
Drug Tolerance to EGFR Tyrosine Kinase Inhibitors in Lung Cancers with EGFR Mutations



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
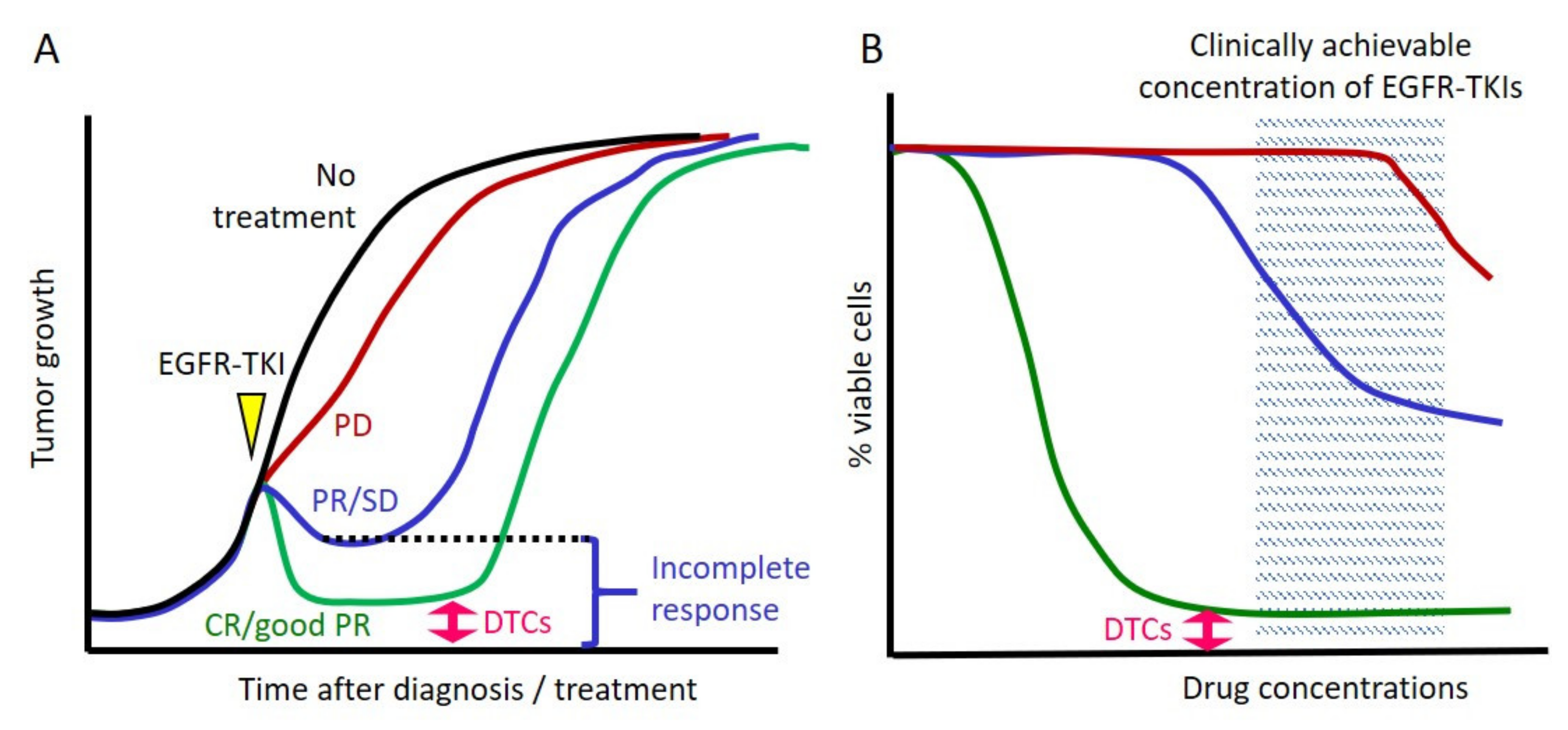
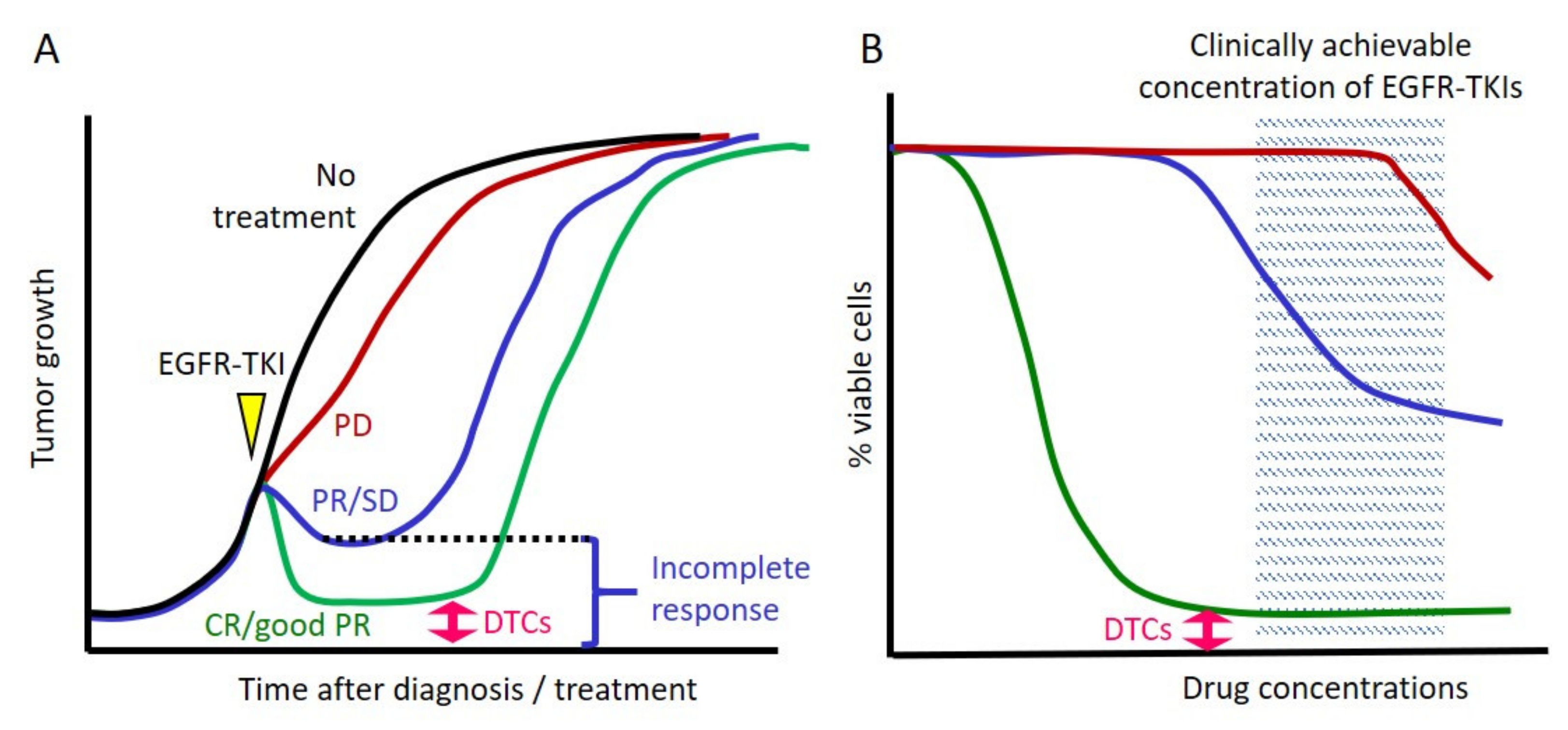
2. Incomplete Response to EGFR TKIs and the Concept of Drug-Tolerant Cells
2.1. Cancer Cells Always Survive after Exposure to EGFR TKIs in Clinical Settings
2.2. Cell Line Models That Mimic Clinical Responses to EGFR TKIs
2.3. Inherent EGFR TKI Insensitivity in Cell Line Models
2.4. The Concept of DTCs
3. Features of DTCs
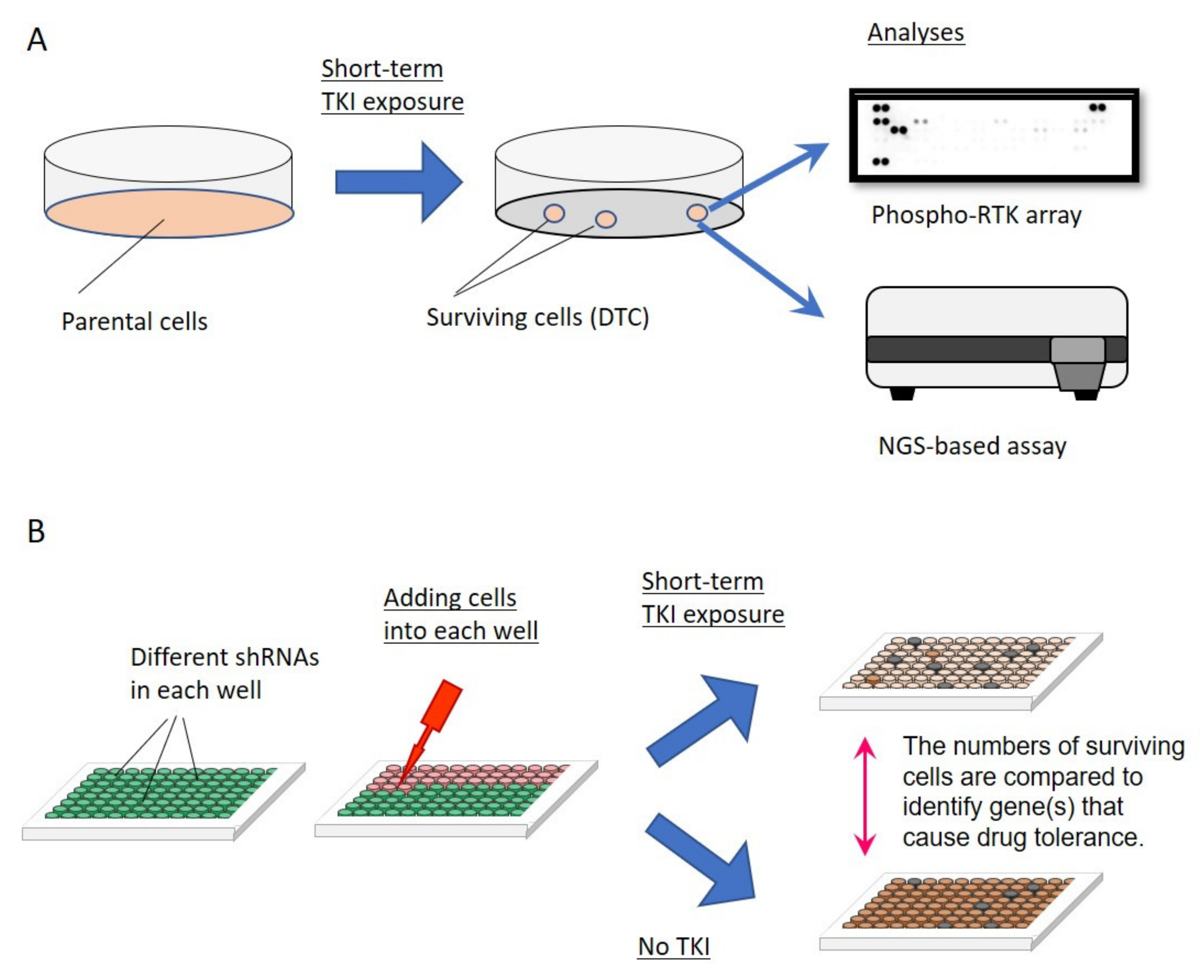
3.1. How Are DTCs Induced upon Exposure to an EGFR TKI?
3.2. Which Cells Will Become DTCs upon Exposure to EGFR TKIs?
3.3. Establishment of DTCs and Optimizing the Concentrations of EGFR TKIs
3.4. “Preference” for Drug Tolerance Mechanisms of Each Cell Line
3.5. Diversity of Molecular Mechanisms That Mediate Drug Tolerance
4. Summary of Molecular Mechanisms Conferring Drug Tolerance in EGFR-Mutated Lung Cancer Cell Lines
4.1. Search Criteria for Published Studies
4.2. Mechanisms of Drug Tolerance—Activation of Bypass Signaling
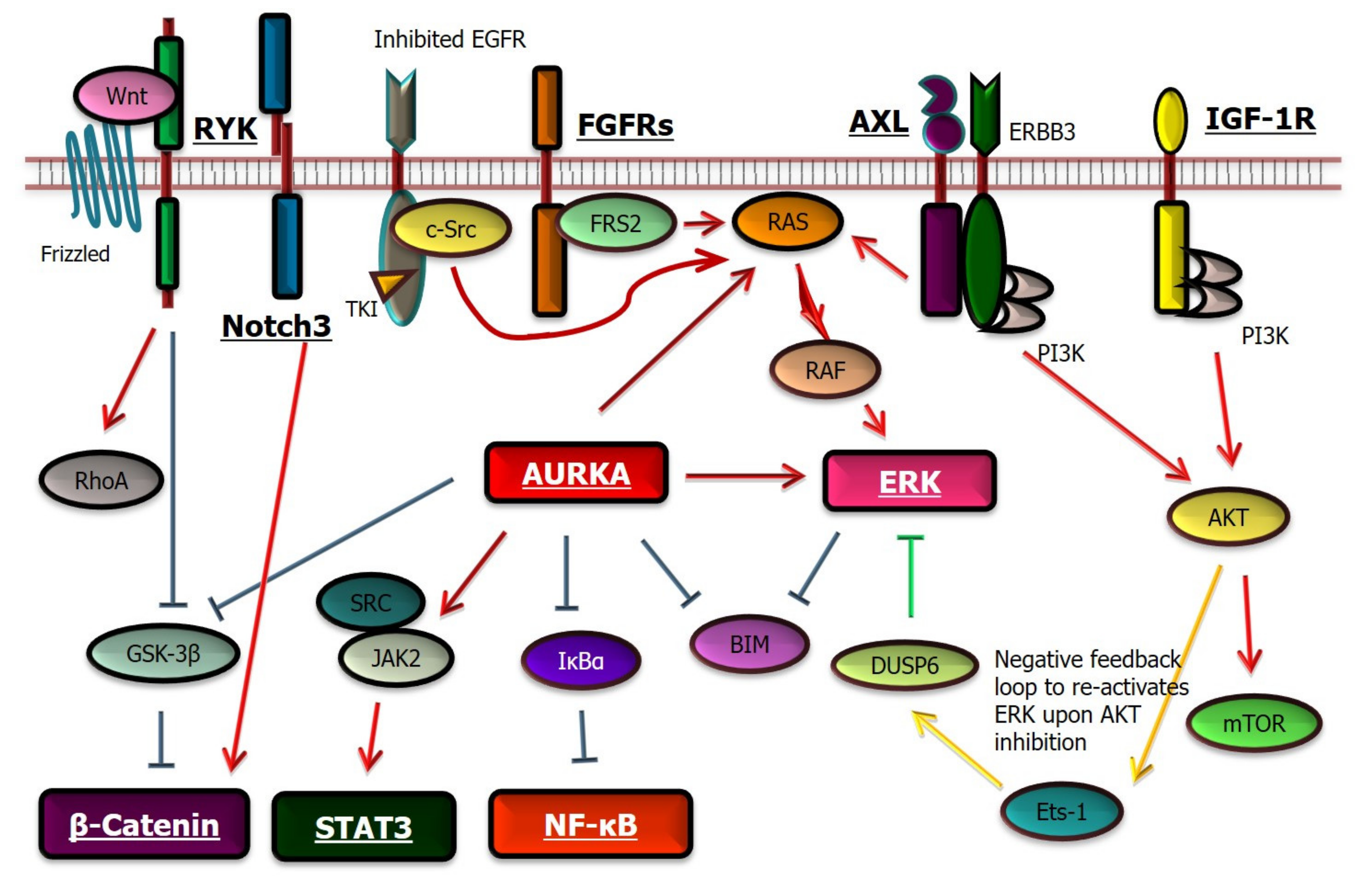
4.3. Mechanisms of Drug Tolerance—Dysregulation of the Apoptotic and Other Pathways
5. Summary
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hirsch, F.R.; Suda, K.; Wiens, J.; Bunn, P.A., Jr. New and emerging targeted treatments in advanced non-small-cell lung cancer. Lancet 2016, 388, 1012–1024. [Google Scholar] [CrossRef]
- Suda, K.; Tomizawa, K.; Mitsudomi, T. Biological and clinical significance of KRAS mutations in lung cancer: An oncogenic driver that contrasts with EGFR mutation. Cancer Metastasis Rev. 2010, 29, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Suda, K.; Bunn, P.A., Jr.; Rivard, C.J.; Mitsudomi, T.; Hirsch, F.R. Primary Double-Strike Therapy for Cancers to Overcome EGFR Kinase Inhibitor Resistance: Proposal from the Bench. J. Thorac. Oncol. 2017, 12, 27–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsudomi, T.; Morita, S.; Yatabe, Y.; Negoro, S.; Okamoto, I.; Tsurutani, J.; Seto, T.; Satouchi, M.; Tada, H.; Hirashima, T.; et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet Oncol. 2010, 11, 121–128. [Google Scholar] [CrossRef]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- Wu, Y.L.; Zhou, C.; Hu, C.P.; Feng, J.; Lu, S.; Huang, Y.; Li, W.; Hou, M.; Shi, J.H.; Lee, K.Y.; et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): An open-label, randomised phase 3 trial. Lancet Oncol. 2014, 15, 213–222. [Google Scholar] [CrossRef]
- Wu, Y.L.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Tsuji, F.; Linke, R.; Rosell, R.; Corral, J.; et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): A randomised, open-label, phase 3 trial. Lancet Oncol. 2017, 18, 1454–1466. [Google Scholar] [CrossRef]
- Soria, J.-C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef]
- Nakagawa, K.; Garon, E.B.; Seto, T.; Nishio, M.; Aix, S.P.; Paz-Ares, L.; Chiu, C.-H.; Park, K.; Novello, S.; Nadal, E.; et al. Ramucirumab plus erlotinib in patients with untreated, EGFR-mutated, advanced non-small-cell lung cancer (RELAY): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 1655–1669. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Janne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef]
- Suda, K.; Mizuuchi, H.; Maehara, Y.; Mitsudomi, T. Acquired resistance mechanisms to tyrosine kinase inhibitors in lung cancer with activating epidermal growth factor receptor mutation-diversity, ductility, and destiny. Cancer Metastasis Rev. 2012, 31, 807–814. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.L.; Ahn, M.J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N. Engl. J. Med. 2016. [Google Scholar] [CrossRef] [Green Version]
- Suda, K.; Tomizawa, K.; Osada, H.; Maehara, Y.; Yatabe, Y.; Sekido, Y.; Mitsudomi, T. Conversion from the “oncogene addiction” to “drug addiction” by intensive inhibition of the EGFR and MET in lung cancer with activating EGFR mutation. Lung Cancer 2012, 76, 292–299. [Google Scholar] [CrossRef]
- Mizuuchi, H.; Suda, K.; Murakami, I.; Sakai, K.; Sato, K.; Kobayashi, Y.; Shimoji, M.; Chiba, M.; Sesumi, Y.; Tomizawa, K.; et al. Oncogene swap as a novel mechanism of acquired resistance to epidermal growth factor receptor-tyrosine kinase inhibitor in lung cancer. Cancer Sci. 2016, 107, 461–468. [Google Scholar] [CrossRef]
- Yatabe, Y.; Matsuo, K.; Mitsudomi, T. Heterogeneous distribution of EGFR mutations is extremely rare in lung adenocarcinoma. J. Clin. Oncol. 2011, 29, 2972–2977. [Google Scholar] [CrossRef]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [Green Version]
- Suda, K.; Sakai, K.; Obata, K.; Ohara, S.; Fujino, T.; Koga, T.; Hamada, A.; Soh, J.; Nishio, K.; Mitsudomi, T. Inter- and Intratumor Heterogeneity of EGFR Compound Mutations in Non-Small Cell Lung Cancers: Analysis of Five Cases. Clin. Lung Cancer 2021, 22, e141–e145. [Google Scholar] [CrossRef]
- Nakagawa, T.; Takeuchi, S.; Yamada, T.; Ebi, H.; Sano, T.; Nanjo, S.; Ishikawa, D.; Sato, M.; Hasegawa, Y.; Sekido, Y.; et al. EGFR-TKI Resistance Due to BIM Polymorphism Can Be Circumvented in Combination with HDAC Inhibition. Cancer Res. 2013, 73, 2428–2434. [Google Scholar] [CrossRef] [Green Version]
- Ohara, S.; Suda, K.; Mitsudomi, T. Cell Line Models for Acquired Resistance to First-Line Osimertinib in Lung Cancers-Applications and Limitations. Cells 2021, 10, 354. [Google Scholar] [CrossRef]
- Ng, K.P.; Hillmer, A.M.; Chuah, C.T.; Juan, W.C.; Ko, T.K.; Teo, A.S.; Ariyaratne, P.N.; Takahashi, N.; Sawada, K.; Fei, Y.; et al. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat. Med. 2012, 18, 521–528. [Google Scholar] [CrossRef]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.E.; Krishnamurthy Radhakrishna, V.; et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med. 2016, 22, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, M.; Rajaram, S.; Steininger, R.J.; Osipchuk, D.; Roth, M.A.; Morinishi, L.S.; Evans, L.; Ji, W.; Hsu, C.H.; Thurley, K.; et al. Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat. Commun. 2016, 7, 10690. [Google Scholar] [CrossRef]
- Bivona, T.G.; Hieronymus, H.; Parker, J.; Chang, K.; Taron, M.; Rosell, R.; Moonsamy, P.; Dahlman, K.; Miller, V.A.; Costa, C.; et al. FAS and NF-kappaB signalling modulate dependence of lung cancers on mutant EGFR. Nature 2011, 471, 523–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakuma, Y.; Yamazaki, Y.; Nakamura, Y.; Yoshihara, M.; Matsukuma, S.; Koizume, S.; Miyagi, Y. NF-kappaB signaling is activated and confers resistance to apoptosis in three-dimensionally cultured EGFR-mutant lung adenocarcinoma cells. Biochem. Biophys. Res. Commun. 2012, 423, 667–671. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Kwon, O.J.; Hong, Y.K.; Kim, J.H.; Solca, F.; Ha, S.J.; Soo, R.A.; Christensen, J.G.; Lee, J.H.; Cho, B.C. Activation of IL-6R/JAK1/STAT3 signaling induces De Novo resistance to irreversible EGFR inhibitors in non-small cell lung cancer with T790M resistance mutation. Mol. Cancer Ther. 2012, 11, 2254–2264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casas-Selves, M.; Kim, J.; Zhang, Z.; Helfrich, B.A.; Gao, D.; Porter, C.C.; Scarborough, H.A.; Bunn, P.A., Jr.; Chan, D.C.; Tan, A.C.; et al. Tankyrase and the canonical Wnt pathway protect lung cancer cells from EGFR inhibition. Cancer Res. 2012, 72, 4154–4164. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Zhuang, G.; Cao, Y.; Du, P.; Kim, H.J.; Settleman, J. Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell 2014, 26, 207–221. [Google Scholar] [CrossRef] [Green Version]
- Blakely, C.M.; Pazarentzos, E.; Olivas, V.; Asthana, S.; Yan, J.J.; Tan, I.; Hrustanovic, G.; Chan, E.; Lin, L.; Neel, D.S.; et al. NF-kappaB-activating complex engaged in response to EGFR oncogene inhibition drives tumor cell survival and residual disease in lung cancer. Cell Rep. 2015, 11, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Lantermann, A.B.; Chen, D.; McCutcheon, K.; Hoffman, G.; Frias, E.; Ruddy, D.; Rakiec, D.; Korn, J.; McAllister, G.; Stegmeier, F.; et al. Inhibition of Casein Kinase 1 Alpha Prevents Acquired Drug Resistance to Erlotinib in EGFR-Mutant Non-Small Cell Lung Cancer. Cancer Res. 2015, 75, 4937–4948. [Google Scholar] [CrossRef] [Green Version]
- Phuchareon, J.; McCormick, F.; Eisele, D.W.; Tetsu, O. EGFR inhibition evokes innate drug resistance in lung cancer cells by preventing Akt activity and thus inactivating Ets-1 function. Proc. Natl. Acad. Sci. USA 2015, 112, E3855–E3863. [Google Scholar] [CrossRef] [Green Version]
- Kunimasa, K.; Nagano, T.; Shimono, Y.; Dokuni, R.; Kiriu, T.; Tokunaga, S.; Tamura, D.; Yamamoto, M.; Tachihara, M.; Kobayashi, K.; et al. Glucose metabolism-targeted therapy and withaferin A are effective for epidermal growth factor receptor tyrosine kinase inhibitor-induced drug-tolerant persisters. Cancer Sci. 2017, 108, 1368–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, K.A.; Hosono, Y.; Turner, C.; Jacob, S.; Lochmann, T.L.; Murakami, Y.; Patel, N.U.; Ham, J.; Hu, B.; Powell, K.M.; et al. Increased Synthesis of MCL-1 Protein Underlies Initial Survival of EGFR-Mutant Lung Cancer to EGFR Inhibitors and Provides a Novel Drug Target. Clin. Cancer Res. 2018, 24, 5658–5672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terai, H.; Kitajima, S.; Potter, D.S.; Matsui, Y.; Quiceno, L.G.; Chen, T.; Kim, T.J.; Rusan, M.; Thai, T.C.; Piccioni, F.; et al. ER Stress Signaling Promotes the Survival of Cancer “Persister Cells” Tolerant to EGFR Tyrosine Kinase Inhibitors. Cancer Res. 2018, 78, 1044–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuoka, M.; Yoshioka, K.; Hohjoh, H. NF-kappaB activation is an early event of changes in gene regulation for acquiring drug resistance in human adenocarcinoma PC-9 cells. PLoS ONE 2018, 13, e0201796. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.N.; Bhatt, R.; Rotow, J.; Rohrberg, J.; Olivas, V.; Wang, V.E.; Hemmati, G.; Martins, M.M.; Maynard, A.; Kuhn, J.; et al. Aurora kinase A drives the evolution of resistance to third-generation EGFR inhibitors in lung cancer. Nat. Med. 2019, 25, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, H.; Yamada, T.; Wang, R.; Tanimura, K.; Adachi, Y.; Nishiyama, A.; Tanimoto, A.; Takeuchi, S.; Araujo, L.H.; Boroni, M.; et al. AXL confers intrinsic resistance to osimertinib and advances the emergence of tolerant cells. Nat. Commun. 2019, 10, 259. [Google Scholar] [CrossRef]
- Raoof, S.; Mulford, I.J.; Frisco-Cabanos, H.; Nangia, V.; Timonina, D.; Labrot, E.; Hafeez, N.; Bilton, S.J.; Drier, Y.; Ji, F.; et al. Targeting FGFR overcomes EMT-mediated resistance in EGFR mutant non-small cell lung cancer. Oncogene 2019, 38, 6399–6413. [Google Scholar] [CrossRef]
- Zhang, W.C.; Wells, J.M.; Chow, K.H.; Huang, H.; Yuan, M.; Saxena, T.; Melnick, M.A.; Politi, K.; Asara, J.M.; Costa, D.B.; et al. miR-147b-mediated TCA cycle dysfunction and pseudohypoxia initiate drug tolerance to EGFR inhibitors in lung adenocarcinoma. Nat. Metab. 2019, 1, 460–474. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, J.; Ren, S.; Sun, D.; Huang, H.Y.; Wang, H.; Jin, Y.; Li, F.; Zheng, C.; Yang, L.; et al. Branched-Chain Amino Acid Metabolic Reprogramming Orchestrates Drug Resistance to EGFR Tyrosine Kinase Inhibitors. Cell Rep. 2019, 28, 512–525.e6. [Google Scholar] [CrossRef]
- Howell, M.C.; Green, R.; Khalil, R.; Foran, E.; Quarni, W.; Nair, R.; Stevens, S.; Grinchuk, A.; Hanna, A.; Mohapatra, S.; et al. Lung cancer cells survive epidermal growth factor receptor tyrosine kinase inhibitor exposure through upregulation of cholesterol synthesis. FASEB Bioadvances 2019, 2, 90–105. [Google Scholar] [CrossRef] [Green Version]
- Kurppa, K.J.; Liu, Y.; To, C.; Zhang, T.; Fan, M.; Vajdi, A.; Knelson, E.H.; Xie, Y.; Lim, K.; Cejas, P.; et al. Treatment-Induced Tumor Dormancy through YAP-Mediated Transcriptional Reprogramming of the Apoptotic Pathway. Cancer Cell 2020, 37, 104–122.e12. [Google Scholar] [CrossRef]
- Giron, P.; Eggermont, C.; Noeparast, A.; Vandenplas, H.; Teugels, E.; Forsyth, R.; De Wever, O.; Aza-Blanc, P.; Gutierrez, G.J.; De Greve, J. Targeting USP13-mediated drug tolerance increases the efficacy of EGFR inhibition of mutant EGFR in non-small cell lung cancer. Int. J. Cancer 2020, 148, 2579–2593. [Google Scholar] [CrossRef]
- Wang, R.; Yamada, T.; Kita, K.; Taniguchi, H.; Arai, S.; Fukuda, K.; Terashima, M.; Ishimura, A.; Nishiyama, A.; Tanimoto, A.; et al. Transient IGF-1R inhibition combined with osimertinib eradicates AXL-low expressing EGFR mutated lung cancer. Nat. Commun. 2020, 11, 4607. [Google Scholar] [CrossRef]
- Ohara, S.; Suda, K.; Fujino, T.; Hamada, A.; Koga, T.; Nishino, M.; Chiba, M.; Shimoji, M.; Takemoto, T.; Soh, J.; et al. Dose-dependence in acquisition of drug tolerant phenotype and high RYK expression as a mechanism of osimertinib tolerance in lung cancer. Lung Cancer 2021, 154, 84–91. [Google Scholar] [CrossRef]
- Guler, G.D.; Tindell, C.A.; Pitti, R.; Wilson, C.; Nichols, K.; KaiWai Cheung, T.; Kim, H.J.; Wongchenko, M.; Yan, Y.; Haley, B.; et al. Repression of Stress-Induced LINE-1 Expression Protects Cancer Cell Subpopulations from Lethal Drug Exposure. Cancer Cell 2017, 32, 221–237.e13. [Google Scholar] [CrossRef]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Kuilman, T.; Peeper, D.S. Senescence-messaging secretome: SMS-ing cellular stress. Nat. Rev. Cancer 2009, 9, 81–94. [Google Scholar] [CrossRef]
- Oren, Y.; Tsabar, M.; Cabanos, H.F.; Cuoco, M.S.; Zaganjor, E.; Thakore, P.I.; Tabaka, M.; Fulco, C.P.; Hurvitz, S.A.; Slamon, D.J.; et al. Cycling cancer persister cells arise from lineages with distinct transcriptional and metabolic programs. bioRxiv Prepr. 2020. [Google Scholar] [CrossRef]
- Arasada, R.R.; Amann, J.M.; Rahman, M.A.; Huppert, S.S.; Carbone, D.P. EGFR blockade enriches for lung cancer stem-like cells through Notch3-dependent signaling. Cancer Res. 2014, 74, 5572–5584. [Google Scholar] [CrossRef] [Green Version]
- Arasada, R.R.; Shilo, K.; Yamada, T.; Zhang, J.; Yano, S.; Ghanem, R.; Wang, W.; Takeuchi, S.; Fukuda, K.; Katakami, N.; et al. Notch3-dependent beta-catenin signaling mediates EGFR TKI drug persistence in EGFR mutant NSCLC. Nat. Commun. 2018, 9, 3198. [Google Scholar] [CrossRef] [PubMed]
- Suda, K. Targeting the reversible drug-tolerant state: Aurora kinase A, is that the final answer? Transl. Cancer Res. 2019, in press. [Google Scholar] [CrossRef]
- Rusan, M.; Li, K.; Li, Y.; Christensen, C.L.; Abraham, B.J.; Kwiatkowski, N.; Buczkowski, K.A.; Bockorny, B.; Chen, T.; Li, S.; et al. Suppression of Adaptive Responses to Targeted Cancer Therapy by Transcriptional Repression. Cancer Discov. 2018, 8, 59–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suda, K.; Mitsudomi, T. Drug Tolerance to EGFR Tyrosine Kinase Inhibitors in Lung Cancers with EGFR Mutations. Cells 2021, 10, 1590. https://doi.org/10.3390/cells10071590
Suda K, Mitsudomi T. Drug Tolerance to EGFR Tyrosine Kinase Inhibitors in Lung Cancers with EGFR Mutations. Cells. 2021; 10(7):1590. https://doi.org/10.3390/cells10071590
Chicago/Turabian StyleSuda, Kenichi, and Tetsuya Mitsudomi. 2021. "Drug Tolerance to EGFR Tyrosine Kinase Inhibitors in Lung Cancers with EGFR Mutations" Cells 10, no. 7: 1590. https://doi.org/10.3390/cells10071590
APA StyleSuda, K., & Mitsudomi, T. (2021). Drug Tolerance to EGFR Tyrosine Kinase Inhibitors in Lung Cancers with EGFR Mutations. Cells, 10(7), 1590. https://doi.org/10.3390/cells10071590