
Acute RhoA/Rho Kinase Inhibition Is Sufficient to Restore Phagocytic Capacity to Retinal Pigment Epithelium Lacking the Engulfment Receptor MerTK



Abstract
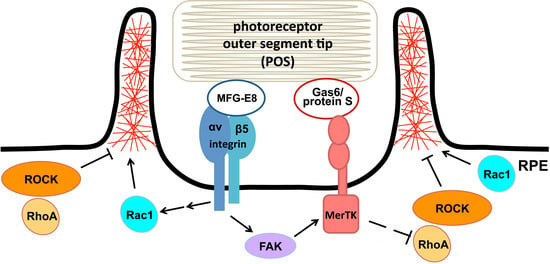
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. GTPase Activity Assays
2.3. Primary RPE Cell Culture
2.4. POS Phagocytosis Assays
2.5. RhoA Pathway Manipulations in RPE in Culture and RPE Ex Vivo
2.6. Immunofluorescence, F-Actin and Live Lysosome Microscopy
2.7. SDS-PAGE and Immunoblotting
2.8. Statistical Analysis
3. Results
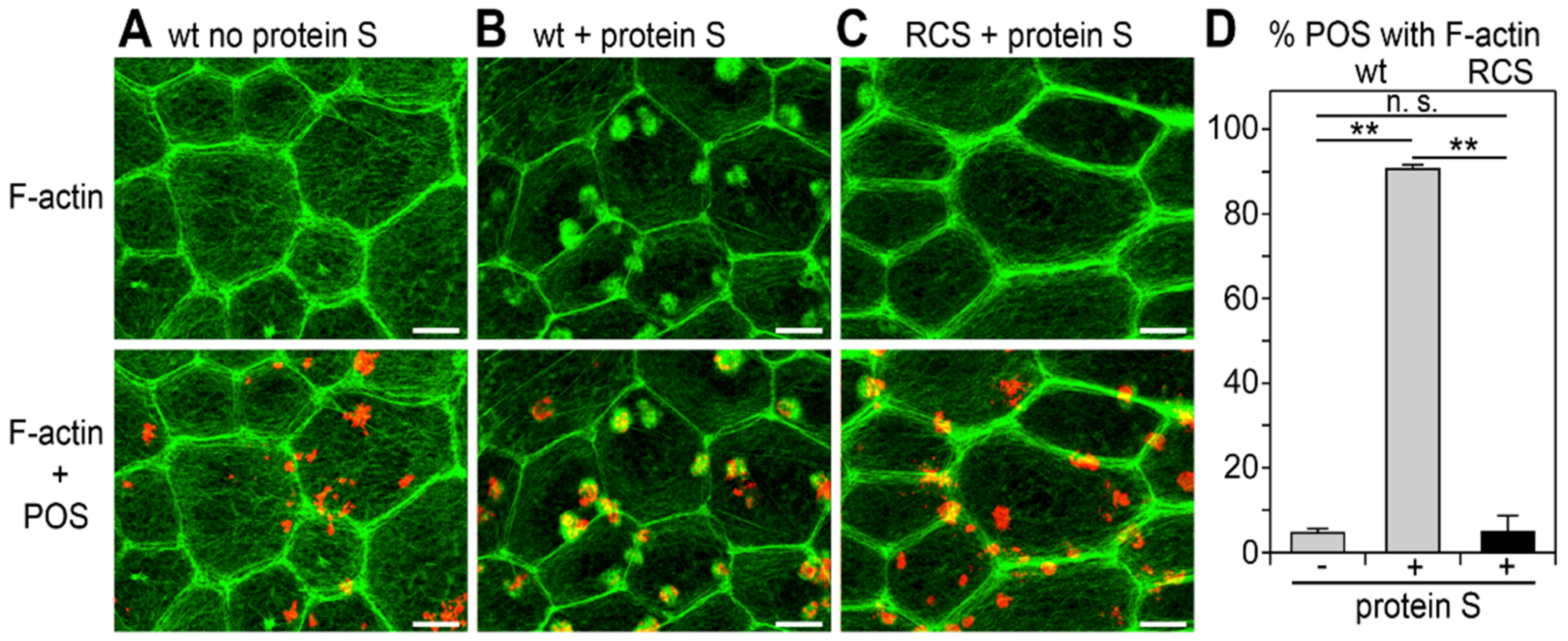
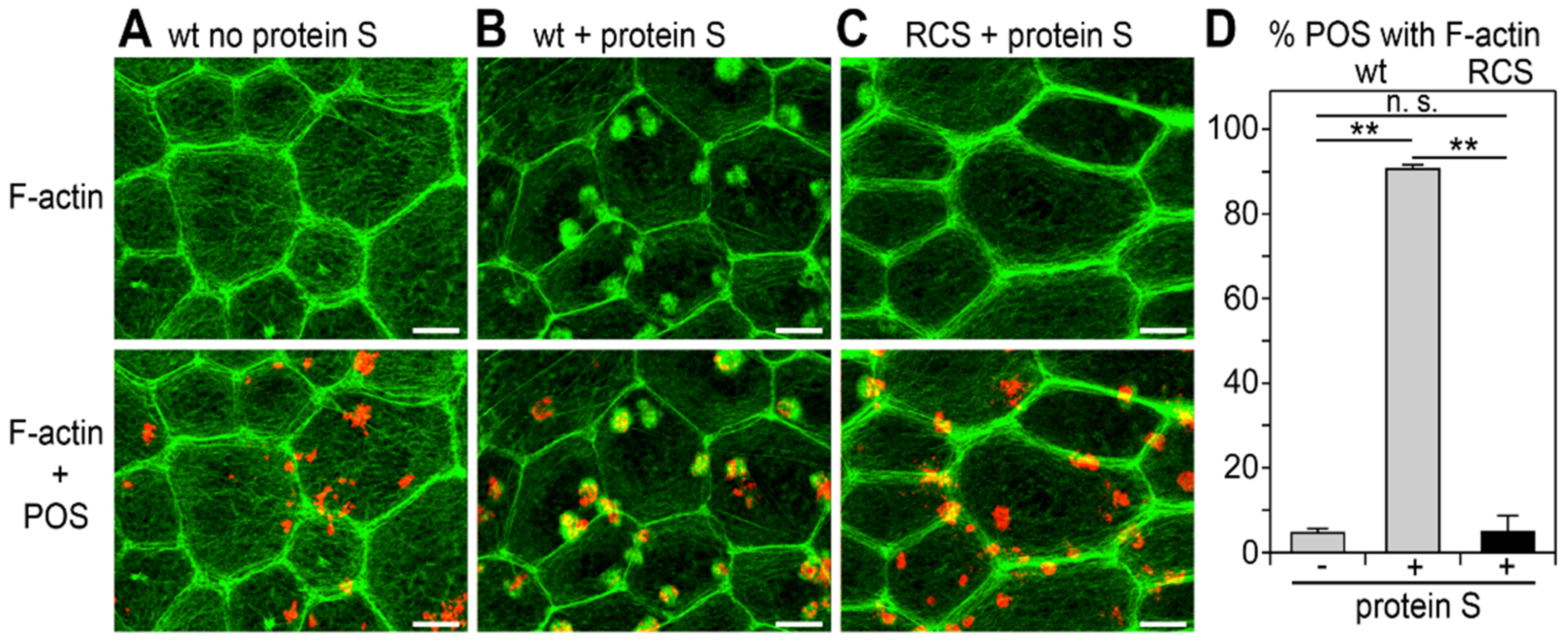
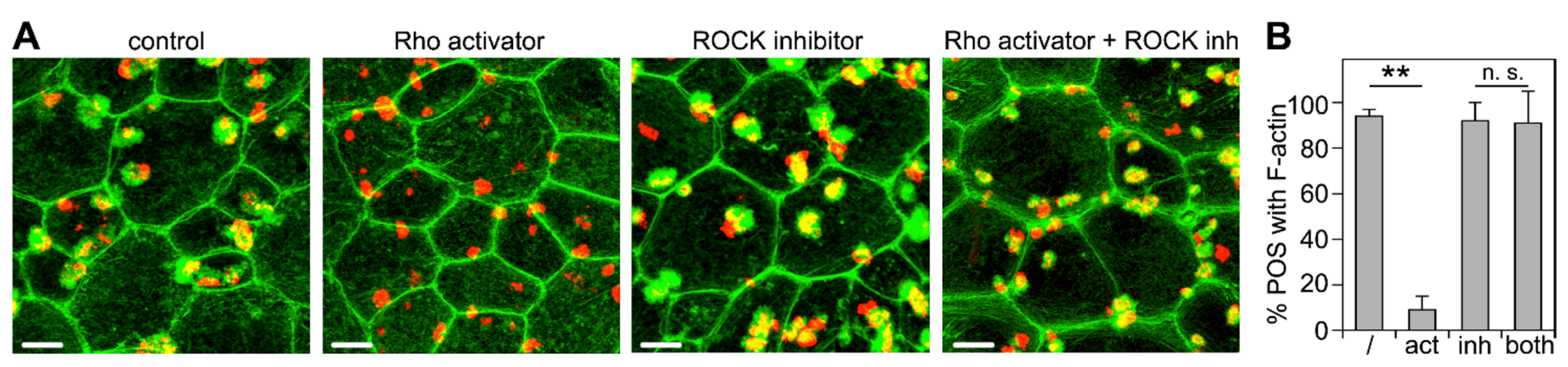
3.1. Recruitment of F-Actin Phagocytic Cups Beneath Surface-Bound POS Requires MerTK Receptor Ligation
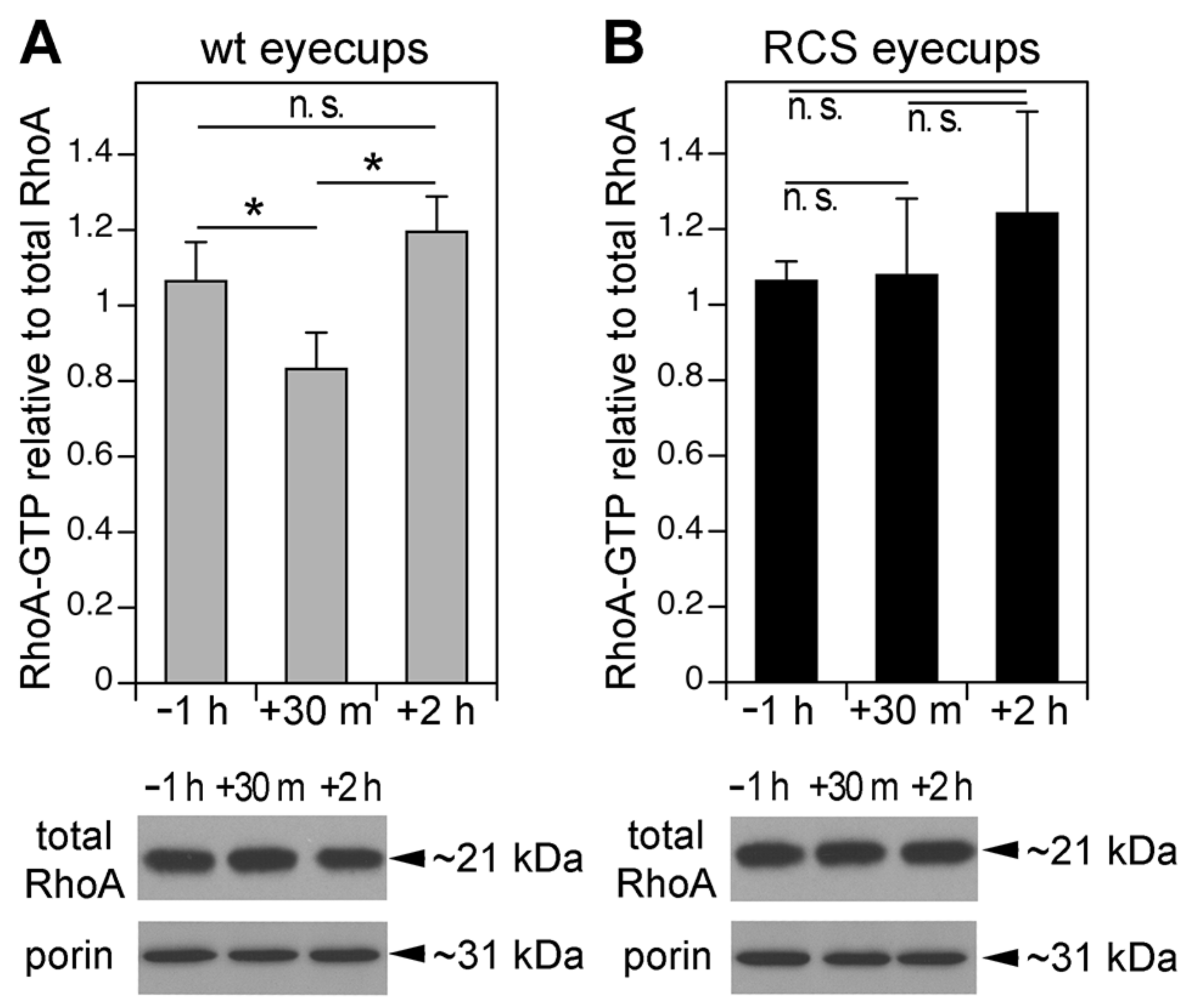
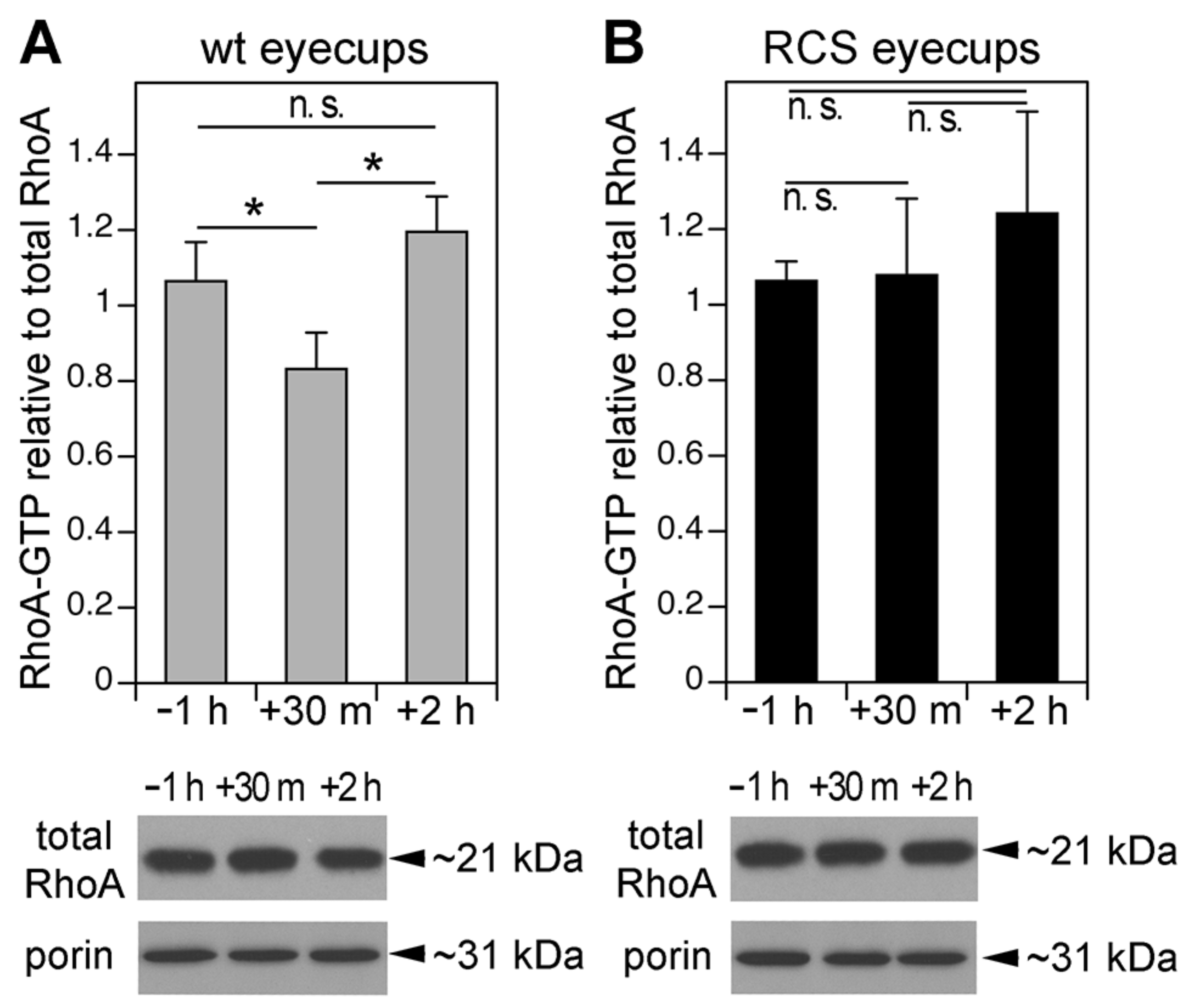
3.2. MerTK-Deficient RPE In Vivo Lacks a Dip in RhoA Activity that Coincides with the Peak of Diurnal POS Phagocytosis in wt RPE
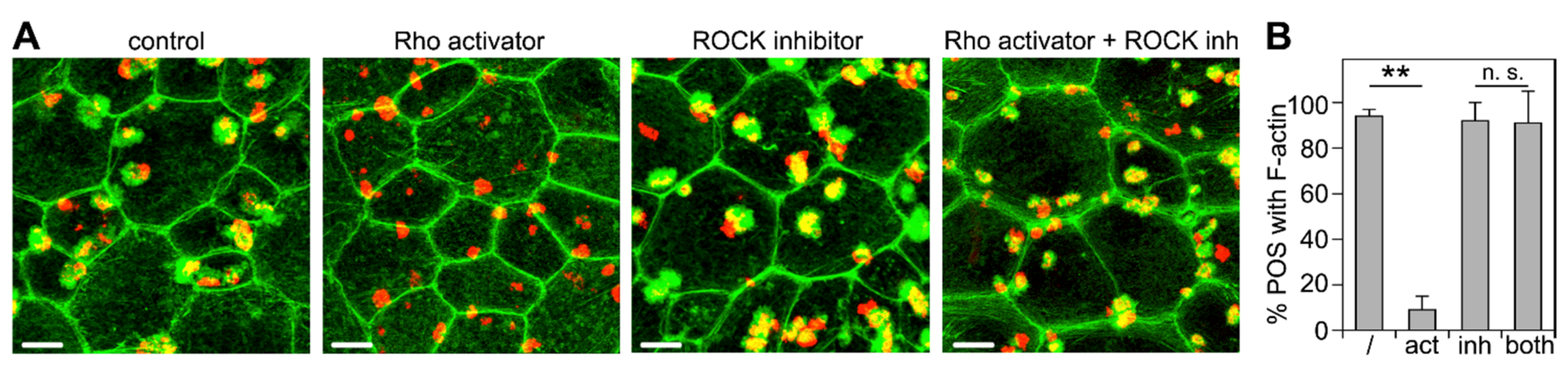
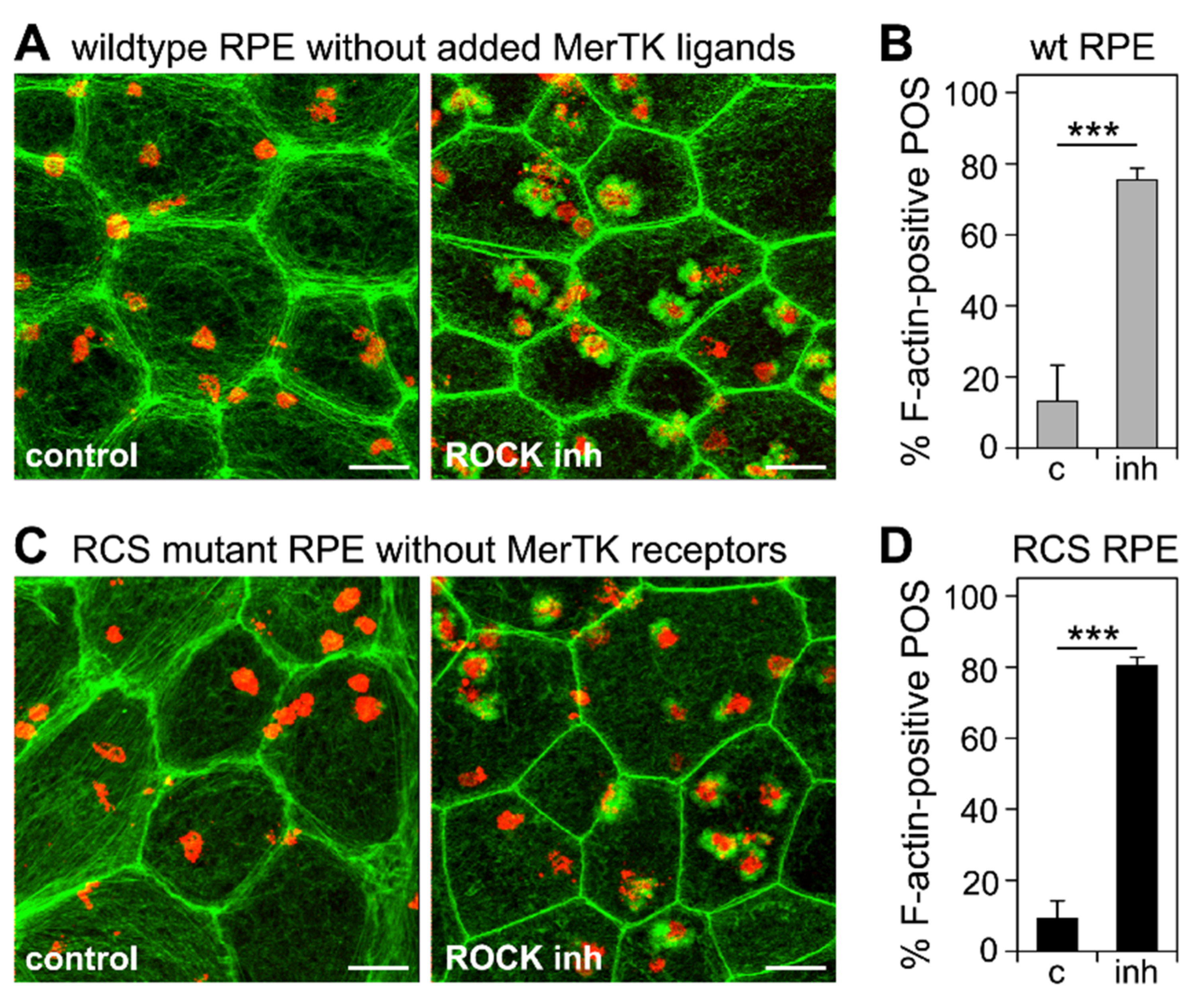
3.3. ROCK Inhibition Rescues Phagocytic Cup Formation by RPE Cells in Which RhoA Is Inhibited
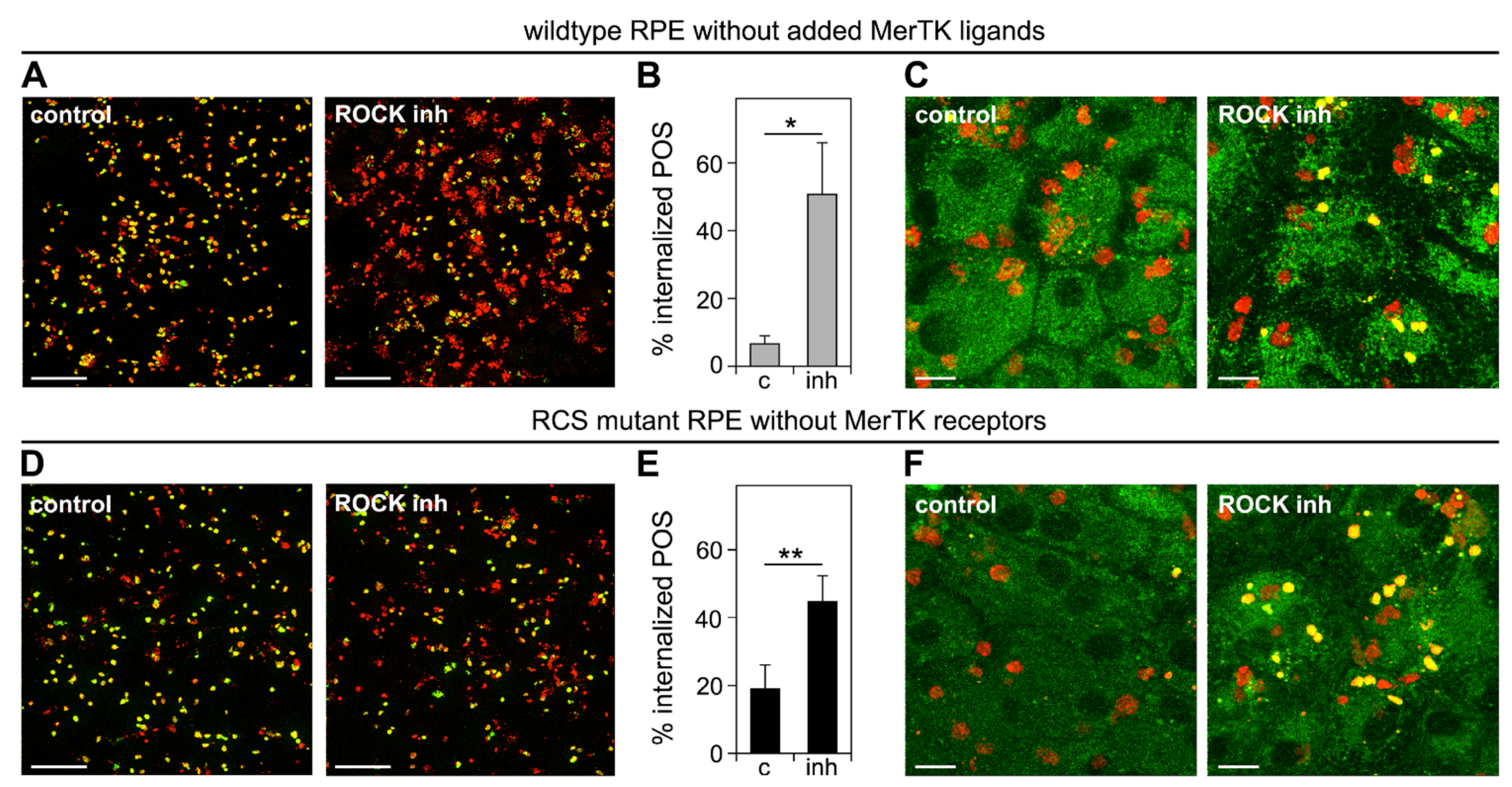
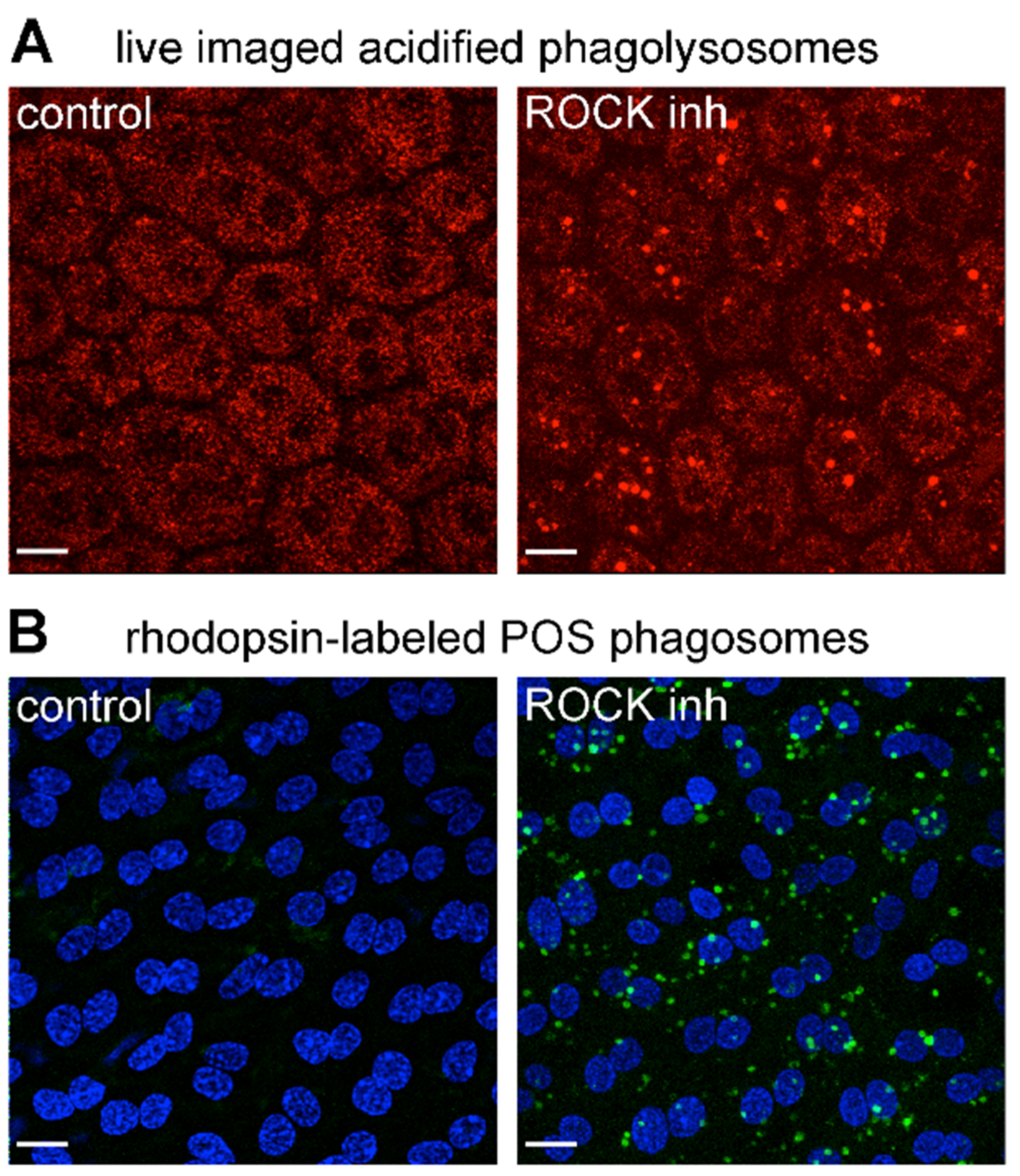
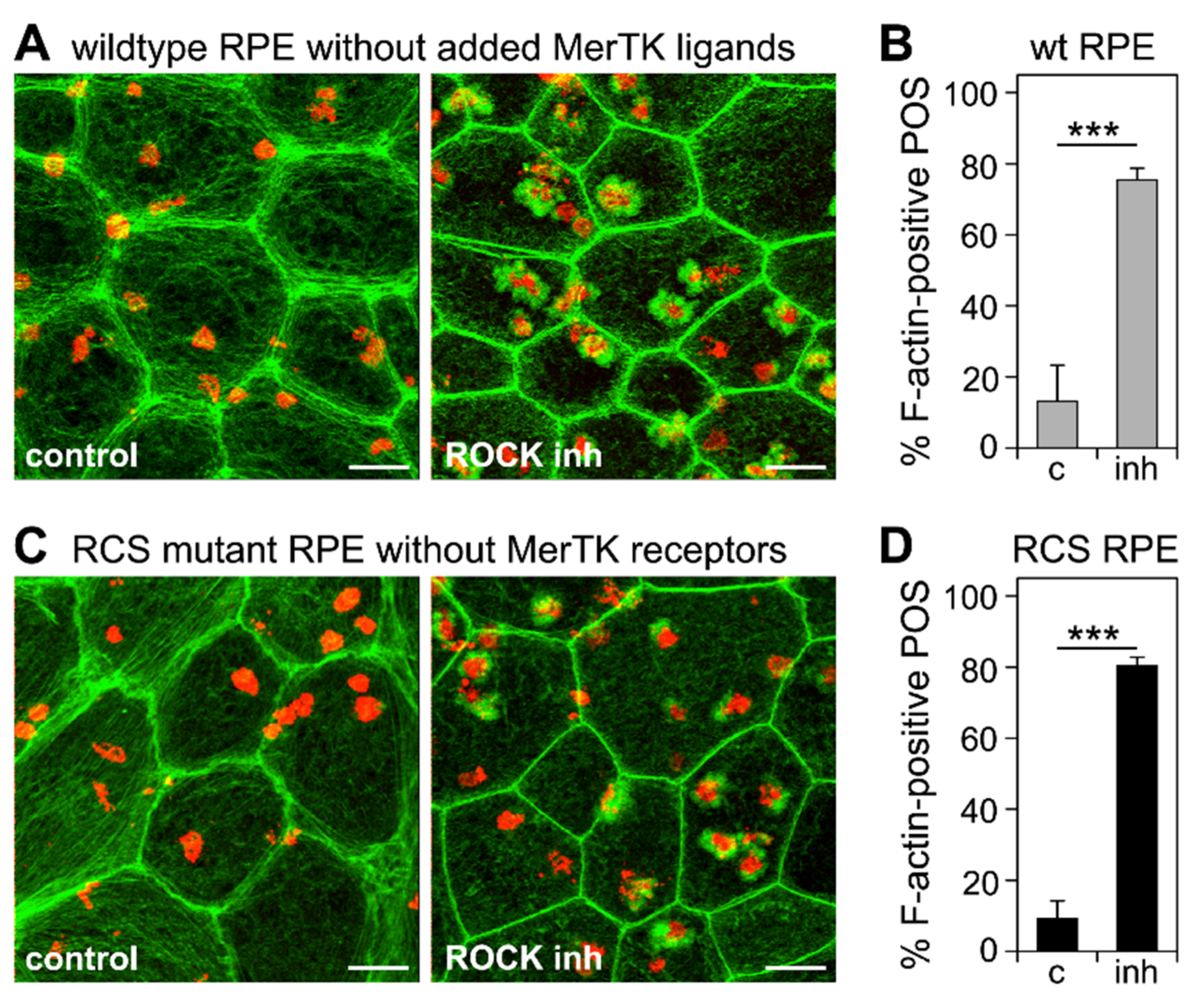
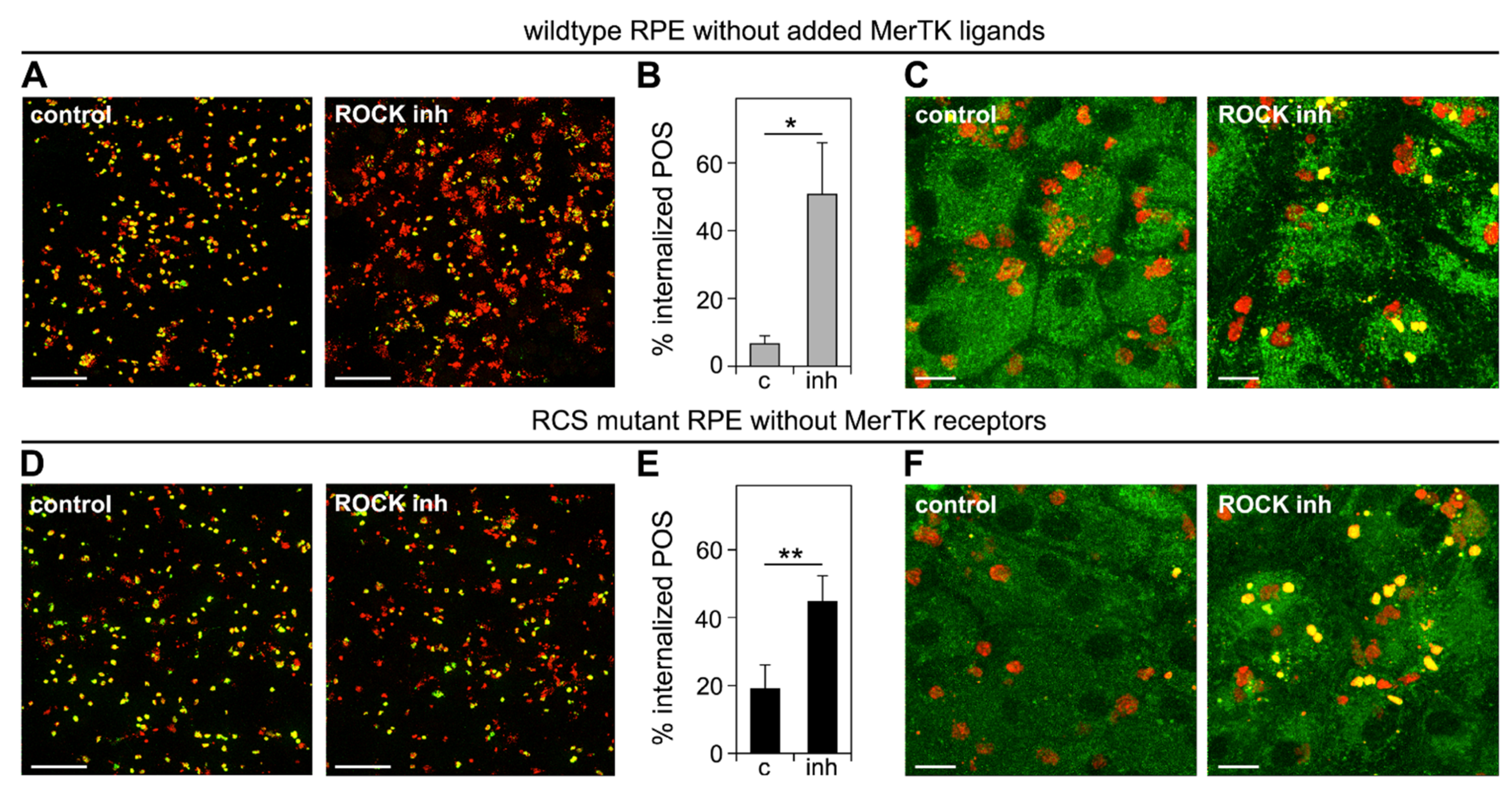
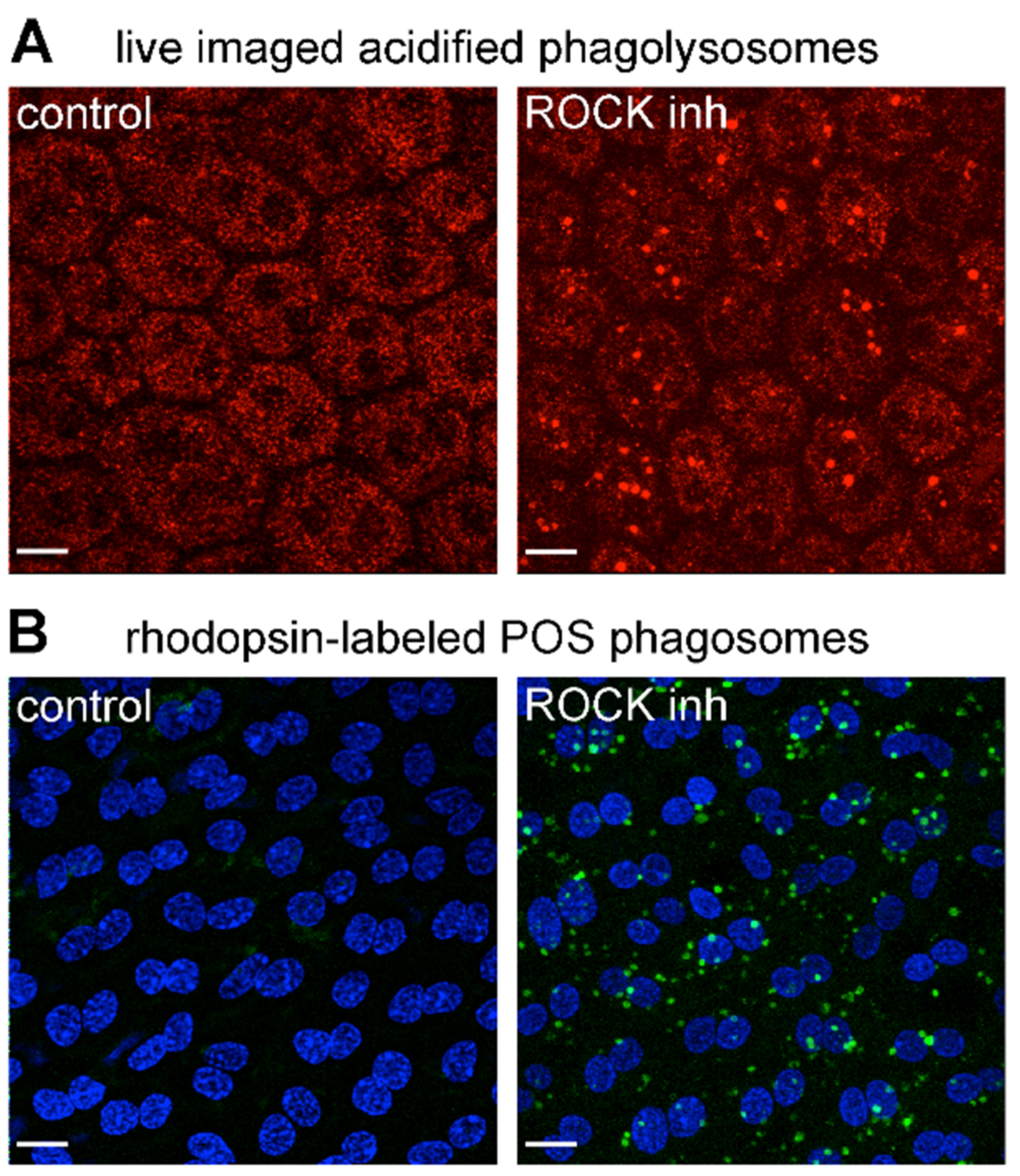
3.4. Inhibition of the RhoA-ROCK Pathway Restores Phagocytic Cup Formation and POS Internalization into Acidified Phagolysosomes to RPE Cells that Lack MerTK Activity
3.5. ROCK Inhibition Promotes Phagocytosis of Endogenous POS by MerTK-Deficient RCS RPE Cells Ex Vivo
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Young, R.W. The renewal of photoreceptor cell outer segments. J. Cell Biol. 1967, 33, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Young, R.W.; Bok, D. Participation of the retinal pigment epithelium in the rod outer segment renewal process. J. Cell Biol. 1969, 42, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Sparrow, J.R.; Boulton, M. RPE lipofuscin and its role in retinal pathobiology. Exp. Eye Res. 2005, 80, 595–606. [Google Scholar] [CrossRef]
- Donato, L.; Abdalla, E.; Scimone, C.; Alibrandi, S.; Rinaldi, C.; Nabil, K.; D’Angelo, R.; Sidoti, A. Impairments of photoreceptor outer segments renewal and phototransduction due to a peripherin rare haplotype variant: Insights from molecular modeling. Int. J. Mol. Sci. 2021, 22, 3484. [Google Scholar] [CrossRef]
- Donato, L.; Scimone, C.; Alibrandi, S.; Pitruzzella, A.; Scalia, F.; D’Angelo, R.; Sidoti, A. Possible A2E mutagenic effects on RPE mitochondrial DNA from innovative RNA-seq bioinformatics pipeline. Antioxidants 2020, 9, 1158. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Muñoz, L.E.; Mallavarapu, M.; Herrmann, M.; Finnemann, S.C. Annexin A5 regulates surface αvβ5 integrin for retinal clearance phagocytosis. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef]
- Donato, L.; Scimone, C.; Alibrandi, S.; Abdalla, E.M.; Nabil, K.M.; D’Angelo, R.; Sidoti, A. New omics–derived perspectives on retinal dystrophies: Could ion channels-encoding or related genes act as modifier of pathological phenotype? Int. J. Mol. Sci. 2020, 22, 70. [Google Scholar] [CrossRef]
- Johansson, J.K.; Karema-Jokinen, V.I.; Hakanen, S.; Jylhä, A.; Uusitalo, H.; Vihinen-Ranta, M.; Skottman, H.; Ihalainen, T.O.; Nymark, S. Sodium channels enable fast electrical signaling and regulate phagocytosis in the retinal pigment epithelium. BMC Biol. 2019, 17, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karl, M.O.; Kroeger, W.; Wimmers, S.; Milenkovic, V.M.; Valtink, M.; Engelmann, K.; Strauss, O. Endogenous Gas6 and Ca2+-channel activation modulate phagocytosis by retinal pigment epithelium. Cell. Signal. 2008, 20, 1159–1168. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.; Gómez, N.M.; Ruth, P.; Strauß, O. CaV1.3 L-type channels, maxiK Ca2+-dependent K+ channels and bestrophin-1 regulate rhythmic photoreceptor outer segment phagocytosis by retinal pigment epithelial cells. Cell. Signal. 2014, 26, 968–978. [Google Scholar] [CrossRef]
- Ruggiero, L.; Connor, M.P.; Chen, J.; Langen, R.; Finnemann, S.C. Diurnal, localized exposure of phosphatidylserine by rod outer segment tips in wild-type but not Itgb5−/− or Mfge8−/− mouse retina. Proc. Natl. Acad. Sci. USA 2012, 109, 8145–8148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finnemann, S.C. Focal adhesion kinase signaling promotes phagocytosis of integrin-bound photoreceptors. EMBO J. 2003, 22, 4143–4154. [Google Scholar] [CrossRef] [Green Version]
- Finnemann, S.C.; Bonilha, V.L.; Marmorstein, A.D.; Rodriguez-Boulan, E. Phagocytosis of rod outer segments by retinal pig-ment epithelial cells requires αvβ5 integrin for binding but not for internalization. Proc. Natl. Acad. Sci. USA 1997, 94, 12932–12937. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.; Finnemann, S.C. Essential diurnal Rac1 activation during retinal phagocytosis requires αvβ5 integrin but not tyrosine kinases focal adhesion kinase or Mer tyrosine kinase. Mol. Biol. Cell 2012, 23, 1104–1114. [Google Scholar] [CrossRef]
- Nandrot, E.F.; Kim, Y.; Brodie, S.; Huang, X.; Sheppard, D.; Finnemann, S.C. Loss of synchronized retinal phagocytosis and age-related blindness in mice lacking αvβ5 integrin. J. Exp. Med. 2004, 200, 1539–1545. [Google Scholar] [CrossRef]
- Burstyn-Cohen, T.; Lew, E.D.; Traves, P.G.; Burrola, P.G.; Hash, J.C.; Lemke, G. Genetic dissection of TAM receptor-ligand interaction in retinal pigment epithelial cell phagocytosis. Neuron 2012, 76, 1123–1132. [Google Scholar] [CrossRef] [Green Version]
- Audo, I.; Mohand-Said, S.; Boulanger-Scemama, E.; Zanlonghi, X.; Condroyer, C.; Démontant, V.; Boyard, F.; Antonio, A.; Méjécase, C.; El Shamieh, S.; et al. MERTK mutation update in inherited retinal diseases. Hum. Mutat. 2018, 39, 887–913. [Google Scholar] [CrossRef] [PubMed]
- Gal, A.; Li, Y.; Thompson, D.; Weir, J.; Orth, U.; Jacobson, S.; Apfelstedt-Sylla, E.; Vollrath, D. Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosa. Nat. Genet. 2000, 26, 270–271. [Google Scholar] [CrossRef]
- Parinot, C.; Nandrot, E.F. A comprehensive review of mutations in the MERTK proto-oncogene. Adv. Exp. Med. Biol. 2015, 854, 259–265. [Google Scholar] [CrossRef]
- D’Cruz, P.M.; Yasumura, D.; Weir, J.; Matthes, M.T.; Abderrahim, H.; LaVail, M.M.; Vollrath, D. Mutation of the receptor tyro-sine kinase gene Mertk in the retinal dystrophic RCS rat. Hum. Mol. Genet. 2000, 9, 645–651. [Google Scholar] [CrossRef] [Green Version]
- Nandrot, E.; Dufour, E.M.; Provost, A.C.; Pequignot, M.O.; Bonnel, S.; Gogat, K.; Marchant, D.; Rouillac, C.; Sepulchre de Conde, B.; Bihoreau, M.T.; et al. Homozygous deletion in the coding sequence of the c-mer gene in RCS rats unravels general mech-anisms of physiological cell adhesion and apoptosis. Neurobiol. Dis. 2000, 7, 586–599. [Google Scholar] [CrossRef] [Green Version]
- Bok, D.; Hall, M.O. The role of the pigment epithelium in the etiology of inherited retinal dystrophy in the rat. J. Cell Biol. 1971, 49, 664–682. [Google Scholar] [CrossRef]
- Duncan, J.L.; Lavail, M.M.; Yasumura, U.; Matthes, M.T.; Yang, H.; Trautmann, N.; Chappelow, A.V.; Feng, W.; Earp, H.S.; Matsushima, G.K.; et al. An RCS-like retinal dystrophy phenotype in MerKnockout mice. Investig. Opthalmol. Vis. Sci. 2003, 44, 826–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vollrath, D.; Yasumura, D.; Benchorin, G.; Matthes, M.T.; Feng, W.; Nguyen, N.M.; Sedano, C.D.; Calton, M.A.; Lavail, M.M. Tyro3 modulates Mertk-associated retinal degeneration. PLoS Genet. 2015, 11, e1005723. [Google Scholar] [CrossRef]
- Shelby, S.J.; Colwill, K.; Dhe-Paganon, S.; Pawson, T.; Thompson, D. MERTK interactions with SH2-domain proteins in the retinal pigment epithelium. PLoS ONE 2013, 8, e53964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todt, J.C.; Hu, B.; Curtis, J.L. The receptor tyrosine kinase MerTK activates phospholipase C gamma2 during recognition of apoptotic thymocytes by murine macrophages. J. Leukoc. Biol. 2004, 75, 705–713. [Google Scholar] [CrossRef]
- Mao, Y.; Finnemann, S.C. Regulation of phagocytosis by Rho GTPases. Small GTPases 2015, 6, 89–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Y.; Finnemann, S.C. Live imaging of LysoTracker-labelled phagolysosomes tracks diurnal phagocytosis of photoreceptor outer segment fragments in Rat RPE tissue ex vivo. Adv. Exp. Med. Biol. 2015, 854, 717–723. [Google Scholar] [CrossRef]
- Adamus, G.; Zam, Z.S.; Arendt, A.; Palczewski, K.; McDowell, J.H.; Hargrave, P.A. Anti-rhodopsin monoclonal antibodies of defined specificity: Characterization and application. Vis. Res. 1991, 31, 17–31. [Google Scholar] [CrossRef]
- Mazzoni, F.; Dun, Y.; Vargas, J.A.; Nandrot, E.F.; Finnemann, S.C. Lack of the antioxidant enzyme methionine sulfoxide reductase A in mice impairs RPE phagocytosis and causes photoreceptor cone dysfunction. Redox Biol. 2021, 42, 101918. [Google Scholar] [CrossRef]
- Parinot, C.; Rieu, Q.; Chatagnon, J.; Finnemann, S.C.; Nandrot, E.F. Large-Scale Purification of Porcine or Bovine Photoreceptor Outer Segments for Phagocytosis Assays on Retinal Pigment Epithelial Cells. J. Vis. Exp. 2014, 12, 52100. [Google Scholar] [CrossRef] [Green Version]
- Mazzoni, F.; Mao, Y.; Finnemann, S.C. Advanced analysis of photoreceptor outer segment phagocytosis by RPE cells in culture. Methods Mol. Biol. 2018, 1834, 95–108. [Google Scholar] [CrossRef]
- Finnemann, S.C.; Rodriguez-Boulan, E. Macrophage and retinal pigment epithelium phagocytosis: Apoptotic cells and pho-toreceptors compete for αvβ3 and αvβ5 integrins, and protein kinase C regulates αvβ5 binding and cytoskeletal linkage. J. Exp. Med. 1999, 190, 861–874. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, G.; Sehr, P.; Wilm, M.; Selzer, J.; Mann, M.; Aktories, K. Gln 63 of Rho is deamidated by Escherichia coli cytotoxic necrotizing factor-1. Nat. Cell Biol. 1997, 387, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Uehata, M.; Ishizaki, T.; Satoh, H.; Ono, T.; Kawahara, T.; Morishita, T.; Tamakawa, H.; Yamagami, K.; Inui, J.; Maekawa, M.; et al. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nat. Cell Biol. 1997, 389, 990–994. [Google Scholar] [CrossRef] [PubMed]
- Moolenaar, W.H.; van Meeteren, L.; Giepmans, B. The ins and outs of lysophosphatidic acid signaling. Bioessays 2004, 26, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Schoenwaelder, S.M.; Burridge, K. Evidence for a calpeptin-sensitive protein-tyrosine phosphatase upstream of the small GTPase rho. A novel role for the calpain inhibitor calpeptin in the inhibition of protein-tyrosine phosphatases. J. Biol. Chem. 1999, 274, 14359–14367. [Google Scholar] [CrossRef] [Green Version]
- Shelby, S.J.; Feathers, K.L.; Ganios, A.M.; Jia, L.; Miller, J.; Thompson, D. MERTK signaling in the retinal pigment epithelium regulates the tyrosine phosphorylation of GDP dissociation inhibitor alpha from the GDI/CHM family of RAB GTPase effectors. Exp. Eye Res. 2015, 140, 28–40. [Google Scholar] [CrossRef] [Green Version]
- Rogers, A.E.J.; Le, J.P.; Sather, S.; Pernu, B.M.; Graham, D.K.; Pierce, A.M.; Keating, A.K. Mer receptor tyrosine kinase inhibition impedes glioblastoma multiforme migration and alters cellular morphology. Oncogene 2011, 31, 4171–4181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penberthy, K.K.; Rival, C.; Shankman, L.S.; Raymond, M.H.; Zhang, J.; Perry, J.S.A.; Lee, C.S.; Han, C.Z.; Onengut-Gumuscu, S.; Palczewski, K.; et al. Context-dependent compensation among phosphatidylserine-recognition receptors. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Allen, L.-A.; Aderem, A. Mechanisms of phagocytosis. Curr. Opin. Immunol. 1996, 8, 36–40. [Google Scholar] [CrossRef]
- Chimini, G.; Chavrier, P. Function of Rho family proteins in actin dynamics during phagocytosis and engulfment. Nat. Cell Biol. 2000, 2, E191–E196. [Google Scholar] [CrossRef]
- Leverrier, Y.; Ridley, A. Requirement for Rho GTPases and PI 3-kinases during apoptotic cell phagocytosis by macrophages. Curr. Biol. 2001, 11, 195–199. [Google Scholar] [CrossRef] [Green Version]
- Nakaya, M.; Tanaka, M.; Okabe, Y.; Hanayama, R.; Nagata, S. Opposite effects of Rho family GTPases on engulfment of apoptotic cells by macrophages. J. Biol. Chem. 2006, 281, 8836–8842. [Google Scholar] [CrossRef] [Green Version]
- Tosello-Trampont, A.-C.; Nakada-Tsukui, K.; Ravichandran, K.S. Engulfment of apoptotic cells is negatively regulated by Rho-mediated signaling. J. Biol. Chem. 2003, 278, 49911–49919. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Singh, S.; Georgescu, M.-M.; Birge, R.B. A role for Mer tyrosine kinase in αvβ5 integrin-mediated phagocytosis of apoptotic cells. J. Cell Sci. 2005, 118, 539–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuji, T.; Ishizaki, T.; Okamoto, M.; Higashida, C.; Kimura, K.; Furuyashiki, T.; Arakawa, Y.; Birge, R.B.; Nakamoto, T.; Hirai, H.; et al. ROCK and mDia1 antagonize in Rho-dependent Rac activation in Swiss 3T3 fibroblasts. J. Cell Biol. 2002, 157, 819–830. [Google Scholar] [CrossRef] [Green Version]
- Maekawa, M.; Ishizaki, T.; Boku, S.; Watanabe, N.; Fujita, A.; Iwamatsu, A.; Obinata, T.; Ohashi, K.; Mizuno, K.; Narumiya, S. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science 1999, 285, 895–898. [Google Scholar] [CrossRef]
- Narumiya, S.; Thumkeo, D. Rho signaling research: History, current status and future directions. FEBS Lett. 2018, 592, 1763–1776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faure, S.; Salazar-Fontana, L.I.; Semichon, M.; Tybulewicz, V.; Bismuth, G.; Trautmann, A.; Germain, R.N.; Delon, J. ERM proteins regulate cytoskeleton relaxation promoting T cell–APC conjugation. Nat. Immunol. 2004, 5, 272–279. [Google Scholar] [CrossRef]
- Ghazi, N.; Abboud, E.B.; Nowilaty, S.R.; Alkuraya, H.; Alhommadi, A.; Cai, H.; Hou, R.; Deng, W.-T.; Boye, S.L.; Almaghamsi, A.; et al. Treatment of retinitis pigmentosa due to MERTK mutations by ocular subretinal injection of adeno-associated virus gene vector: Results of a phase I trial. Qual. Life Res. 2016, 135, 327–343. [Google Scholar] [CrossRef]
- Garafalo, A.V.; Cideciyan, A.V.; Héon, E.; Sheplock, R.; Pearson, A.; Yu, C.W.; Sumaroka, A.; Aguirre, G.D.; Jacobson, S.G. Progress in treating inherited retinal diseases: Early subretinal gene therapy clinical trials and candidates for future initiatives. Prog. Retin. Eye Res. 2020, 77, 100827. [Google Scholar] [CrossRef]
- Castro, A.A.; Long, K.; Bassett, A.; Machuca, C.; León, M.; Ávila-Fernandez, A.; Corton, M.; Vidal-Puig, T.; Ayuso, C.; Lukovic, D.; et al. Generation of gene-corrected human induced pluripotent stem cell lines derived from retinitis pigmentosa patient with Ser331Cysfs*5 mutation in MERTK. Stem Cell Res. 2019, 34, 101341. [Google Scholar] [CrossRef]
- Artero-Castro, A.; Long, K.; Bassett, A.; Ávila-Fernandez, A.; Cortón, M.; Vidal-Puig, A.; Jendelova, P.; Rodriguez-Jimenez, F.; Clemente, E.; Ayuso, C.; et al. Gene correction recovers phagocytosis in retinal pigment epithelium derived from retinitis pigmentosa-human-induced pluripotent stem cells. Int. J. Mol. Sci. 2021, 22, 2092. [Google Scholar] [CrossRef] [PubMed]
- Ramsden, C.M.; Nommiste, B.; Lane, A.R.; Carr, A.-J.F.; Powner, M.B.; Smart, M.J.K.; Chen, L.L.; Muthiah, M.N.; Webster, A.R.; Moore, A.T.; et al. Rescue of the MERTK phagocytic defect in a human iPSC disease model using translational read-through inducing drugs. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Humimat, G.; Marashdeh, I.; Daradkeh, D.; Kooner, K. Investigational Rho kinase inhibitors for the treatment of glaucoma. J. Exp. Pharmacol. 2021, 13, 197–212. [Google Scholar] [CrossRef]
- Lew, D.S.; Mazzoni, F.; Finnemann, S.C. microglia inhibition delays retinal degeneration due to MerTK phagocytosis receptor deficiency. Front. Immunol. 2020, 11, 1463. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mao, Y.; Finnemann, S.C. Acute RhoA/Rho Kinase Inhibition Is Sufficient to Restore Phagocytic Capacity to Retinal Pigment Epithelium Lacking the Engulfment Receptor MerTK. Cells 2021, 10, 1927. https://doi.org/10.3390/cells10081927
Mao Y, Finnemann SC. Acute RhoA/Rho Kinase Inhibition Is Sufficient to Restore Phagocytic Capacity to Retinal Pigment Epithelium Lacking the Engulfment Receptor MerTK. Cells. 2021; 10(8):1927. https://doi.org/10.3390/cells10081927
Chicago/Turabian StyleMao, Yingyu, and Silvia C. Finnemann. 2021. "Acute RhoA/Rho Kinase Inhibition Is Sufficient to Restore Phagocytic Capacity to Retinal Pigment Epithelium Lacking the Engulfment Receptor MerTK" Cells 10, no. 8: 1927. https://doi.org/10.3390/cells10081927
APA StyleMao, Y., & Finnemann, S. C. (2021). Acute RhoA/Rho Kinase Inhibition Is Sufficient to Restore Phagocytic Capacity to Retinal Pigment Epithelium Lacking the Engulfment Receptor MerTK. Cells, 10(8), 1927. https://doi.org/10.3390/cells10081927