4,5)P2-mediated anchoring of cytoplasmic dynein to the cortex. Proc. Natl. Acad. Sci. USA 2017, 114, E2672–E2681. [Google Scholar] [CrossRef] [PubMed]
- Castellano-Pozo, M.; Pacheco, S.; Sioutas, G.; Jaso-Tamame, A.L.; Dore, M.H.; Karimi, M.M.; Martinez-Perez, E. Surveillance of cohesin-supported chromosome structure controls meiotic progression. Nat. Commun. 2020, 11, 4345. [Google Scholar] [CrossRef] [PubMed]
- Chikashige, Y.; Yamane, M.; Okamasa, K.; Mori, C.; Fukuta, N.; Matsuda, A.; Haraguchi, T.; Hiraoka, Y. Chromosomes rein back the spindle pole body during horsetail movement in fission yeast meiosis. Cell Struct. Funct. 2014, 39, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Mary, H.; Fouchard, J.; Gay, G.; Reyes, C.; Gauthier, T.; Gruget, C.; Pecreaux, J.; Tournier, S.; Gachet, Y. Fission yeast kinesin-8 controls chromosome congression independently of oscillations. J. Cell Sci. 2015, 128, 3720–3730. [Google Scholar] [CrossRef]
- Tanaka, K.; Kohda, T.; Yamashita, A.; Nonaka, N.; Yamamoto, M. Hrs1p/Mcp6p on the meiotic SPB organizes astral microtubule arrays for oscillatory nuclear movement. Curr. Biol. 2005, 15, 1479–1486. [Google Scholar] [CrossRef]
- Yamada, S.; Kugou, K.; Ding, D.Q.; Fujita, Y.; Hiraoka, Y.; Murakami, H.; Ohta, K.; Yamada, T. The histone variant H2A.Z promotes initiation of meiotic recombination in fission yeast. Nucleic Acids Res. 2018, 46, 609–620. [Google Scholar] [CrossRef]
- Ding, D.Q.; Matsuda, A.; Okamasa, K.; Nagahama, Y.; Haraguchi, T.; Hiraoka, Y. Meiotic cohesin-based chromosome structure is essential for homologous chromosome pairing in Schizosaccharomyces pombe. Chromosoma 2016, 125, 205–214. [Google Scholar] [CrossRef]
- Mine-Hattab, J.; Chiolo, I. Complex chromatin motions for DNA repair. Front. Genet. 2020, 11, 800. [Google Scholar] [CrossRef]
- Ryu, T.; Spatola, B.; Delabaere, L.; Bowlin, K.; Hopp, H.; Kunitake, R.; Karpen, G.H.; Chiolo, I. Heterochromatic breaks move to the nuclear periphery to continue recombinational repair. Nat. Cell Biol. 2015, 17, 1401–1411. [Google Scholar] [CrossRef]
- Lavielle, M. Detection of multiple changes in a sequence of dependent variables. Stoch. Process. Appl. 1999, 83, 79–102. [Google Scholar] [CrossRef]
- Patin, R.; Etienne, M.P.; Lebarbier, E.; Chamaillé-Jammes, S.; Benhamou, S. Identifying stationary phases in multivariate time series for highlighting behavioural modes and home range settlements. J. Anim. Ecol. 2020, 89, 44–56. [Google Scholar] [CrossRef]
- Machné, R.; Murray, D.B.; Stadler, P.F. Similarity-based segmentation of multi-dimensional signals. Sci. Rep. 2017, 7, 12355. [Google Scholar] [CrossRef]
- Scrucca, L.; Fop, M.; Murphy, T.B.; Raftery, A.E. Mclust 5: Clustering, classification and density estimation using Gaussian finite mixture models. R J. 2016, 8, 289–317. [Google Scholar] [CrossRef] [PubMed]
- Hastie, T.; Tibshirani, R.; Friedman, J. Linear methods for regression. In The Elements of Statistical Learning; Springer: New York, NY, USA, 2009. [Google Scholar]
- Yeh, C.-C.M.; Zhu, Y.; Ulanova, L.; Begum, N.; Ding, Y.; Dau, H.A.; Silva, D.F.; Mueen, A.; Keogh, E. Matrix profile I: All pairs similarity joins for time series: A unifying view that includes motifs, discords and shapelets. In Proceedings of the 2016 IEEE 16th International Conference on Data Mining (ICDM), Barcelona, Spain, 12–15 December 2016; pp. 1317–1322. [Google Scholar]
- Zhu, Y.; Yeh, C.-C.M.; Zimmerman, Z.; Kamgar, K.; Keogh, E. Matrix profile XI: SCRIMP++: Time series motif discovery at interactive speeds. In Proceedings of the 2018 IEEE International Conference on Data Mining (ICDM), Singapore, 17–20 November 2018; pp. 837–846. [Google Scholar]
- Spirtes, P.; Glymour, C.; Scheines, R. Causation, Prediction and Search; Springer: New York, NY, USA, 1993; Volume 81. [Google Scholar]
- Kalisch, M.; Mächler, M.; Colombo, D.; Maathuis, M.H.; Bühlmann, P. Causal inference using graphical models with the R package pcalg. J. Stat. Softw. 2012, 47, 1–26. [Google Scholar] [CrossRef]
- Amornbunchornvej, C.; Zheleva, E.; Berger-Wolf, T.Y. Variable-lag Granger causality for time series analysis. In Proceedings of the 2019 IEEE International Conference on Data Science and Advanced Analytics (DSAA), Washington, DC, USA, 5–8 October 2019; pp. 21–30. [Google Scholar]
- Glymour, C.; Zhang, K.; Spirtes, P. Review of causal discovery methods based on graphical models. Front. Genet. 2019, 10, 524. [Google Scholar] [CrossRef] [PubMed]
- Moreno, S.; Klar, A.; Nurse, P. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzym. 1991, 194, 795–823. [Google Scholar] [CrossRef]
- Thevenaz, P.; Ruttimann, U.E.; Unser, M. A pyramid approach to subpixel registration based on intensity. IEEE Trans. Image Process. 1998, 7, 27–41. [Google Scholar] [CrossRef]
- Allan, D.; Caswell, T.; Keim, N.; Van Der Wel, C. Trackpy: Trackpy v0.3.2. 2016. Available online: zenodo.org/record/60550#.YQkmsEBRVPY (accessed on 26 April 2021).
- Miki, F.; Okazaki, K.; Shimanuki, M.; Yamamoto, A.; Hiraoka, Y.; Niwa, O. The 14-kDa dynein light chain-family protein Dlc1 is required for regular oscillatory nuclear movement and efficient recombination during meiotic prophase in fission yeast. Mol. Biol. Cell 2002, 13, 930–946. [Google Scholar] [CrossRef]
- Saito, T.T.; Tougan, T.; Okuzaki, D.; Kasama, T.; Nojima, H. Mcp6, a meiosis-specific coiled-coil protein of Schizosaccharomyces pombe, localizes to the spindle pole body and is required for horsetail movement and recombination. J. Cell Sci. 2005, 118, 447–459. [Google Scholar] [CrossRef][Green Version]
- Catlett, M.G.; Forsburg, S.L. Schizosaccharomyces pombe Rdh54 (TID1) acts with Rhp54 (RAD54) to repair meiotic double-strand breaks. Mol. Biol. Cell 2003, 14, 4707–4720. [Google Scholar] [CrossRef] [PubMed]
- Fennell, A.; Fernández-álvarez, A.; Tomita, K.; Cooper, J.P. Telomeres and centromeres have interchangeable roles in promoting meiotic spindle formation. J. Cell Biol. 2015, 208, 415–428. [Google Scholar] [CrossRef]
- Van Benschoten, A.H.; Ouyang, A.; Bischoff, F.; Marrs, T.W. MPA: A novel cross-language API for time series analysis. J. Open Source Softw. 2020, 5, 2179. [Google Scholar] [CrossRef]







| Module | Function | Description | Use-Cases |
|---|---|---|---|
| Segments | Discovery | Performs time-series segmentation, per particle ID and per group, and clusters all discovered segments into a common pool, visualized as a clustered segment plot. Then, assesses if the cluster composition (location, abundance) is a good predictor of group using logistic regression models. |
|
| Cluster Summary | Displays a table-like summary of the discovered clusters, and the corresponding features used for clustering. | --- | |
| Spectrum | Calculation of spectral characteristics of the main coordinate variable (only in 1D), per particle and group ID. Displays global and per-cluster spectral densities, as well as heatmaps of spectral density across time, global and per discovered cluster—needs prior segmentation of data with the function Discovery—. Provides a summary with statistics to assess whether the spectrum is significantly different between strains. |
| |
| Distribution | Calculates the empirical distribution (probability density) of any selected numerical variable (column of the dataset) and displays it on violin and density plots, for the whole time-series, per group, or per group and discovered behavioural mode. Additionally, it calculates the 2D density of the selected feature over time. It is possible to set up a moving average to smooth the data. Provides a summary with statistics to assess whether the probability density is significantly different between strains. |
| |
| Displacement | Calculate the Mean Squared Displacement (MSD) of the data selected as coordinate variables, per group, and per discovered cluster. |
| |
| Individual | Tools to visualize the time-series of each particle, as well as the individual spectrograms for any variable. | --- | |
| Motifs | Global | After selecting a variable (1D) and groups to analyse, motifs (and discords) are calculated for the concatenated time-series. It is possible to parametrise the window-length for which motifs will be discovered, as well as other tuneable parameters (see tsmp reference guide on these [71]. |
|
| Per cluster | Analogous to Global motif analysis, for each discovered behavioural cluster. Displays the distribution of discovered motifs across time. |
| |
| Causality | PC-alg VLTE VLGC | Calculates a causality graph using the PC-algorithm, Variable-Lag Transfer Entropy, or Variable-Lag Granger Causality, for selected variables, per particle ID and per group. Then, it builds a global graph of all the observed relationships in all the particles. Configurable parameters are the significance threshold (p-value) to consider a relationship, the maximum lags to explore, and the presence of connection, with respect to how many times a relationship must appear in the dataset to be considered positive. |
|
| Correlation | Calculates a pairwise correlation between all selected variables and shows the corresponding scatterplots. |
| |
| Matrix | Shows the adjacency matrix for the calculated causality graphs | --- |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
León-Periñán, D.; Fernández-Álvarez, A. ChroMo, an Application for Unsupervised Analysis of Chromosome Movements in Meiosis. Cells 2021, 10, 2013. https://doi.org/10.3390/cells10082013
León-Periñán D, Fernández-Álvarez A. ChroMo, an Application for Unsupervised Analysis of Chromosome Movements in Meiosis. Cells. 2021; 10(8):2013. https://doi.org/10.3390/cells10082013
Chicago/Turabian StyleLeón-Periñán, Daniel, and Alfonso Fernández-Álvarez. 2021. "ChroMo, an Application for Unsupervised Analysis of Chromosome Movements in Meiosis" Cells 10, no. 8: 2013. https://doi.org/10.3390/cells10082013
APA StyleLeón-Periñán, D., & Fernández-Álvarez, A. (2021). ChroMo, an Application for Unsupervised Analysis of Chromosome Movements in Meiosis. Cells, 10(8), 2013. https://doi.org/10.3390/cells10082013