
The Role of Immunotherapy in a Tolerogenic Environment: Current and Future Perspectives for Hepatocellular Carcinoma

 , , , ,
, , , ,  ,
,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
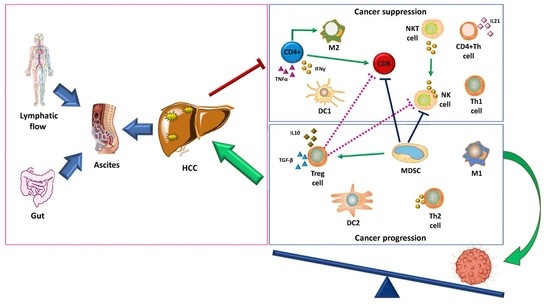
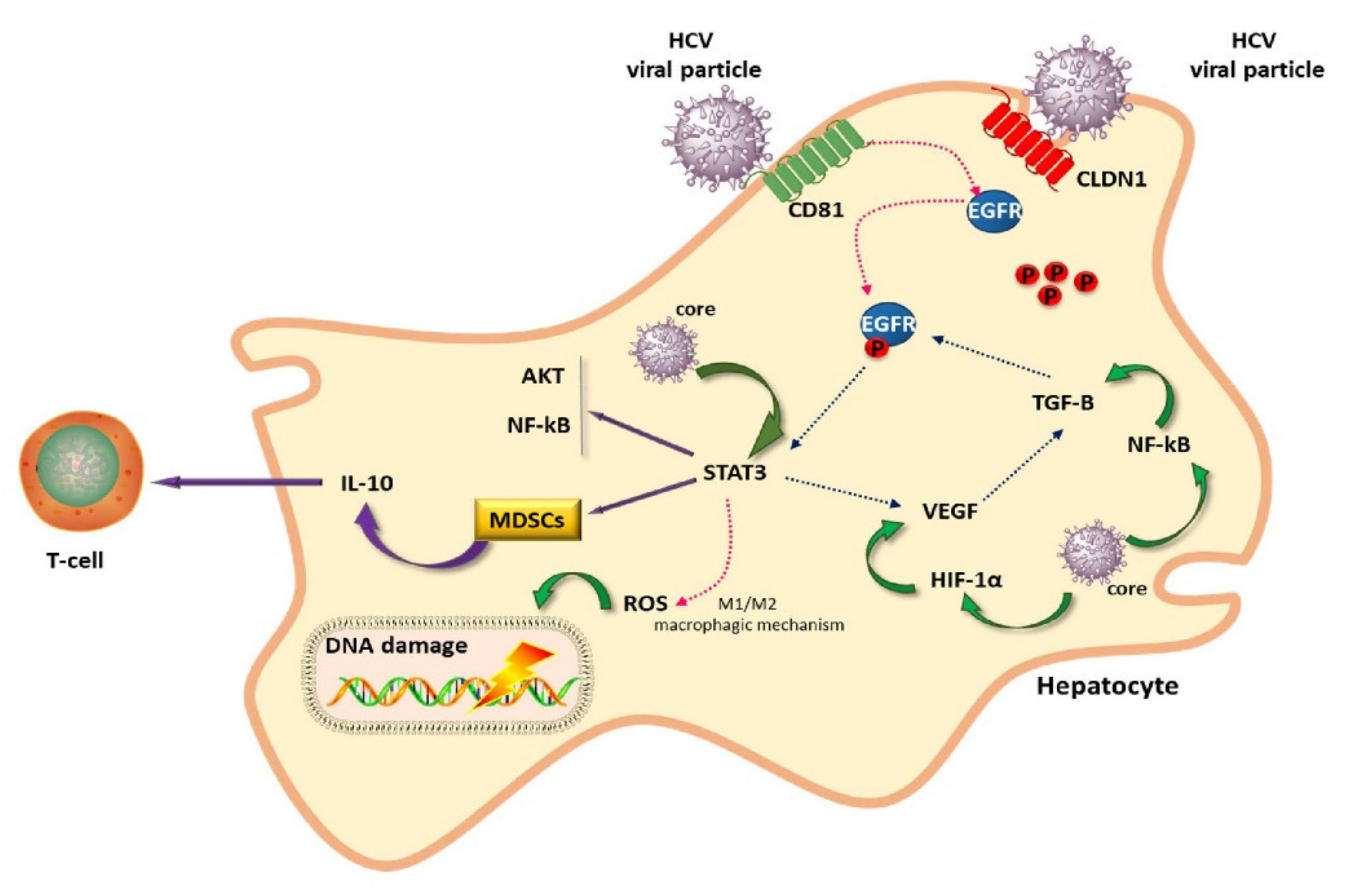
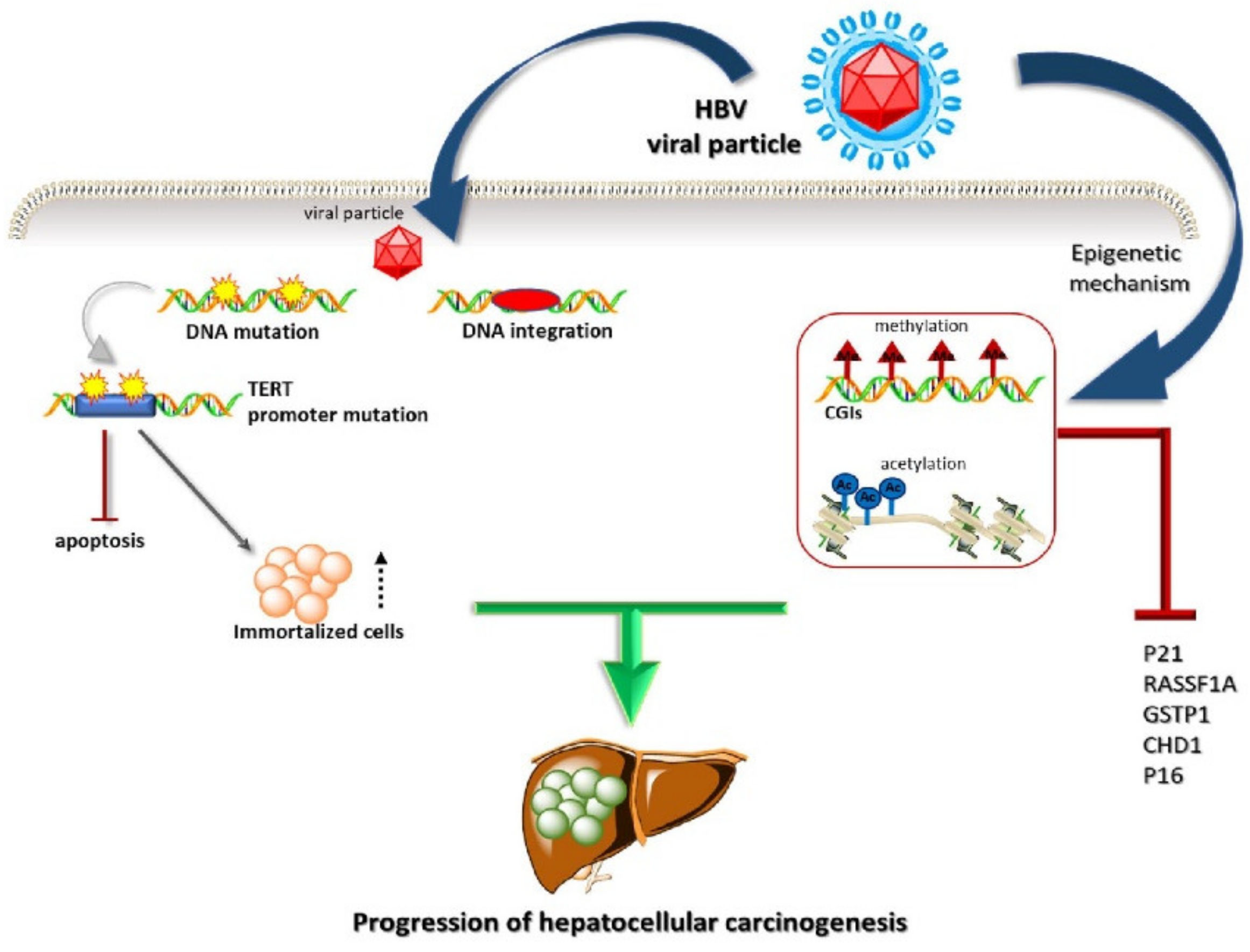
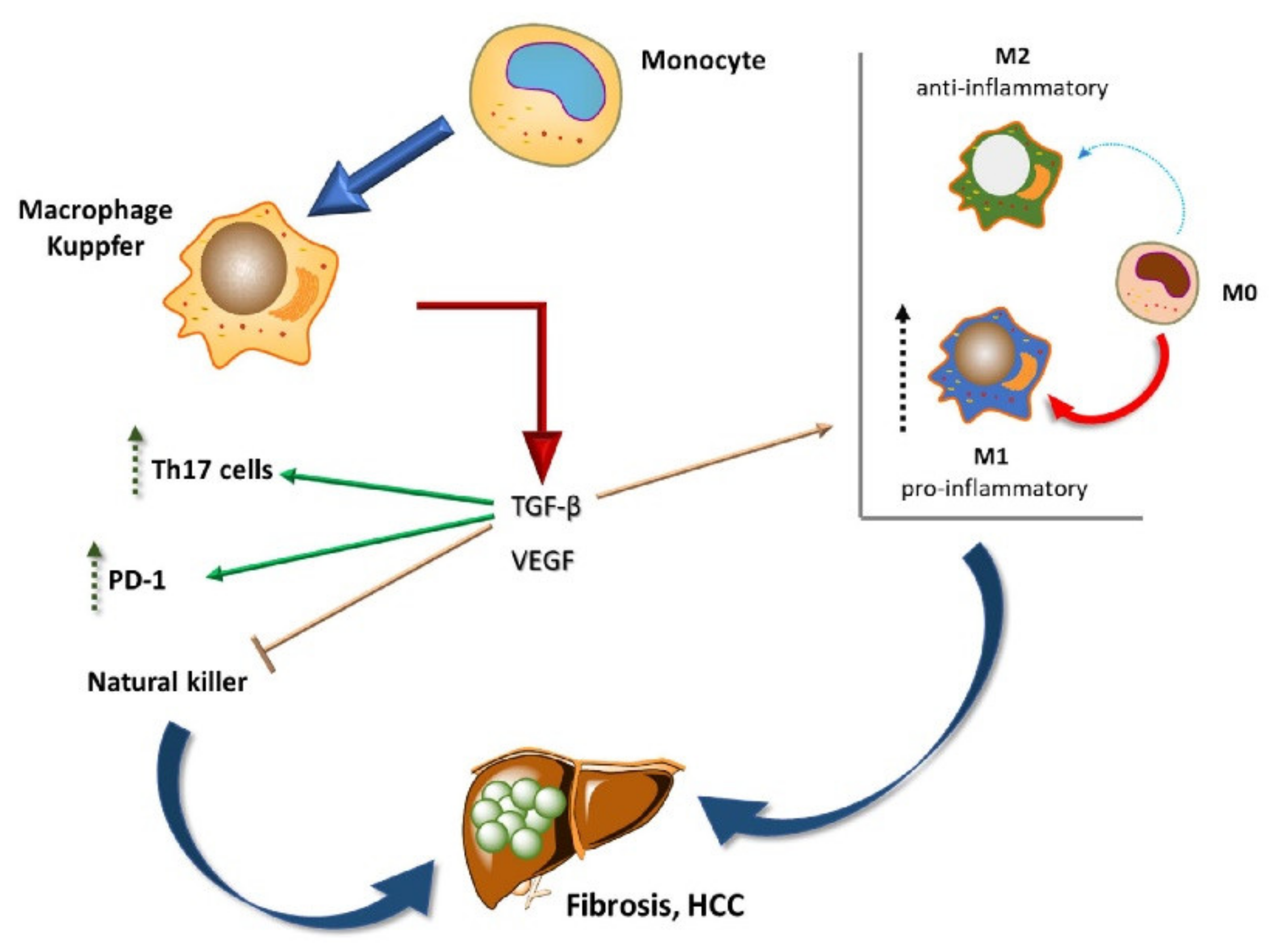
2. Liver Cancer Pathogenesis and Immunological Tolerance as the Basis for HCC Development
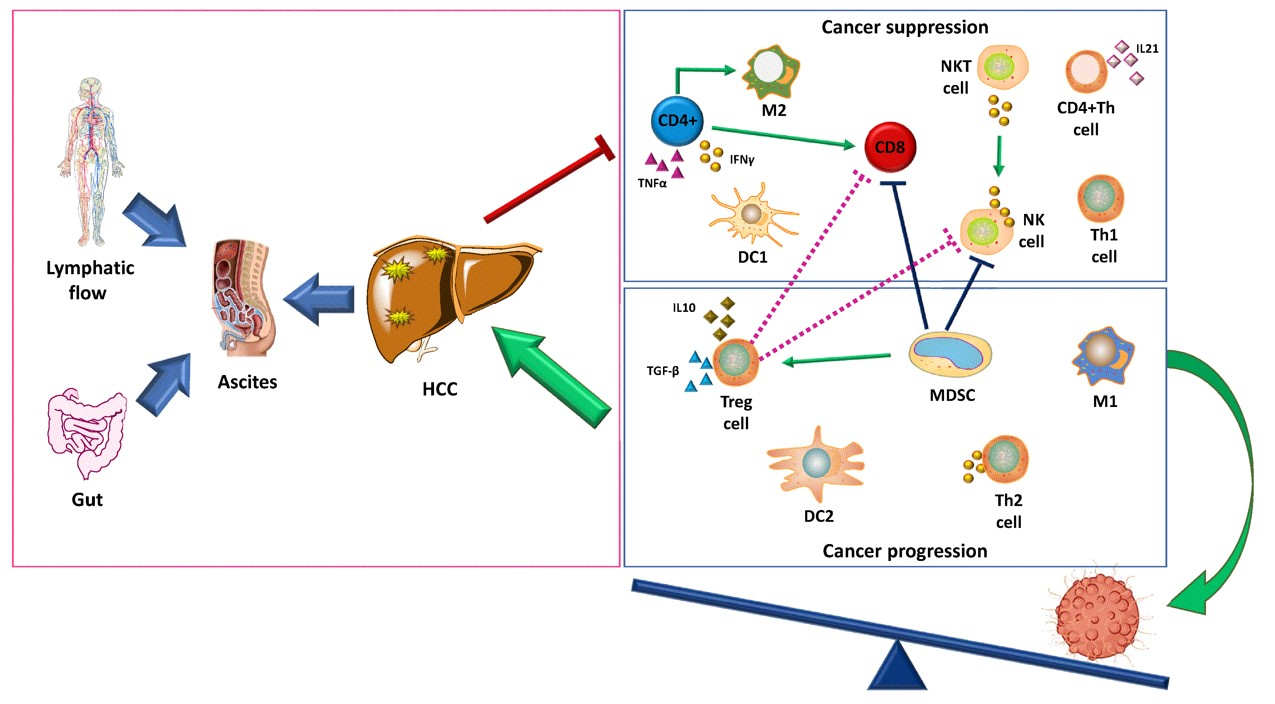
Dynamics of Immune Cells in HCC
3. Targeting Immunosuppressive Cells in the Tumor Microenvironment
3.1. Targeting TAMs
3.2. Targeting MDSCs
3.3. Drugs in Phase I Trials
4. Looking at Predictive Factors for Response to Immunotherapy in HCC
5. Immune Checkpoint Inhibitors in HCC
6. Vaccines
7. Adoptive Cell Therapies
8. Locoregional Plus Immunotherapy
9. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- European Association For The Study Of The Liver. European Organisation for Research and Treatment of Cancer. EASL–EORTC clinical practice guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2012, 56, 908–943. [Google Scholar] [CrossRef] [Green Version]
- Gawrieh, S.; Dakhoul, L.; Miller, E.; Scanga, A.; Delemos, A.; Kettler, C.; Burney, H.; Liu, H.; Abu-Sbeih, H.; Chalasani, N.; et al. Characteristics, aetiologies and trends of hepatocellular carcinoma in patients without cirrhosis: A United States multicentre study. Aliment. Pharmacol. Ther. 2019, 50, 809–821. [Google Scholar] [CrossRef]
- Montella, L.; Palmieri, G.; Addeo, R.; Del Prete, S. Hepatocellular carcinoma: Will novel targeted drugs really impact the next future? World J. Gastroenterol. 2016, 22, 6114–26. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; De Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.-W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, G.; Montella, L.; Milo, M.; Fiore, R.; Biondi, E.; Bianco, A.R.; Martignetti, A. Ultra-Low-Dose Interleukin-2 in Unresectable Hepatocellular Carcinoma. Am. J. Clin. Oncol. 2002, 25, 224–226. [Google Scholar] [CrossRef]
- Llovet, J.M.; Sala, M.; Castells, L.; Suarez, Y.; Vilana, R.; Bianchi, L.; Ayuso, C.; Vargas, V.; Rodés, J.; Bruix, J. Randomized controlled trial of interferon treatment for advanced hepatocellular carcinoma. Hepatology 2000, 31, 54–58. [Google Scholar] [CrossRef]
- Rotte, A.; Jin, J.; Lemaire, V. Mechanistic overview of immune checkpoints to support the rational design of their combinations in cancer immunotherapy. Ann. Oncol. 2018, 29, 71–83. [Google Scholar] [CrossRef] [Green Version]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.C.C.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Fishman, S.L.; Factor, S.H.; Balestrieri, C.; Fan, X.; DiBisceglie, A.M.; Desai, S.M.; Benson, G.; Branch, A.D. Mutations in the Hepatitis C Virus core Gene Are Associated with Advanced Liver Disease and Hepatocellular Carcinoma. Clin. Cancer Res. 2009, 15, 3205–3213. [Google Scholar] [CrossRef] [Green Version]
- Shirvani-Dastgerdi, E.; E Schwartz, R.; Ploss, A. Hepatocarcinogenesis associated with hepatitis B, delta and C viruses. Curr. Opin. Virol. 2016, 20, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Mailly, L.; Baumert, T.F. Hepatitis C virus infection and tight junction proteins: The ties that bind. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183296. [Google Scholar] [CrossRef]
- Goto, K.; Suarez, A.A.R.R.; Wrensch, F.; Baumert, T.F.; Lupberger, J. Hepatitis C Virus and Hepatocellular Carcinoma: When the Host Loses Its Grip. Int. J. Mol. Sci. 2020, 21, 3057. [Google Scholar] [CrossRef]
- Korkut, A.; Zaidi, S.; Kanchi, R.S.; Rao, S.; Gough, N.R.; Schultz, A.; Li, X.; Lorenzi, P.L.; Ashton C Berger, A.C.; Robertson, G.; et al. A pan-cancer analysis reveals high-frequency genetic alterations in mediators of signaling by the TGF-b superfamily. Cell Syst. 2018, 7, 422–437.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Gingold, J.A.; Su, X. Immunomodulatory TGF-β Signaling in Hepatocellular Carcinoma. Trends Mol. Med. 2019, 25, 1010–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perugorria, M.J.; Olaizola, P.; Labiano, I.; Esparza-Baquer, A.; Marzioni, M.; Marin, J.J.G.; Bujanda, L.; Banales, J.M. Wnt–β-catenin signalling in liver development, health and disease. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 121–136. [Google Scholar] [CrossRef]
- Yang, Y.; Ye, Y.-C.; Chen, Y.; Zhao, J.-L.; Gao, C.-C.; Han, H.; Liu, W.-C.; Qin, H.-Y. Crosstalk between hepatic tumor cells and macrophages via Wnt/β-catenin signaling promotes M2-like macrophage polarization and reinforces tumor malignant behaviors. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Bauer, T.; Sprinzl, M.; Protzer, U. Immune Control of Hepatitis B Virus. Dig. Dis. 2011, 29, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.K.K.; Andrisani, O. Hepatitis B Virus-Associated Hepatocellular Carcinoma and Hepatic Cancer Stem Cells. Genes 2018, 9, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai-Kitahata, F.; Asahina, Y.; Tanaka, S.; Kakinuma, S.; Murakawa, M.; Nitta, S.; Watanabe, T.; Otani, S.; Taniguchi, M.; Goto, F.; et al. Comprehensive analyses of mutations and hepatitis B virus integration in hepatocellular carcinoma with clinicopathological features. J. Gastroenterol. 2015, 51, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, B.; Kumar, R. TERT promoter mutations in telomere biology. Mutat. Res. Mutat. Res. 2017, 771, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- Block, T.M.; Mehta, A.S.; Fimmel, C.J.; Jordan, R. Molecular viral oncology of hepatocellular carcinoma. Oncogene 2003, 22, 5093–5107. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Ou, J.-H.J. Genetic and epigenetic alterations in hepatitis B virus-associated hepatocellular carcinoma. Virol. Sin. 2015, 30, 85–91. [Google Scholar] [CrossRef] [Green Version]
- Large, M.K.; Kittlesen, D.J.; Hahn, Y.S. Suppression of Host Immune Response by the Core Protein of Hepatitis C Virus: Possible Implications for Hepatitis C Virus Persistence. J. Immunol. 1999, 162, 931–938. [Google Scholar]
- Chen, S.; Wu, Z.; Wang, M.; Cheng, A. Innate Immune Evasion Mediated by Flaviviridae Non-Structural Proteins. Viruses 2017, 9, 291. [Google Scholar] [CrossRef]
- Sun, S.; Li, Y.; Han, S.; Jia, H.; Li, X.; Li, X. A comprehensive genome-wide profiling comparison between HBV and HCV infected hepatocellular carcinoma. BMC Med. Genom. 2019, 12, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diehl, L.; Schurich, A.; Grochtmann, R.; Hegenbarth, S.; Chen, L.; Knolle, P.A. Tolerogenic maturation of liver sinusoidal endothelial cells promotes B7-homolog 1-dependent CD8+ T cell tolerance. Hepatology 2007, 47, 296–305. [Google Scholar] [CrossRef]
- Schurich, A.; Khanna, P.; Lopes, A.R.; Han, K.J.; Peppa, D.; Micco, L.; Nebbia, G.; Kennedy, P.T.; Geretti, A.-M.; Dusheiko, G.; et al. Role of the coinhibitory receptor cytotoxic T lymphocyte antigen-4 on apoptosis-Prone CD8 T cells in persistent hepatitis B virus infection. Hepatology 2011, 53, 1494–1503. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, N.; Cho, H.; Shaked, A.; Olthoff, K.; Valiga, M.E.; Kaminski, M.; Gostick, E.; Price, D.; Freeman, G.J.; Wherry, E.J.; et al. Synergistic Reversal of Intrahepatic HCV-Specific CD8 T Cell Exhaustion by Combined PD-1/CTLA-4 Blockade. PLoS Pathog. 2009, 5, e1000313. [Google Scholar] [CrossRef]
- Dou, L.; Shi, X.; He, X.; Gao, Y. Macrophage Phenotype and Function in Liver Disorder. Front. Immunol. 2020, 10, 3112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buonaguro, L.; Mauriello, A.; Cavalluzzo, B.; Petrizzo, A.; Tagliamonte, M. Immunotherapy in hepatocellular carcinoma. Ann. Hepatol. 2019, 18, 291–297. [Google Scholar] [CrossRef]
- Ding, W.; Tan, Y.; Qian, Y.; Xue, W.; Wang, Y.; Jiang, P.; Xu, X. Clinicopathologic and prognostic significance of tumor-associated macrophages in patients with hepatocellular carcinoma: A meta-analysis. PLoS ONE 2019, 14, e0223971. [Google Scholar] [CrossRef]
- Yang, R.; Xu, Y.; Dai, Z.; Lin, X.; Wang, H. The Immunologic Role of Gut Microbiota in Patients with Chronic HBV Infection. J. Immunol. Res. 2018, 2018, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.-X.; Schwabe, L.-X.Y.R.F. The gut microbiome and liver cancer: Mechanisms and clinical translation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 527–539. [Google Scholar] [CrossRef]
- Behary, J.; Amorim, N.; Jiang, X.-T.; Raposo, A.; Gong, L.; McGovern, E.; Ibrahim, R.; Chu, F.; Stephens, C.; Jebeili, H.; et al. Gut microbiota impact on the peripheral immune response in non-alcoholic fatty liver disease related hepatocellular carcinoma. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Qin, N.; Yang, F.; Li, A.; Prifti, E.; Chen, Y.; Shao, L.; Guo, J.; Le Chatelier, E.; Yao, J.; Wu, L.; et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014, 513, 59–64. [Google Scholar] [CrossRef] [PubMed]
- El-Mowafy, M.; Elgaml, A.; El-Mesery, M.; Sultan, S.; Ahmed, T.A.E.; Gomaa, A.I.; Aly, M.; Mottawea, W. Changes of Gut-Microbiota-Liver Axis in Hepatitis C Virus Infection. Biology 2021, 10, 55. [Google Scholar] [CrossRef] [PubMed]
- Daillère, R.; Routy, B.; Goubet, A.-G.; Cogdill, A.; Ferrere, G.; Silva, C.A.-C.; Fluckiger, A.; Ly, P.; Haddad, Y.; Pizzato, E.; et al. Elucidating the gut microbiota composition and the bioactivity of immunostimulatory commensals for the optimization of immune checkpoint inhibitors. OncoImmunology 2020, 9, 1794423. [Google Scholar] [CrossRef] [PubMed]
- Routy, B.; Le Chatelier, E.; DeRosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Ye, J. Characterization of gut microbiota in patients with primary hepatocellular carcinoma received immune checkpoint inhibitors. Medicine 2020, 99, e21788. [Google Scholar] [CrossRef]
- Zhang, Q.; He, Y.; Luo, N.; Patel, S.J.; Han, Y.; Gao, R.; Modak, M.; Carotta, S.; Haslinger, C.; Kind, D.; et al. Landscape and Dynamics of Single Immune Cells in Hepatocellular Carcinoma. Cell 2019, 179, 829–845.e20. [Google Scholar] [CrossRef]
- Sun, Y.; Wu, L.; Zhong, Y.; Zhou, K.; Hou, Y.; Wang, Z.; Zhang, Z.; Xie, J.; Wang, C.; Chen, D.; et al. Single-cell landscape of the ecosystem in early-relapse hepatocellular carcinoma. Cell 2021, 184, 404–421.e16. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Puig, O.; Daniele, B.; Kudo, M.; Merle, P.; Park, J.-W.; Ross, P.; Peron, J.-M.; Ebert, O.; Chan, S.; et al. Randomized phase II placebo controlled study of codrituzumab in previously treated patients with advanced hepatocellular carcinoma. J. Hepatol. 2016, 65, 289–295. [Google Scholar] [CrossRef]
- Ao, J.-Y.; Zhu, X.-D.; Chai, Z.-T.; Cai, H.; Zhang, Y.-Y.; Zhang, K.-Z.; Kong, L.-Q.; Zhang, N.; Ye, B.-G.; Ma, D.-N.; et al. Colony-Stimulating Factor 1 Receptor Blockade Inhibits Tumor Growth by Altering the Polarization of Tumor-Associated Macrophages in Hepatocellular Carcinoma. Mol. Cancer Ther. 2017, 16, 1544–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- 32nd Annual Meeting and Pre-Conference Programs of the Society for Immunotherapy of Cancer (SITC 2017): Late-Breaking Abstracts. J. Immunother. Cancer 2017, 5, 89. [CrossRef] [Green Version]
- Chen, L.-M.; Tseng, H.-Y.; Chen, Y.-A.; Al Haq, A.T.; Hwang, P.-A.; Hsu, H.-L. Oligo-Fucoidan Prevents M2 Macrophage Differentiation and HCT116 Tumor Progression. Cancers 2020, 12, 421. [Google Scholar] [CrossRef] [Green Version]
- Available online: Clinicaltrials.gov (accessed on 16 April 2021).
- Lam, W.; Ren, Y.; Guan, F.; Jiang, Z.; Cheng, W.; Xu, C.-H.; Liu, S.-H.; Cheng, Y.-C. Mechanism Based Quality Control (MBQC) of Herbal Products: A Case Study YIV-906 (PHY906). Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef]
- Changou, C.A.; Shiah, H.; Chen, L.; Liu, S.; Luh, F.; Liu, S.; Cheng, Y.; Yen, Y. A Phase II Clinical Trial on the Combination Therapy of PHY906 Plus Capecitabine in Hepatocellular Carcinoma. Oncology 2021, 26, e367–e373. [Google Scholar] [CrossRef]
- Laport, G.; Powderly, J.D.; Chokshi, S.; Luke, J.J.; Bendell, J.C.; Amanda Enstrom, A.; Whiting, C.C.; Dubensky, T.W. Phase 1/1b multicenter trial of TPST-1120, a peroxisome proliferator-activated receptor alpha (PPARα) antagonist as a single agent (SA) or in combination in patients with advanced solid tumors. J. Clin. Oncol. 2019, 37 (Suppl. 15). [Google Scholar] [CrossRef]
- Bilusic, M.; Heery, C.R.; Collins, J.M.; Donahue, R.N.; Palena, C.; Madan, R.A.; Karzai, F.; Marté, J.L.; Strauss, J.; Gatti-Mays, M.E.; et al. Phase I trial of HuMax-IL8 (BMS-986253), an anti-IL-8 monoclonal antibody, in patients with metastatic or unresectable solid tumors. J. Immunother. Cancer 2019, 7, 240. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Huang, H.; Sorrelle, N.; Brekken, R.A. Sitravatinib potentiates immune checkpoint blockade in refractory cancer models. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.; Keam, S.J. Tislelizumab: First Approval. Drugs 2020, 80, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Rasco, D.W.; Bendell, J.C.; Wang-Gillam, A.; Park, W.; O’Reilly, E.M.; Zhou, L.; Galkin, A.; Carter, L.L.; Nickle, D.; Li, J.; et al. A phase I/II study of GB1275, a first-in-class oral CD11b modulator, alone, and combined with pembrolizumab in specified advanced solid tumors or with chemotherapy in metastatic pancreatic cancer (KEYNOTE-A36). J. Clin. Oncol. 2020, 38, 3085. [Google Scholar] [CrossRef]
- Ding, W.; Xu, X.; Qian, Y.; Xue, W.; Wang, Y.; Du, J.; Jin, L.; Tan, Y. Prognostic value of tumor-infiltrating lymphocytes in hepatocellular carcinoma. Medicine 2018, 97, e13301. [Google Scholar] [CrossRef]
- Xu, X.; Tan, Y.; Qian, Y.; Xue, W.; Wang, Y.; Du, J.; Jin, L.; Ding, W. Clinicopathologic and prognostic significance of tumor-infiltrating CD8+ T cells in patients with hepatocellular carcinoma. Medicine 2019, 98, e13923. [Google Scholar] [CrossRef]
- Yang, H.; Zhou, X.; Sun, L.; Mao, Y. Correlation Between PD-L2 Expression and Clinical Outcome in Solid Cancer Patients: A Meta-Analysis. Front. Oncol. 2019, 9, 47. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Qin, S. Immune Checkpoint Inhibitors in Hepatocellular Carcinoma: Opportunities and Challenges. Oncology 2019, 24, S3–S10. [Google Scholar] [CrossRef] [Green Version]
- Liao, H.; Chen, W.; Dai, Y.; Richardson, J.J.; Guo, J.; Yuan, K.; Zeng, Y.; Xie, K. Expression of Programmed Cell Death-Ligands in Hepatocellular Carcinoma: Correlation With Immune Microenvironment and Survival Outcomes. Front. Oncol. 2019, 9, 883. [Google Scholar] [CrossRef]
- Chan, T.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef]
- Löffler, M.W.; HEPAVAC Consortium; Mohr, C.; Bichmann, L.; Freudenmann, L.K.; Walzer, M.; Schroeder, C.M.; Trautwein, N.; Hilke, F.J.; Zinser, R.S.; et al. Multi-omics discovery of exome-derived neoantigens in hepatocellular carcinoma. Genome Med. 2019, 11, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Yarchoan, M.; Albacker, L.A.; Hopkins, A.C.; Montesion, M.; Murugesan, K.; Vithayathil, T.T.; Zaidi, N.; Azad, N.S.; Laheru, D.A.; Frampton, G.M.; et al. PD-L1 expression and tumor mutational burden are independent biomarkers in most cancers. JCI Insight 2019, 4, 126908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.; Ding, T.; Guo, Z.-W.; Yu, X.-J.; Hu, Y.-Z.; Zheng, L.; Xu, J. Expression pattern of tumour-associated antigens in hepatocellular carcinoma: Association with immune infiltration and disease progression. Br. J. Cancer 2013, 109, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Shimada, S.; Mogushi, K.; Akiyama, Y.; Furuyama, T.; Watanabe, S.; Ogura, T.; Ogawa, K.; Ono, H.; Mitsunori, Y.; Ban, D.; et al. Comprehensive molecular and immunological characterization of hepatocellular carcinoma. EBioMedicine 2019, 40, 457–470. [Google Scholar] [CrossRef] [Green Version]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.S.; Ryoo, B.-Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab As Second-Line Therapy in Patients With Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef]
- Yau, T.; Kang, Y.-K.; Kim, T.-Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.-M.; Matilla, A.; et al. Efficacy and Safety of Nivolumab Plus Ipilimumab in Patients With Advanced Hepatocellular Carcinoma Previously Treated With Sorafenib. JAMA Oncol. 2020, 6, e204564. [Google Scholar] [CrossRef]
- Ziogas, I.A.; Evangeliou, A.P.; Giannis, D.; Hayat, M.H.; Mylonas, K.S.; Tohme, S.; Geller, D.A.; Elias, N.; Goyal, L.; Tsoulfas, G. The Role of Immunotherapy in Hepatocellular Carcinoma: A Systematic Review and Pooled Analysis of 2,402 Patients. Oncology 2021, 26, e1036–e1049. [Google Scholar] [CrossRef]
- Nakano, S.; Eso, Y.; Okada, H.; Takai, A.; Takahashi, K.; Seno, H. Recent Advances in Immunotherapy for Hepatocellular Carcinoma. Cancers 2020, 12, 775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, H.; Suri, P.; Gupta, J.C.; Talwar, G.P.; Dubey, S. Survivin: A unique target for tumor therapy. Cancer Cell Int. 2016, 16, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Berinstein, N.L.; Karkada, M.; Oza, A.; Odunsi, K.; Villella, J.A.; Nemunaitis, J.J.; Morse, M.A.; Pejovic, T.; Bentley, J.; Buyse, M.; et al. Survivin-targeted immunotherapy drives robust polyfunctional T cell generation and differentiation in advanced ovarian cancer patients. OncoImmunology 2015, 4, e1026529. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://immuno-oncologynews.com/ino9012/ (accessed on 16 April 2021).
- Xie, Y.; Xiang, Y.; Sheng, J.; Zhang, D.; Yao, X.; Yang, Y.; Zhang, X. Immunotherapy for Hepatocellular Carcinoma: Current Advances and Future Expectations. J. Immunol. Res. 2018, 2018, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Abken, H. TRUCKS, the fourth-generation CAR T cells: Current developments and clinical translation. Adv. CELL GENE Ther. 2020, 3, 384. [Google Scholar] [CrossRef]
- Jiang, W.; Chan, C.K.; Weissman, I.L.; Kim, B.Y.; Hahn, S.M. Immune Priming of the Tumor Microenvironment by Radiation. Trends Cancer 2016, 2, 638–645. [Google Scholar] [CrossRef]
- Kachikwu, E.L.; Iwamoto, K.S.; Liao, Y.-P.; DeMarco, J.J.; Agazaryan, N.; Economou, J.S.; McBride, W.H.; Schaue, D. Radiation Enhances Regulatory T Cell Representation. Int. J. Radiat. Oncol. 2011, 81, 1128–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, L.; Liang, H.; Burnette, B.; Beckett, M.; Darga, T.; Weichselbaum, R.R.; Fu, Y.-X. Irradiation and anti–PD-L1 treatment synergistically promote antitumor immunity in mice. J. Clin. Investig. 2014, 124, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.I.; Lee, S.J.; Lee, J.; Lim, H.Y.; Paik, S.W.; Yoo, G.S.; Choi, C.; Park, H.C. Clinical significance of radiotherapy before and/or during nivolumab treatment in hepatocellular carcinoma. Cancer Med. 2019, 8, 6986–6994. [Google Scholar] [CrossRef] [PubMed]
- Flynn, M.J.; Sayed, A.A.; Sharma, R.; Siddique, A.; Pinato, D.J. Challenges and Opportunities in the Clinical Development of Immune Checkpoint Inhibitors for Hepatocellular Carcinoma. Hepatology 2019, 69, 2258–2270. [Google Scholar] [CrossRef] [Green Version]
- Vogel, A.; Cervantes, A.; Chau, I.; Daniele, B.; Llovet, J.M.; Meyer, T.; Nault, J.-C.; Neumann, U.; Ricke, J.; Sangro, B.; et al. Hepatocellular carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv238–iv255. [Google Scholar] [CrossRef]
- Shen, L.; Xi, M.; Zhao, L.; Zhang, X.; Wang, X.; Huang, Z.; Chen, Q.; Zhang, T.; Shen, J.; Liu, M.; et al. Combination Therapy after TACE for Hepatocellular Carcinoma with Macroscopic Vascular Invasion: Stereotactic Body Radiotherapy versus Sorafenib. Cancers 2018, 10, 516. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.M.; Ryoo, B.-Y.; Lee, S.J.; Kim, J.H.; Shin, J.H.; An, J.; Lee, H.C.; Lim, Y.-S. Efficacy and Safety of Transarterial Chemoembolization Plus External Beam Radiotherapy vs Sorafenib in Hepatocellular Carcinoma with Macroscopic Vascular Invasion. JAMA Oncol. 2018, 4, 661–669. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Lim, H.Y.; Kudo, M.; Breder, V.V.; Merle, P.; et al. IMbrave150: Updated overall survival (OS) data from a global, randomized, open-label phase III study of atezolizumab (atezo) + bevacizumab (bev) versus sorafenib (sor) in patients (pts) with unresectable hepatocellular carcinoma (HCC). J. Clin. Oncol. 2021, 39, 267. [Google Scholar] [CrossRef]
- Del Prete, S.; Montella, L.; Caraglia, M.; Maiorino, L.; Cennamo, G.; Montesarchio, V.; Piai, G.; Febbraro, A.; Tarantino, L.; Capasso, E.; et al. Sorafenib plus octreotide is an effective and safe treatment in advanced hepatocellular carcinoma: Multicenter phase II So.LAR. study. Cancer Chemother. Pharmacol. 2009, 66, 837–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santini, D.; Addeo, R.; Vincenzi, B.; Calvieri, A.; Montella, L.; Silletta, M.; Caraglia, M.; Vespasiani, U.; Picardi, A.; Del Prete, S.; et al. Exploring the efficacy and safety of single-agent sorafenib in a cohort of Italian patients with hepatocellular carcinoma. Expert Rev. Anticancer. Ther. 2012, 12, 1283–1288. [Google Scholar] [CrossRef]
- Montella, L.; Addeo, R.; Cennamo, G.; Vincenzi, B.; Palmieri, R.; Sperlongano, P.; Sperlongano, R.; Iodice, P.; Russo, P.; Del Prete, S. Sorafenib in Elderly Patients with Advanced Hepatocellular Carcinoma: A Case Series. Oncology 2013, 84, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Huang, H.; Wang, Y.; Pan, C.; Yin, S.; Zhou, L.; Zheng, S. Tumor Immune Microenvironment Characterization in Hepatocellular Carcinoma Identifies Four Prognostic and Immunotherapeutically Relevant Subclasses. Front. Oncol. 2021, 10. [Google Scholar] [CrossRef]
- Dai, Y.; Qiang, W.; Lin, K.; Gui, Y.; Lan, X.; Wang, D. An immune-related gene signature for predicting survival and immunotherapy efficacy in hepatocellular carcinoma. Cancer Immunol. Immunother. 2021, 70, 967–979. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, Z.; Li, F. Survival prediction and response to immune checkpoint inhibitors: A prognostic immune signature for hepatocellular carcinoma. Transl. Oncol. 2021, 14, 100957. [Google Scholar] [CrossRef] [PubMed]
- Harding, J.J.; Nandakumar, S.; Armenia, J.; Khalil, D.N.; Albano, M.; Ly, M.; Shia, J.; Hechtman, J.F.; Kundra, R.; El Dika, I.; et al. Prospective Genotyping of Hepatocellular Carcinoma: Clinical Implications of Next-Generation Sequencing for Matching Patients to Targeted and Immune Therapies. Clin. Cancer Res. 2019, 25, 2116–2126. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trial Identifier | Drugs | Phase | Treatment Arms | Estimated Enrollment | Estimated Study Completion Date |
|---|---|---|---|---|---|
| NCT03298451 (Himalaya) | Durvalumab Tremelimumab Sorafenib | III |
| 1504 participants | 30 April 2022 |
| NCT04194775 | CS1003 (anti-PD-1 antibody) Lenvatinib | III |
| 525 participants | 30 June 2023 |
| CheckMate 9DW NCT04039607 | Nivolumab Ipilimumab Sorafenib Lenvatinib | III |
| 650 participants | 30 September 2023 |
| NCT04720716 | IBI310 (anti-CTLA4) Sintilimab Sorafenib | III |
| 490 participants | 1 December 2023 |
| NCT04723004 | Toripalimab (anti-PD1) Bevacizumab Sorafenib | III |
| 280 participants | 31 December 2024 |
| NCT04523493 | Toripalimab Lenvatinib | III |
| 486 participants | 24 August 2024 |
| NCT04560894 | SCT-I10A (anti- PD-1) SCT510 (bevacizumab biosimilar) Sorafenib | II/III |
| 621 participants | September 2024 |
| NCT03605706 | SHR-1210 (Camrelizumab, anti-PD-1) | III |
| 396 participants | December 2021 |
| NCT03755791 (COSMIC-312) | Cabozantinib Sorafenib Atezolizumab | III |
| 740 participants | 1 December 2021 |
| NCT04310709 (RENOBATE) | Regorafenib Nivolumab | II | Regorafenib + Nivolumab | 42 participants | 30 May 2023 |
| NCT03695250 | BMS-986205 (IDO1 inhibitor) Nivolumab | I/II | BMS-986205 + Nivolumab | 23 participants | 1 June 2022 |
| NCT03680508 | TSR-022 (cobolimab, TIM-3-binding antibody) and TSR-042 (anti PD-1 dostarlimab) | II | TSR-022 + TSR-042 | 42 participants | October 2023 |
| Trial Identifier | Drugs | Phase | Treatment Arms | Main Patient Characteristics | Estimated Enrollment | Estimated Study Completion Date |
|---|---|---|---|---|---|---|
| NCT04167293 | Anti PD-1 sintilimab | III |
| Portal vein invasion No previous treatment | 116 participants | 31 October 2022 Arms and interventions |
| NCT04709380 | Toripalimab Sorafenib | III |
| BCLC stage C with portal vein/hepatic vein tumor thrombosis | 85 participants | 28 February 2023 |
| NCT04712643 | Atezolizumab Bevacizumab | III |
| No prior systemic therapy | 342 participants | 26 February 2027 |
| NCT04268888 (TACE-3) | Nivolumab | III |
| 522 participants | June 2026 | |
| NCT04340193 (CheckMate 74W) | Nivolumab Ipilimumab | III |
| 765 participants | 10 June 2028 | |
| NCT03778957 (Emerald) | Durvalumab Bevacizumab | III |
| 710 participants | 30 August 2024 | |
| NCT04229355 | Sorafenib Lenvatinib anti-PD-1 | III |
| 90 participants | 30 December 2022 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montella, L.; Sarno, F.; Ambrosino, A.; Facchini, S.; D’Antò, M.; Laterza, M.M.; Fasano, M.; Quarata, E.; Ranucci, R.A.N.; Altucci, L.; et al. The Role of Immunotherapy in a Tolerogenic Environment: Current and Future Perspectives for Hepatocellular Carcinoma. Cells 2021, 10, 1909. https://doi.org/10.3390/cells10081909
Montella L, Sarno F, Ambrosino A, Facchini S, D’Antò M, Laterza MM, Fasano M, Quarata E, Ranucci RAN, Altucci L, et al. The Role of Immunotherapy in a Tolerogenic Environment: Current and Future Perspectives for Hepatocellular Carcinoma. Cells. 2021; 10(8):1909. https://doi.org/10.3390/cells10081909
Chicago/Turabian StyleMontella, Liliana, Federica Sarno, Annamaria Ambrosino, Sergio Facchini, Maria D’Antò, Maria Maddalena Laterza, Morena Fasano, Ermelinda Quarata, Raffaele Angelo Nicola Ranucci, Lucia Altucci, and et al. 2021. "The Role of Immunotherapy in a Tolerogenic Environment: Current and Future Perspectives for Hepatocellular Carcinoma" Cells 10, no. 8: 1909. https://doi.org/10.3390/cells10081909
APA StyleMontella, L., Sarno, F., Ambrosino, A., Facchini, S., D’Antò, M., Laterza, M. M., Fasano, M., Quarata, E., Ranucci, R. A. N., Altucci, L., Berretta, M., & Facchini, G. (2021). The Role of Immunotherapy in a Tolerogenic Environment: Current and Future Perspectives for Hepatocellular Carcinoma. Cells, 10(8), 1909. https://doi.org/10.3390/cells10081909