
Liquid Biopsy for Disease Monitoring in Non-Small Cell Lung Cancer: The Link between Biology and the Clinic

 , ,
, ,  , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
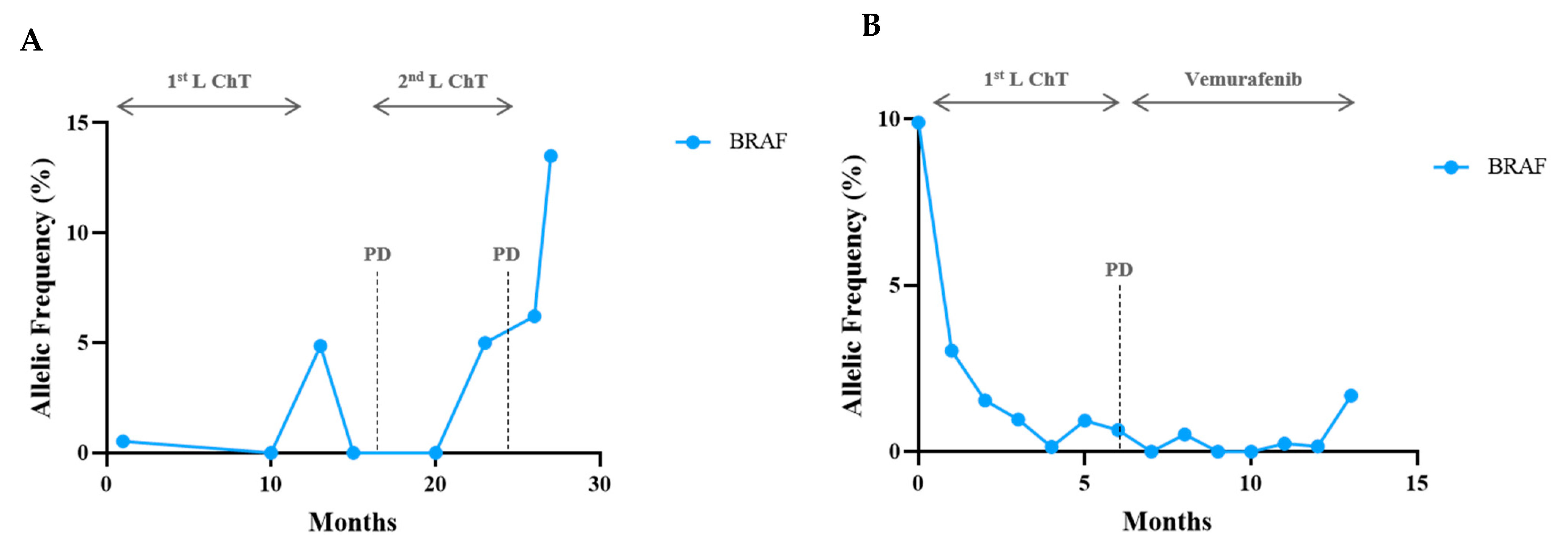
3. Results
Sequencing Results and Correlation between Molecular Monitoring and Clinical Outcomes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kris, M.G.; Johnson, B.E.; Berry, L.D.; Kwiatkowski, D.J.; Iafrate, A.J.; Wistuba, I.I.; Varella-Garcia, M.; Franklin, W.A.; Aronson, S.L.; Su, P.-F.; et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014, 311, 1998–2006. [Google Scholar] [CrossRef] [PubMed]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, iv192–iv237. [Google Scholar] [CrossRef]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001, 61, 1659–1665. [Google Scholar]
- Luo, J.; Shen, L.; Zheng, D. Diagnostic value of circulating free DNA for the detection of EGFR mutation status in NSCLC: A systematic review and meta-analysis. Sci. Rep. 2014, 4, 6269. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zhang, X.; Wang, F.; Zhang, M.; Sun, B.; Yin, W.; Deng, S.; Wan, Y.; Lu, W. The Diagnostic Accuracy of Liquid Biopsy in EGFR-Mutated NSCLC: A Systematic Review and Meta-Analysis of 40 Studies. Slas Technol 2021, 26, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhuo, M.; Ye, X.; Bai, H.; Wang, Z.; Sun, Y.; Zhao, J.; An, T.; Duan, J.; Wu, M.; et al. Quantification of mutant alleles in circulating tumor DNA can predict survival in lung cancer. Oncotarget 2016, 7, 20810–20824. [Google Scholar] [CrossRef]
- Li, Y.S.; Jiang, B.Y.; Yang, J.J.; Zhang, X.C.; Zhang, Z.; Ye, J.Y.; Zhong, W.Z.; Tu, H.Y.; Chen, H.J.; Wang, Z.; et al. Unique genetic profiles from cerebrospinal fluid cell-free DNA in leptomeningeal metastases of EGFR-mutant non-small-cell lung cancer: A new medium of liquid biopsy. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 945–952. [Google Scholar] [CrossRef]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Fernandes, M.G.O.; Cruz-Martins, N.; Souto Moura, C.; Guimarães, S.; Pereira Reis, J.; Justino, A.; Pina, M.J.; Magalhães, A.; Queiroga, H.; Machado, J.C.; et al. Clinical Application of Next-Generation Sequencing of Plasma Cell-Free DNA for Genotyping Untreated Advanced Non-Small Cell Lung Cancer. Cancers 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.; Wu, Y.L.; Lee, J.S.; Yu, C.J.; Sriuranpong, V.; Sandoval-Tan, J.; Ladrera, G.; Thongprasert, S.; Srimuninnimit, V.; Liao, M.; et al. Detection and Dynamic Changes of EGFR Mutations from Circulating Tumor DNA as a Predictor of Survival Outcomes in NSCLC Patients Treated with First-line Intercalated Erlotinib and Chemotherapy. Clin. Cancer Res. 2015, 21, 3196–3203. [Google Scholar] [CrossRef]
- Douillard, J.-Y.; Ostoros, G.; Cobo, M.; Ciuleanu, T.; Cole, R.; McWalter, G.; Walker, J.; Dearden, S.; Webster, A.; Milenkova, T.; et al. Gefitinib treatment in EGFR mutated caucasian NSCLC: Circulating-free tumor DNA as a surrogate for determination of EGFR status. J. Thorac. Oncol. 2014, 9, 1345–1353. [Google Scholar] [CrossRef] [PubMed]
- Sueoka-Aragane, N.; Katakami, N.; Satouchi, M.; Yokota, S.; Aoe, K.; Iwanaga, K.; Otsuka, K.; Morita, S.; Kimura, S.; Negoro, S.; et al. Monitoring EGFR T790M with plasma DNA from lung cancer patients in a prospective observational study. Cancer Sci. 2016, 107, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Ye, X.; Zhang, M.Z.; Sun, Y.; Wang, J.Y.; Ni, J.; Zhang, H.P.; Zhang, L.; Luo, J.; Zhang, J.; et al. Plasma EGFR T790M ctDNA status is associated with clinical outcome in advanced NSCLC patients with acquired EGFR-TKI resistance. Sci. Rep. 2016, 6, 20913. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, Z.; Niederst, M.J.; Karlovich, C.A.; Wakelee, H.A.; Neal, J.W.; Mino-Kenudson, M.; Fulton, L.; Hata, A.N.; Lockerman, E.L.; Kalsy, A.; et al. Heterogeneity Underlies the Emergence of EGFRT790 Wild-Type Clones Following Treatment of T790M-Positive Cancers with a Third-Generation EGFR Inhibitor. Cancer Discov. 2015, 5, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Helman, E.; Nguyen, M.; Karlovich, C.A.; Despain, D.; Choquette, A.K.; Spira, A.I.; Yu, H.A.; Camidge, D.R.; Harding, T.C.; Lanman, R.B.; et al. Cell-Free DNA Next-Generation Sequencing Prediction of Response and Resistance to Third-Generation EGFR Inhibitor. Clin. Lung Cancer 2018, 19, 518–530.e517. [Google Scholar] [CrossRef]
- Papadimitrakopoulou, V.A.; Wu, Y.L.; Han, J.Y.; Ahn, M.J.; Ramalingam, S.S.; John, T.; Okamoto, I.; Yang, J.C.H.; Bulusu, K.C.; Laus, G.; et al. Analysis of resistance mechanisms to osimertinib in patients with EGFR T790M advanced NSCLC from the AURA3 study. Ann. Oncol. 2018, 29, viii741. [Google Scholar] [CrossRef]
- Bordi, P.; Del Re, M.; Minari, R.; Rofi, E.; Buti, S.; Restante, G.; Squadrilli, A.; Crucitta, S.; Casartelli, C.; Gnetti, L.; et al. From the beginning to resistance: Study of plasma monitoring and resistance mechanisms in a cohort of patients treated with osimertinib for advanced T790M-positive NSCLC. Lung Cancer 2019, 131, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.; Ladrera, G.; Srimuninnimit, V.; Sriuranpong, V.; Yu, C.J.; Thongprasert, S.; Sandoval-Tan, J.; Lee, J.S.; Fuerte, F.; Shames, D.S.; et al. Tumor marker analyses from the phase III, placebo-controlled, FASTACT-2 study of intercalated erlotinib with gemcitabine/platinum in the first-line treatment of advanced non-small-cell lung cancer. Lung Cancer 2016, 98, 1–8. [Google Scholar] [CrossRef][Green Version]
- Thress, K.S.; Markovets, A.; Barrett, J.C.; Chmielecki, J.; Goldberg, S.B.; Shepherd, F.A.; Vowler, S.; Oxnard, G.R. Complete clearance of plasma EGFR mutations as a predictor of outcome on osimertinib in the AURA trial. J. Clin. Oncol. 2017, 35, 9018. [Google Scholar] [CrossRef]
- Phallen, J.; Leal, A.; Woodward, B.D.; Forde, P.M.; Naidoo, J.; Marrone, K.A.; Brahmer, J.R.; Fiksel, J.; Medina, J.E.; Cristiano, S.; et al. Early Noninvasive Detection of Response to Targeted Therapy in Non–Small Cell Lung Cancer. Cancer Res. 2019, 79, 1204–1213. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Paweletz, C.P.; Kuang, Y.; Mach, S.L.; O’Connell, A.; Messineo, M.M.; Luke, J.J.; Butaney, M.; Kirschmeier, P.; Jackman, D.M.; et al. Noninvasive Detection of Response and Resistance in EGFR-Mutant Lung Cancer Using Quantitative Next-Generation Genotyping of Cell-Free Plasma DNA. Clin. Cancer Res. 2014, 20, 1698–1705. [Google Scholar] [CrossRef]
- Karlovich, C.; Goldman, J.W.; Sun, J.-M.; Mann, E.; Sequist, L.V.; Konopa, K.; Wen, W.; Angenendt, P.; Horn, L.; Spigel, D.; et al. Assessment of EGFR Mutation Status in Matched Plasma and Tumor Tissue of NSCLC Patients from a Phase I Study of Rociletinib (CO-1686). Clin. Cancer Res. 2016, 22, 2386–2395. [Google Scholar] [CrossRef] [PubMed]
- Ahlborn, L.B.; Tuxen, I.V.; Mouliere, F.; Kinalis, S.; Schmidt, A.Y.; Rohrberg, K.S.; Santoni-Rugiu, E.; Nielsen, F.C.; Lassen, U.; Yde, C.W.; et al. Circulating tumor DNA as a marker of treatment response in BRAF V600E mutated non-melanoma solid tumors. Oncotarget 2018, 9, 32570–32579. [Google Scholar] [CrossRef] [PubMed]
- Malapelle, U.; Buono, M.; Pisapia, P.; Russo, G.; Tufano, R.; Pepe, F.; Rolfo, C.; Troncone, G. Circulating tumor DNA in cancer: Predictive molecular pathology meets mathematics. Crit. Rev. Oncol. Hematol. 2021, 163, 103394. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Yu, C.-J.; Kim, S.-W.; Lin, M.-C.; Sriuranpong, V.; Tsai, C.-M.; Lee, J.-S.; Kang, J.-H.; Chan, K.C.A.; Perez-Moreno, P.; et al. First-Line Erlotinib Therapy Until and Beyond Response Evaluation Criteria in Solid Tumors Progression in Asian Patients with Epidermal Growth Factor Receptor Mutation-Positive Non-Small-Cell Lung Cancer: The ASPIRATION Study. JAMA Oncol. 2016, 2, 305–312. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | Gender | Age | Genomics | Aa | Tissue DNA VAF% | ctDNA VAF % | Progression Pattern | cfDNA Concentration (µg/µL) | Tissue Rebiopsy at Progression | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Baseline | Best Response | Progression | Baseline | Best Response | Progression | ||||||||
| 2 | F | 38 | EGFR | c.2240_2257del18 | 40.00 | 0.72 | 0.00 | 24.30 | VAF increase | 3.36 | 1.03 | 15.60 | T790M negative |
| 15 | M | 60 | KRAS | c.182A > G | 8.80 | 0.43 | 0.00 | 19.60 | VAF increase | 2.27 | 31.70 | 3.17 | |
| TP53 | c.344G > T | 25.00 | 0.71 | 0.00 | 36.90 | ||||||||
| STK1 | c.597G > T | 12.80 | 0.53 | 0.00 | 24.00 | ||||||||
| 62 | M | 60 | BRAF | c.1799T > A | 36.30 | 0.53 | 0.00 | 13.50 | VAF increase | 3.93 | 0.29 | 2.22 | |
| 74 | M | 59 | EGFR | c.2236_2250del15 | 11.10 | 0.27 | 0.00 | 2.55 | VAF increase andde novo T790M | 17.70 | 1.12 | 0.75 | T790M positive |
| EGFR | c.2369C > T | 0.00 | 0.00 | 0.00 | 0.65 | ||||||||
| KRAS | c.182A > G | 0.38 | 0.07 | 0.00 | 0.00 | ||||||||
| 80 | F | 58 | BRAF | c.1799T > A | 50.40 | 9.90 | 0.14 | 1.69 | VAF increase | 1.55 | 0.86 | 0.63 | |
| TP53 | c.476C > G | 39.20 | 4.90 | 0.00 | 0.00 | ||||||||
| 81 | M | 40 | EGFR | c.2236_2250del15 | 36.70 | 0.18 | 0.00 | 11.85 | VAF increase andde novo T790M | 1.62 | 0.29 | 1.73 | T790M positive |
| c.2369C > T | 15.00 | 0.14 | 0.00 | 4.67 | |||||||||
| 89 | M | 64 | EGFR | c.2237_2255del18 | 0.00 | 0.00 | 0.00 | No change | 0.35 | 0.52 | 0.34 | T790M negative | |
| 91 | F | 76 | EGFR | c.2573T > G | 1.72 | 0.00 | 27.55 | VAF increase and de novo T790M | 0.47 | 0.95 | |||
| C.2369C > T | 0.28 | 0.00 | 12.50 | ||||||||||
| 95 | F | 59 | EGFR | c.2240_2257del15 | 96.50 | 0.65 | 0.21 | 1.95 | VAF increase and de novo T790M | 0.55 | 0.58 | 0.59 | |
| c.2369C > T | 0.00 | 0.00 | 0.00 | 1.40 | |||||||||
| 107 | F | 54 | EGFR | c.2240_2257del18 | 67.20 | 5.50 | 3.62 | 0.13 | VAF increase in the T790M | 1.03 | 1.53 | ||
| c.2369C > T | 1.25 | 0.00 | 2.69 | 8.46 | |||||||||
| 130 | M | 63 | EGFR | c.2235_2249del15 | 13.20 | 1.06 | 0.00 | 11.05 | VAF increase and de novo T790M | 0.56 | 0.26 | 4.15 | T790M positive |
| c.2369C > T | 0.00 | 0.00 | 0.00 | 5.88 | |||||||||
| 38 | F | 65 | EGFR | c.2573T > G | 6.30 | 0.91 | 0.12 | 1.94 | VAF increase | 1.98 | 1.39 | 24.10 | |
| 65 | F | 66 | EGFR | c.2235_2249del15 | 7.60 | 2.01 | 0.00 | 0.00 | No change | 0.281 | 0.214 | 0.396 | T790M positive |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandes, M.G.O.; Sousa, C.; Pereira Reis, J.; Cruz-Martins, N.; Souto Moura, C.; Guimarães, S.; Justino, A.; Pina, M.J.; Magalhães, A.; Queiroga, H.; et al. Liquid Biopsy for Disease Monitoring in Non-Small Cell Lung Cancer: The Link between Biology and the Clinic. Cells 2021, 10, 1912. https://doi.org/10.3390/cells10081912
Fernandes MGO, Sousa C, Pereira Reis J, Cruz-Martins N, Souto Moura C, Guimarães S, Justino A, Pina MJ, Magalhães A, Queiroga H, et al. Liquid Biopsy for Disease Monitoring in Non-Small Cell Lung Cancer: The Link between Biology and the Clinic. Cells. 2021; 10(8):1912. https://doi.org/10.3390/cells10081912
Chicago/Turabian StyleFernandes, Maria Gabriela O., Catarina Sousa, Joana Pereira Reis, Natália Cruz-Martins, Conceição Souto Moura, Susana Guimarães, Ana Justino, Maria João Pina, Adriana Magalhães, Henrique Queiroga, and et al. 2021. "Liquid Biopsy for Disease Monitoring in Non-Small Cell Lung Cancer: The Link between Biology and the Clinic" Cells 10, no. 8: 1912. https://doi.org/10.3390/cells10081912
APA StyleFernandes, M. G. O., Sousa, C., Pereira Reis, J., Cruz-Martins, N., Souto Moura, C., Guimarães, S., Justino, A., Pina, M. J., Magalhães, A., Queiroga, H., Marques, J. A., Machado, J. C., Costa, J. L., & Hespanhol, V. (2021). Liquid Biopsy for Disease Monitoring in Non-Small Cell Lung Cancer: The Link between Biology and the Clinic. Cells, 10(8), 1912. https://doi.org/10.3390/cells10081912