
The Role of Alveolar Edema in COVID-19


Abstract
1. Introduction
2. Endothelial Injuries in COVID-19
3. Ventilation-Perfusion Mismatch and Intravascular Coagulation in COVID-19
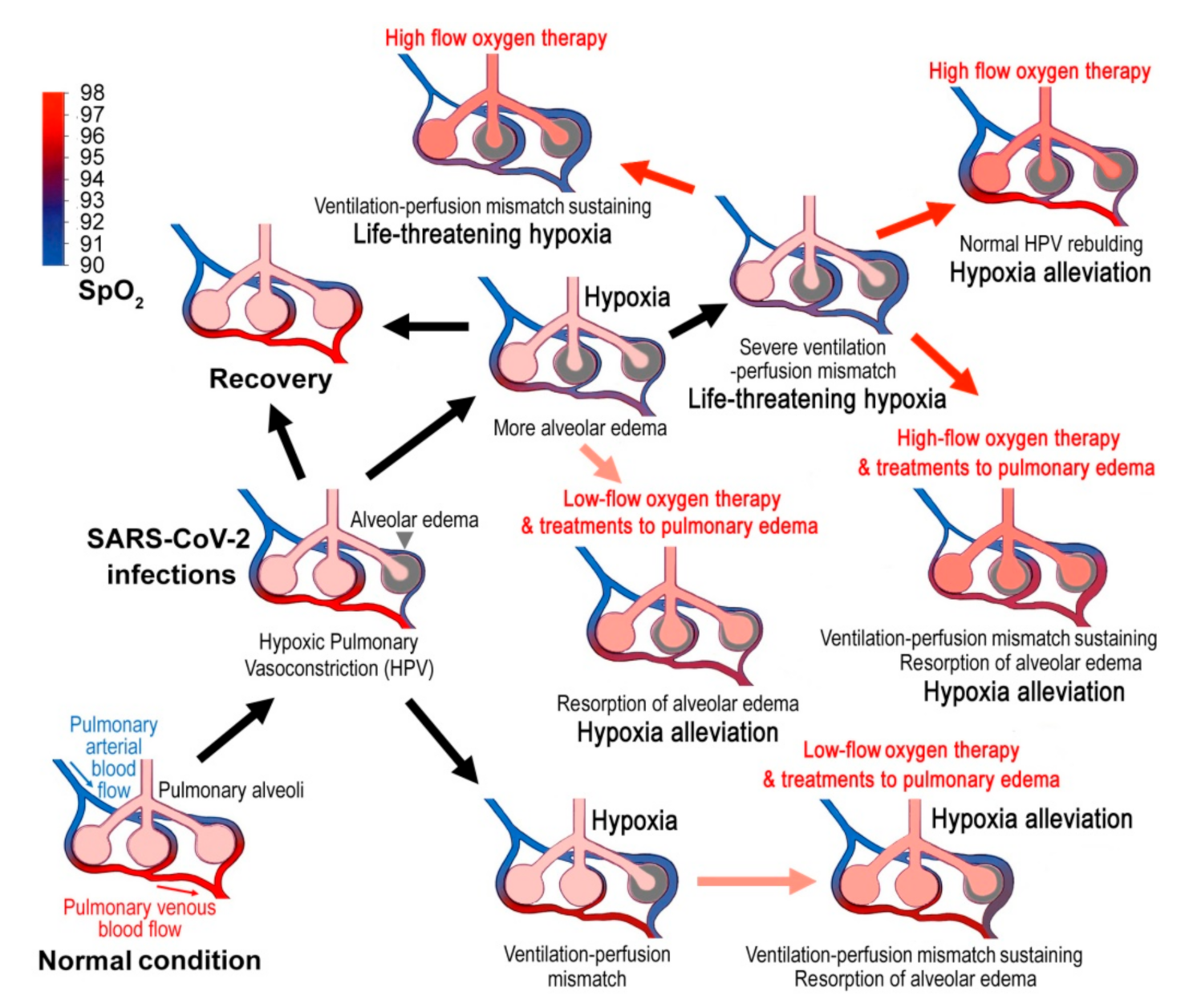
4. Alveolar Edema in COVID-19
5. The Optimal Time and SpO2 Threshold for Oxygen Therapy
6. Four Clinical Comments
- (a)
- For the suspected cases with symptoms, finger SpO2 (with finger oximeter ideally) should be measured at each time of nucleic acid test sampling and daily after symptom onset. However, finger SpO2 varies greatly with the altitude and the age [35], and the finger oximeter itself may have a large deviation, so it is recommended that each oximeter should be calibrated with several healthy people of different ages to get the reference value. If the patient’s SpO2 was lower than the reference value by 3% or more (e.g., if the reference value was 98%, then ≤95% is the threshold for oxygen therapy), it is suggested that the patients were hospitalized immediately for standard low-flow oxygen inhalation possibly combined with 20% ethanol as humidifier. If immediate hospitalization was not possible, the patient was recommended to take oxygen in the home, such as with a portable oxygen respirator. During the in-home oxygen therapy, finger SpO2 should be monitored continually to assure that SpO2 has been restored to 96%, but not higher than that (Table 3). This is because saturation above this level likely causes an increased risk of death without plausible benefits [94,95]. This upper limit may be lower for the patients with chronic respiratory diseases. For instance, the oxygen treatment goal should be 88–92% for patients with chronic type II respiratory failure (Table 3) [92]. Nevertheless, if SpO2 cannot be enhanced afterwards, the patient should seek medical advice or go to the hospital in time. The in-home oxygen therapy may be of great significance for countries with a shortage of medical resources. The COVID-19 patients usually require oxygen long-term oxygen supplies. However, if humidification with 20% ethanol was adopted, long-time ethanol vapor inhalation may generate adverse effects to the respiratory system and the nervous system [96,97]. The optimal length of ethanol-oxygen vapor therapy needs to be investigated in clinical trials.
- (b)
- For the patients with very low SpO2, high-flow oxygen inhalation should be applied. Nevertheless, humidification with 20% ethanol might be also recommended on this occasion.
- (c)
- Fluid management might be considered for all COVID-19 patients and conservative fluid management might be applied to severe cases. Some patients may be dehydrated with evolving acute kidney injury at hospital presentation for COVID-19 pneumonia. Therefore, conservative fluid management to these patients should be applied with caution. Detailed guidance of fluid administration in patients with COVID-19 has been discussed elsewhere [98]. For the in-home patients, appropriate reduction in water intake might be an expedient measure.
- (d)
- The prone position could reduce the risk of ventilation-associated lung injuries by the combined effects of more uniform distribution of breathing and less compression of the left lower pulmonary lobe by the heart [5,99,100,101]. Therefore, patients with low SpO2 are advised to use prone position as much as possible. In addition, the patients should avoid any vigorous activity that may increase respiratory rate and tidal volume because pulmonary injury will be worsened by the mechanical stretch during the strained breathing [5,99,100,101]. The benefit of prone ventilation is larger than that for typical ARDS. HPV is regionally variable, resulting in heterogenous ventilation-perfusion matching. Prone ventilation may minimize the heterogeneity and allow HPV to divert blood flow to the caudal/dorsal regions of the lung. Although HPV is considered to be weak in COVID-19, residual HPV might be optimized when prone [38].
7. Clinical Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACE2 | angiotensin-converting enzyme 2 |
| AEI | alveolar epithelial cells type I |
| AEII | alveolar epithelial cells type II |
| ALI | acute lung injury |
| Ang II | angiotensin II |
| ARDS | acute respiratory distress syndrome |
| COPD | chronic obstructive pulmonary disease |
| COVID-19 | coronavirus disease 2019 |
| CT | computed tomography |
| DAD | diffuse alveolar damage |
| ECMO | extracorporeal membrane oxygenation |
| FiO2 | fractional inspired oxygen |
| GAGs | glycosaminoglycans |
| HIF-1α | hypoxia inducible factor-1α |
| HPV | hypoxic pulmonary vasoconstriction |
| ICU | intensive care unit |
| IPF | idiopathic pulmonary fibrosis |
| NO | nitric oxide |
| PaO2 | arterial oxygen tension |
| PASMC | pulmonary artery smooth muscle cells |
| PDK | pyruvate dehydrogenase kinase |
| PH | pulmonary hypertension |
| PKC | protein kinase C |
| RAAS | renin-angiotensin-aldosterone system |
| RBD | receptor-binding domain |
| ROS | reactive oxygen species |
| SARS-CoV-2 | SARS-like coronavirus 2 |
| Sicam | soluble intercellular adhesion molecule |
| SpO2 | blood oxygen saturation |
| TMPRSS2 | trans-membrane protease/serine subfamily 2 |
| VEGF | vascular endothelial growth factor |
| vWF | von Willebrand factor |
References
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef]
- Wölfel, R.; Corman, V.M.; Guggemos, W.; Seilmaier, M.; Zange, S.; Müller, M.A.; Niemeyer, D.; Jones, T.C.; Vollmar, P.; Rothe, C.; et al. Virological assessment of hospitalized patients with COVID-2019. Nature 2020, 581, 465–469. [Google Scholar] [CrossRef]
- Fan, E.; Beitler, J.R.; Brochard, L.; Calfee, C.S.; Ferguson, N.D.; Slutsky, A.S.; Brodie, D. COVID-19-associated acute respiratory distress syndrome: Is a different approach to management warranted? Lancet Respir. Med. 2020, 8, 816–821. [Google Scholar] [CrossRef]
- Thompson, B.T.; Chambers, R.C.; Liu, K.D. Acute respiratory distress syndrome. N. Engl. J. Med. 2017, 377, 562–572. [Google Scholar] [CrossRef]
- Meduri, G.U.; Annane, D.; Confalonieri, M.; Chrousos, G.P.; Rochwerg, B.; Busby, A.; Ruaro, B.; Meibohm, B. Pharmacological principles guiding prolonged glucocorticoid treatment in ARDS. Intensiv. Care Med. 2020, 46, 2284–2296. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Coppola, S.; Cressoni, M.; Busana, M.; Rossi, S.; Chiumello, D. COVID-19 does not lead to a “typical” Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2020, 201, 1299–1300. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir. Med. 2020, 8, 475–481. [Google Scholar] [CrossRef]
- Ruaro, B.; Salton, F.; Braga, L.; Wade, B.; Confalonieri, P.; Volpe, M.C.; Baratella, E.; Maiocchi, S.; Confalonieri, M. The history and mystery of alveolar epithelial type II cells: Focus on their physiologic and pathologic role in lung. Int. J. Mol. Sci. 2021, 22, 2566. [Google Scholar] [CrossRef]
- Geri, P.; Salton, F.; Zuccatosta, L.; Tamburrini, M.; Biolo, M.; Busca, A.; Santagiuliana, M.; Zuccon, U.; Confalonieri, P.; Ruaro, B.; et al. Limited role for bronchoalveolar lavage to exclude COVID-19 after negative upper respiratory tract swabs: A multicentre study. Eur. Respir. J. 2020, 56, 2001733. [Google Scholar] [CrossRef]
- Morris, G.; Bortolasci, C.C.; Puri, B.K.; Olive, L.; Marx, W.; O’Neil, A.; Athan, E.; Carvalho, A.F.; Maes, M.; Walder, K.; et al. The pathophysiology of SARS-CoV-2: A suggested model and therapeutic approach. Life Sci. 2020, 258, 118166. [Google Scholar] [CrossRef]
- Chilosi, M.; Poletti, V.; Ravaglia, C.; Rossi, G.; Dubini, A.; Piciucchi, S.; Pedica, F.; Bronte, V.; Pizzolo, G.; Martignoni, G.; et al. The pathogenic role of epithelial and endothelial cells in early-phase COVID-19 pneumonia: Victims and partners in crime. Mod. Pathol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Pannone, G.; Caponio, V.C.A.; De Stefano, I.S.; Ramunno, M.A.; Meccariello, M.; Agostinone, A.; Pedicillo, M.C.; Troiano, G.; Zhurakivska, K.; Cassano, T.; et al. Lung histopathological findings in COVID-19 disease—A systematic review. Infect. Agent Cancer 2021, 16, 34. [Google Scholar] [CrossRef]
- Salton, F.; Confalonieri, P.; Meduri, G.U.; Santus, P.; Harari, S.; Scala, R.; Lanini, S.; Vertui, V.; Oggionni, T.; Caminati, A.; et al. Prolonged low-dose methylprednisolone in patients with severe COVID-19 pneumonia. Open Forum Infect. Dis. 2020, 7, ofaa421. [Google Scholar] [CrossRef] [PubMed]
- Dada, L.A.; Sznajder, J.I. Hypoxic inhibition of alveolar fluid reabsorption. Adv. Exp. Med. Biol. 2007, 618, 159–168. [Google Scholar] [CrossRef]
- Archer, S.L.; Weir, E.K.; Reeve, H.L.; Michelakis, E. Molecular identification of O2 sensors and O2-sensitive potassium channels in the pulmonary circulation. Adv. Exp. Med. Biol. 2000, 475, 219–240. [Google Scholar] [CrossRef]
- Yuan, S. Drugs to cure avian influenza infection—Multiple ways to prevent cell death. Cell Death Dis. 2013, 4, e835. [Google Scholar] [CrossRef]
- Kylhammar, D.; Rådegran, G. The principal pathways involved in the in vivo modulation of hypoxic pulmonary vasoconstriction, pulmonary arterial remodelling and pulmonary hypertension. Acta Physiol. 2017, 219, 728–756. [Google Scholar] [CrossRef] [PubMed]
- Sommer, N.; Strielkov, I.; Pak, O.; Weissmann, N. Oxygen sensing and signal transduction in hypoxic pulmonary vasoconstriction. Eur. Respir. J. 2016, 47, 288–303. [Google Scholar] [CrossRef]
- Dunham-Snary, K.J.; Wu, D.; Sykes, E.A.; Thakrar, A.; Parlow, L.R.G.; Mewburn, J.D.; Parlow, J.L.; Archer, S.L. Hypoxic pulmonary vasoconstriction: From molecular mechanisms to medicine. Chest 2017, 151, 181–192. [Google Scholar] [CrossRef]
- Villamor, E.; Moreno, L.; Mohammed, R.; Pérez-Vizcaíno, F.; Cogolludo, A. Reactive oxygen species as mediators of oxygen signaling during fetal-to-neonatal circulatory transition. Free Radic. Biol. Med. 2019, 142, 82–96. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, A.; Wu, D.; Tian, L.; Xiong, P.Y.; Dunham-Snary, K.J.; Chen, K.H.; Alizadeh, E.; Motamed, M.; Potus, F.; Hindmarch, C.C.T.; et al. Mitochondria in the pulmonary vasculature in health and disease: Oxygen-sensing, metabolism, and dynamics. Compr. Physiol. 2020, 10, 713–765. [Google Scholar] [CrossRef]
- Sommer, N.; Alebrahimdehkordi, N.; Pak, O.; Knoepp, F.; Strielkov, I.; Scheibe, S.; Dufour, E.; Andjelković, A.; Sydykov, A.; Saraji, A.; et al. Bypassing mitochondrial complex III using alternative oxidase inhibits acute pulmonary oxygen sensing. Sci. Adv. 2020, 6, eaba0694. [Google Scholar] [CrossRef] [PubMed]
- Kreutz, R.; Algharably, E.A.E.; Azizi, M.; Dobrowolski, P.; Guzik, T.; Januszewicz, A.; Persu, A.; Prejbisz, A.; Riemer, T.G.; Wang, J.G.; et al. Hypertension, the renin-angiotensin system, and the risk of lower respiratory tract infections and lung injury: Implications for COVID-19. Cardiovasc. Res. 2020, 116, 1688–1699. [Google Scholar] [CrossRef] [PubMed]
- Kai, H.; Kai, M. Interactions of coronaviruses with ACE2, angiotensin II, and RAS inhibitors-lessons from available evidence and insights into COVID-19. Hypertens. Res. 2020, 43, 648–654. [Google Scholar] [CrossRef]
- Hussman, J.P. Cellular and molecular pathways of COVID-19 and potential points of therapeutic intervention. Front. Pharmacol. 2020, 11, 1169. [Google Scholar] [CrossRef]
- Gao, Y.L.; Du, Y.; Zhang, C.; Cheng, C.; Yang, H.Y.; Jin, Y.F.; Duan, G.C.; Chen, S.Y. Role of renin-angiotensin system in acute lung injury caused by viral infection. Infect. Drug Resist. 2020, 13, 3715–3725. [Google Scholar] [CrossRef]
- Cai, L.; Guo, X.; Cao, Y.; Ying, P.; Hong, L.; Zhang, Y.; Yi, G.; Fu, M. Determining available strategies for prevention and therapy: Exploring COVID-19 from the perspective of ACE2 (Review). Int. J. Mol. Med. 2021, 47, 43. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 2020, 181, 894–904.e9. [Google Scholar] [CrossRef]
- Costa, L.B.; Perez, L.G.; Palmeira, V.A.; Macedo, E.; e Cordeiro, T.M.; Ribeiro, V.T.; Lanza, K.; e Silva, A.C.S. Insights on SARS-CoV-2 molecular interactions with the renin-angiotensin system. Front. Cell. Dev. Biol. 2020, 8, 559841. [Google Scholar] [CrossRef]
- Gan, R.; Rosoman, N.P.; Henshaw, D.J.E.; Noble, E.P.; Georgius, P.; Sommerfeld, N. COVID-19 as a viral functional ACE2 deficiency disorder with ACE2 related multi-organ disease. Med. Hypotheses 2020, 144, 110024. [Google Scholar] [CrossRef]
- Petersson, J.; Glenny, R.W. Gas exchange and ventilation-perfusion relationships in the lung. Eur. Respir. J. 2014, 44, 1023–1041. [Google Scholar] [CrossRef] [PubMed]
- Yanamandra, U.; Nair, V.; Singh, S. Managing high-altitude pulmonary edema with oxygen alone: Results of a randomized con- trolled trial. High Alt. Med. Biol. 2016, 17, 294–299. [Google Scholar] [CrossRef]
- Rello, J.; Storti, E.; Belliato, M.; Serrano, R. Clinical phenotypes of SARS-CoV-2: Implications for clinicians and researchers. Eur. Respir. J. 2020, 55, 2001028. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L.; Sharp, W.W.; Weir, E.K. Differentiating COVID-19 pneumonia from acute respiratory distress syndrome and high altitude pulmonary edema: Therapeutic implications. Circulation 2020, 142, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Pagnesi, M.; Baldetti, L.; Beneduce, A.; Calvo, F.; Gramegna, M.; Pazzanese, V.; Ingallina, G.; Napolano, A.; Finazzi, R.; Ruggeri, A.; et al. Pulmonary hypertension and right ventricular involvement in hospitalised patients with COVID-19. Heart 2020, 106, 1324–1331. [Google Scholar] [CrossRef]
- Van Dongen, C.M.; Janssen, M.T.; van der Horst, R.P.; van Kraaij, D.J.; Peeters, R.H.; van den Toorn, L.M.; Mostard, R.L. Unusually rapid development of pulmonary hypertension and right ventricular failure after COVID-19 pneumonia. Eur. J. Case Rep. Intern. Med. 2020, 7, 001784. [Google Scholar] [CrossRef]
- Kubánková, M.; Hohberger, B.; Hoffmanns, J.; Fürst, J.; Herrmann, M.; Guck, J.; Kräter, M. Physical phenotype of blood cells is altered in COVID-19. Biophys. J. 2021. [Google Scholar] [CrossRef]
- Herrmann, J.; Mori, V.; Bates, J.H.T.; Suki, B. Modeling lung perfusion abnormalities to explain early COVID-19 hypoxemia. Nat. Commun. 2020, 11, 4883. [Google Scholar] [CrossRef]
- Mauri, T.; Spinelli, E.; Scotti, E.; Colussi, G.; Basile, M.C.; Crotti, S.; Tubiolo, D.; Tagliabue, P.; Zanella, A.; Grasselli, G.; et al. Potential for lung recruitment and ventilation-perfusion mismatch in patients with the acute respiratory distress syndrome from coronavirus disease 2019. Crit. Care Med. 2020, 48, 1129–1134. [Google Scholar] [CrossRef]
- Ospina-Tascón, G.A.; Bautista, D.F.; Madriñán, H.J.; Valencia, J.D.; Bermúdez, W.F.; Quiñones, E.; Calderón-Tapia, L.E.; Hernandez, G.; Bruhn, A.; De Backer, D. Microcirculatory dysfunction and dead-space ventilation in early ARDS: A hypothesis-generating observational study. Ann. Intensiv. Care 2020, 10, 35. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in COVID-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Mesa, J.E.; Galindo-Coral, S.; Montes, M.C.; Muñoz Martin, A.J. Thrombosis and coagulopathy in COVID-19. Curr. Probl. Cardiol. 2021, 46, 100742. [Google Scholar] [CrossRef] [PubMed]
- Adusumilli, N.C.; Zhang, D.; Friedman, J.M.; Friedman, A.J. Harnessing nitric oxide for preventing, limiting and treating the severe pulmonary consequences of COVID-19. Nitric Oxide 2020, 103, 4–8. [Google Scholar] [CrossRef]
- Ricciardolo, F.L.M.; Bertolini, F.; Carriero, V.; Högman, M. Nitric oxide’s physiologic effects and potential as a therapeutic agent against COVID-19. J. Breath Res. 2020, 15, 014001. [Google Scholar] [CrossRef]
- Mohamed-Hussein, A.A.R.; Aly, K.M.E.; Ibrahim, M.A.A. Should aspirin be used for prophylaxis of COVID-19-induced coagulopathy? Med. Hypotheses 2020, 144, 109975. [Google Scholar] [CrossRef] [PubMed]
- Haque, S.; Jawed, A.; Akhter, N.; Dar, S.A.; Khan, F.; Mandal, R.K.; Areeshi, M.Y.; Lohani, M.; Wahid, M. Acetylsalicylic acid (Aspirin): A potent medicine for preventing COVID-19 deaths caused by thrombosis and pulmonary embolism. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 9244–9245. [Google Scholar] [CrossRef] [PubMed]
- Chow, J.H.; Khanna, A.K.; Kethireddy, S.; Yamane, D.; Levine, A.; Jackson, A.M.; McCurdy, M.T.; Tabatabai, A.; Kumar, G.; Park, P.; et al. Aspirin use is associated with decreased mechanical ventilation, ICU admission, and in-hospital mortality in hospitalized patients with COVID-19. Anesth. Analg. 2021, 132, 930–941. [Google Scholar] [CrossRef] [PubMed]
- Pavoni, V.; Gianesello, L.; Pazzi, M.; Stera, C.; Meconi, T.; Frigieri, F.C. Venous thromboembolism and bleeding in critically ill COVID-19 patients treated with higher than standard low molecular weight heparin doses and aspirin: A call to action. Thromb. Res. 2020, 196, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Falcone, M.; Tiseo, G.; Barbieri, G.; Galfo, V.; Russo, A.; Virdis, A.; Forfori, F.; Corradi, F.; Guarracino, F.; Carrozzi, L.; et al. Role of low-molecular-weight heparin in hospitalized patients with severe acute respiratory syndrome coronavirus 2 pneumonia: A prospective observational sudy. Open Forum Infect. Dis. 2020, 7, ofaa563. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, I.; Ciavarella, A.; Abbattista, M.; Aliberti, S.; De Zan, V.; Folli, C.; Panigada, M.; Gori, A.; Artoni, A.; Ierardi, A.M.; et al. Increasing dosages of low-molecular-weight heparin in hospitalized patients with COVID-19. Intern. Emerg. Med. 2021. [Google Scholar] [CrossRef]
- Shen, L.; Qiu, L.; Liu, D.; Wang, L.; Huang, H.; Ge, H.; Xiao, Y.; Liu, Y.; Jin, J.; Liu, X.; et al. The association of low molecular weight heparin use and in-hospital mortality among patients hospitalized with COVID-19. Cardiovasc. Drugs Ther. 2021. [Google Scholar] [CrossRef]
- Di Castelnuovo, A.; Costanzo, S.; Antinori, A.; Berselli, N.; Blandi, L.; Bonaccio, M.; Cauda, R.; Guaraldi, G.; Menicanti, L.; Mennuni, M.; et al. Heparin in COVID-19 patients is associated with reduced in-hospital mortality: The multicenter Italian CORIST study. Thromb. Haemost. 2021. [Google Scholar] [CrossRef]
- Cuker, A.; Tseng, E.K.; Nieuwlaat, R.; Angchaisuksiri, P.; Blair, C.; Dane, K.; Davila, J.; DeSancho, M.T.; Diuguid, D.; Griffin, D.O.; et al. American Society of Hematology 2021 guidelines on the use of anticoagulation for thromboprophylaxis in patients with COVID-19. Blood Adv. 2021, 5, 872–888. [Google Scholar] [CrossRef]
- Mennuni, M.G.; Renda, G.; Grisafi, L.; Rognoni, A.; Colombo, C.; Lio, V.; Foglietta, M.; Petrilli, I.; Pirisi, M.; Spinoni, E.; et al. Clinical outcome with different doses of low-molecular-weight heparin in patients hospitalized for COVID-19. J. Thromb. Thrombolysis 2021. [Google Scholar] [CrossRef]
- Lang, J.; Yang, N.; Deng, J.; Liu, K.; Yang, P.; Zhang, G.; Jiang, C. Inhibition of SARS pseudovirus cell entry by lactoferrin binding to heparan sulfate proteoglycans. PLoS ONE 2011, 6, e23710. [Google Scholar] [CrossRef]
- Clausen, T.M.; Sandoval, D.R.; Spliid, C.B.; Pihl, J.; Perrett, H.R.; Painter, C.D.; Narayanan, A.; Majowicz, S.A.; Kwong, E.M.; McVicar, R.N.; et al. SARS-CoV-2 infection depends on cellular heparan sulfate and ACE2. Cell 2020, 183, 1043–1057.e15. [Google Scholar] [CrossRef]
- Yuan, S.; Jiang, S.; Zhang, Z.; Li, Z.; Wang, C.; Yuan, M.; Chen, Y.; Tao, Q.; Lan, T.; Tang, X.; et al. TMPRSS2 protease inhibitors may prolong but heparins accelerate SARS-CoV-2 clearance. Preprints 2020, 2020060249. [Google Scholar] [CrossRef]
- Perera, R.A.; Mok, C.K.; Tsang, O.T.; Lv, H.; Ko, R.L.; Wu, N.C.; Yuan, M.; Leung, W.S.; Chan, J.M.; Chik, T.S.; et al. Serological assays for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), March 2020. Eurosurveillance 2020, 25, 2000421. [Google Scholar] [CrossRef] [PubMed]
- Montero-Fernández, M.A.; Pardo-Garcia, R. Histopathology features of the lung in COVID-19 patients. Diagn. Histopathol. 2021, 27, 123–127. [Google Scholar] [CrossRef]
- Takahashi, K.; Kajiura, K.; Nasu, M.; Nakamura, K.; Sugata, K.; Matsuzaki, A. Post-mortem biopsy of a patient with late exacerbation of COVID-19 pneumonia. Respirol. Case Rep. 2021, 9, e00724. [Google Scholar] [CrossRef]
- Wang, X.X.; Shao, C.; Huang, X.J.; Sun, L.; Meng, L.J.; Liu, H.; Zhang, S.J.; Li, H.J.; Lv, F.D. Histopathological features of multiorgan percutaneous tissue core biopsy in patients with COVID-19. J. Clin. Pathol. 2021, 74, 522–527. [Google Scholar] [CrossRef]
- Zhou, G.; Dada, L.A.; Sznajder, J.I. Regulation of alveolar epithelial function by hypoxia. Eur. Respir. J. 2008, 31, 1107–1113. [Google Scholar] [CrossRef] [PubMed]
- Schousboe, P.; Wiese, L.; Heiring, C.; Verder, H.; Poorisrisak, P.; Verder, P.; Nielsen, H.B. Assessment of pulmonary surfactant in COVID-19 patients. Crit. Care 2020, 24, 552. [Google Scholar] [CrossRef]
- Islam, A.B.M.M.K.; Khan, M.A. Lung transcriptome of a COVID-19 patient and systems biology predictions suggest impaired surfactant production which may be druggable by surfactant therapy. Sci. Rep. 2020, 10, 19395. [Google Scholar] [CrossRef] [PubMed]
- Piva, S.; DiBlasi, R.M.; Slee, A.E.; Jobe, A.H.; Roccaro, A.M.; Filippini, M.; Latronico, N.; Bertoni, M.; Marshall, J.C.; Portman, M.A. Surfactant therapy for COVID-19 related ARDS: A retrospective case-control pilot study. Respir. Res. 2021, 22, 20. [Google Scholar] [CrossRef]
- Cattel, F.; Giordano, S.; Bertiond, C.; Lupia, T.; Corcione, S.; Scaldaferri, M.; Angelone, L.; De Rosa, F.G. Use of exogenous pulmonary surfactant in acute respiratory distress syndrome (ARDS): Role in SARS-CoV-2-related lung injury. Respir. Physiol. Neurobiol. 2021, 288, 103645. [Google Scholar] [CrossRef] [PubMed]
- Ghati, A.; Dam, P.; Tasdemir, D.; Kati, A.; Sellami, H.; Sezgin, G.C.; Ildiz, N.; Franco, O.L.; Mandal, A.K.; Ocsoy, I. Exogenous pulmonary surfactant: A review focused on adjunctive therapy for severe acute respiratory syndrome coronavirus 2 including SP-A and SP-D as added clinical marker. Curr. Opin. Colloid Interface Sci. 2021, 51, 101413. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, M.A.; Luisada, A.A. Alcohol-oxygen vapor therapy of pulmonary edema. Ann. Intern. Med. 1952, 37, 1221–1231. [Google Scholar] [CrossRef]
- Waugh, W.H. Potential use of warm butyl alcohol vapor as adjunct agent in the emergency treatment of sea water wet near-drowning. Am. J. Emerg. Med. 1993, 11, 20–27. [Google Scholar] [CrossRef]
- Waugh, W.H. Adjuvant use of warm butyl alcohol vapor in experimental pulmonary edema. Life Sci. 1993, 52, 171–182. [Google Scholar] [CrossRef]
- Jiang, X.W.; Gao, M.Z.; Liang, J.H. Experimental study on different concentration alcohol humidifying oxygen supply improving hypoxia caused by pulmonary edema. Zhonghua Hu Li Za Zhi 1996, 31, 373–376. (In Chinese) [Google Scholar] [PubMed]
- The National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network. Comparison of two fluid-management strategies in acute lung injury. N. Engl. J. Med. 2006, 354, 2564–2575. [Google Scholar] [CrossRef]
- Famous, K.R.; Delucchi, K.; Ware, L.B.; Kangelaris, K.N.; Liu, K.D.; Thompson, B.T.; Calfee, C.S.; ARDS Network. Acute respiratory distress syndrome subphenotypes respond differently to randomized fluid management strategy. Am. J. Respir. Crit. Care Med. 2017, 195, 331–338. [Google Scholar] [CrossRef]
- Casey, J.D.; Semler, M.W.; Rice, T.W. Fluid management in acute respiratory distress syndrome. Semin. Respir. Crit. Care Med. 2019, 40, 57–65. [Google Scholar] [CrossRef]
- Barton, L.M.; Duval, E.J.; Stroberg, E.; Ghosh, S.; Mukhopadhyay, S. COVID-19 autopsies, Oklahoma, USA. Am. J. Clin. Pathol. 2020, 153, 725–733. [Google Scholar] [CrossRef]
- Menter, T.; Haslbauer, J.D.; Nienhold, R.; Savic, S.; Hopfer, H.; Deigendesch, N.; Frank, S.; Turek, D.; Willi, N.; Pargger, H.; et al. Postmortem examination of COVID-19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings in lungs and other organs suggesting vascular dysfunction. Histopathology 2020, 77, 198–209. [Google Scholar] [CrossRef]
- Carsana, L.; Sonzogni, A.; Nasr, A.; Rossi, R.S.; Pellegrinelli, A.; Zerbi, P.; Rech, R.; Colombo, R.; Antinori, S.; Corbellino, M.; et al. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: A two-centre descriptive study. Lancet Infect. Dis. 2020, 20, 1135–1140. [Google Scholar] [CrossRef]
- Navarro Conde, P.; Alemany Monraval, P.; Medina Medina, C.; Jiménez Sánchez, A.; Andrés Teruel, J.C.; Ferrando Marco, J.; Puglia Santos, V.; Mayordomo Aranda, E. Autopsy findings from the first known death from Severe Acute Respiratory Syndrome SARS-CoV-2 in Spain. Rev. Esp. Patol. 2020, 53, 188–192. [Google Scholar] [CrossRef]
- Benumof, J.L. One-lung ventilation and hypoxic pulmonary vasoconstriction: Implications for anesthetic management. Anesth. Analg. 1985, 64, 821–833. [Google Scholar] [CrossRef]
- Li, Y.; Tesselaar, E.; Borges, J.B.; Böhm, S.H.; Sjöberg, F.; Janerot-Sjöberg, B. Hyperoxia affects the regional pulmonary ventilation/perfusion ratio: An electrical impedance tomography study. Acta Anaesthesiol. Scand. 2014, 58, 716–725. [Google Scholar] [CrossRef]
- Hartmann, E.K.; Duenges, B.; Boehme, S.; Szczyrba, M.; Liu, T.; Klein, K.U.; Baumgardner, J.E.; Markstaller, K.; David, M. Ventilation/perfusion ratios measured by multiple inert gas elimination during experimental cardiopulmonary resuscitation. Acta Anaesthesiol. Scand. 2014, 58, 1032–1039. [Google Scholar] [CrossRef]
- Jaffal, K.; Six, S.; Zerimech, F.; Nseir, S. Relationship between hyperoxemia and ventilator associated pneumonia. Ann. Transl. Med. 2017, 5, 453. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, B.; Li, Q.; Wen, L.; Zhang, R. Clinical features of 69 cases with coronavirus disease 2019 in Wuhan, China. Clin. Infect. Dis. 2020, 71, 769–777. [Google Scholar] [CrossRef]
- Dai, H.; Zhang, X.; Xia, J.; Zhang, T.; Shang, Y.; Huang, R.; Liu, R.; Wang, D.; Li, M.; Wu, J.; et al. High-resolution chest CT features and clinical characteristics of patients infected with COVID-19 in Jiangsu, China. Int. J. Infect. Dis. 2020, 95, 106–112. [Google Scholar] [CrossRef]
- Li, X.; Marmar, T.; Xu, Q.; Tu, J.; Yin, Y.; Tao, Q.; Chen, H.; Shen, T.; Xu, D. Predictive indicators of severe COVID-19 independent of comorbidities and advanced age: A nested case-control study. Epidemiol. Infect. 2020, 148, e255. [Google Scholar] [CrossRef]
- WHO. Clinical Management of Severe Acute Respiratory Infection when Middle East Respiratory Syndrome Coronavirus (MERS-CoV) Infection is Suspected (Interim Guidance). Available online: https://www.who.int/csr/disease/coronavirus_infections/case-management-ipc/en/ (accessed on 15 April 2020).
- BMJ Best Practice. Coronavirus Disease 2019 (COVID-19). Available online: https://bestpractice.bmj.com/topics/en-gb/3000168/ (accessed on 18 August 2020).
- The First Affiliated Hospital, Zhejiang University School of Medicine. Handbook of COVID-19 Prevention and Treatment. Available online: http://che.zju.edu.cn/cheen/2020/0401/c27758a2021088/page.htm (accessed on 18 August 2020).
- Jouffroy, R.; Jost, D.; Prunet, B. Prehospital pulse oximetry: A red flag for early detection of silent hypoxemia in COVID-19 patients. Crit. Care 2020, 24, 313. [Google Scholar] [CrossRef] [PubMed]
- Siemieniuk, R.; Chu, D.K.; Kim, L.H.; Güell-Rous, M.R.; Alhazzani, W.; Soccal, P.M.; Karanicolas, P.J.; Farhoumand, P.D.; Siemieniuk, J.L.K.; Satia, I.; et al. Oxygen therapy for acutely ill medical patients: A clinical practice guideline. BMJ 2018, 363, k4169. [Google Scholar] [CrossRef]
- Alhazzani, W.; Møller, M.H.; Arabi, Y.M.; Loeb, M.; Gong, M.N.; Fan, E.; Oczkowski, S.; Levy, M.M.; Derde, L.; Dzierba, A.; et al. Surviving sepsis campaign: Guidelines on the management of critically ill adults with coronavirus disease 2019 (COVID-19). Crit. Care Med. 2020, 48, e440–e469. [Google Scholar] [CrossRef]
- Amodeo, L.R.; Wills, D.N.; Sanchez-Alavez, M.; Nguyen, W.; Conti, B.; Ehlers, C.L. Intermittent voluntary ethanol consumption combined with ethanol vapor exposure during adolescence increases drinking and alters other behaviors in adulthood in female and male rats. Alcohol 2018, 73, 57–66. [Google Scholar] [CrossRef]
- Nentwig, T.B.; Starr, E.M.; Chandler, L.J.; Glover, E.J. Absence of compulsive drinking phenotype in adult male rats exposed to ethanol in a binge-like pattern during adolescence. Alcohol 2019, 79, 93–103. [Google Scholar] [CrossRef]
- Hasanin, A.; Mostafa, M. Evaluation of fluid responsiveness during COVID-19 pandemic: What are the remaining choices? J. Anesth. 2020, 34, 758–764. [Google Scholar] [CrossRef]
- Kallet, R.H. A comprehensive review of prone position in ARDS. Respir. Care 2015, 60, 1660–1687. [Google Scholar] [CrossRef] [PubMed]
- Slutsky, A.S.; Ranieri, V.M. Ventilator induced lung injury. N. Engl. J. Med. 2013, 369, 2126–2136. [Google Scholar] [CrossRef]
- Guérin, C.; Albert, R.K.; Beitler, J.; Gattinoni, L.; Jaber, S.; Marini, J.J.; Munshi, L.; Papazian, L.; Pesenti, A.; Vieillard-Baron, A.; et al. Prone position in ARDS patients: Why, when, how and for whom. Intensiv. Care Med. 2020, 46, 2385–2396. [Google Scholar] [CrossRef]
- Hamilton, C.; Steinlechner, B.; Gruber, E.; Simon, P.; Wollenek, G. The oxygen dissociation curve: Quantifying the shift. Perfusion 2004, 19, 141–144. [Google Scholar] [CrossRef]
- Dhont, S.; Derom, E.; Van Braeckel, E.; Depuydt, P.; Lambrecht, B.N. The pathophysiology of ‘happy’ hypoxemia in COVID-19. Respir. Res. 2020, 21, 198. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Lu, L.; Cao, W.; Li, T. Hypothesis for potential pathogenesis of SARS-CoV-2 infection-a review of immune changes in patients with viral pneumonia. Emerg. Microbes Infect. 2020, 9, 727–732. [Google Scholar] [CrossRef]
- Mannino, F.; Bitto, A.; Irrera, N. Severe acute respiratory syndrome coronavirus-2 induces cytokine storm and inflammation during coronavirus disease 19: Perspectives and possible therapeutic approaches. Front. Pharmacol. 2020, 11, 592169. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Jiang, S.C.; Zhang, Z.W.; Fu, Y.F.; Hu, J.; Li, Z.L. Quantification of cytokine storms during virus infections. Front. Immunol. 2021, 12, 659419. [Google Scholar] [CrossRef] [PubMed]
- Tavasolian, F.; Hatam, G.R.; Mosawi, S.H.; Saadi, M.I.; Abdollahi, E.; Jamialahmadi, T.; Sathyapalan, T.; Sahebkar, A. The immune response and effectiveness of COVID-19 therapies. Adv. Exp. Med. Biol. 2021, 1321, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Signorini, C.; Pignatti, P.; Coccini, T. How do inflammatory mediators, immune response and air pollution contribute to COVID-19 disease severity? A lesson to learn. Life 2021, 11, 182. [Google Scholar] [CrossRef] [PubMed]
- Yokota, S.; Miyamae, T.; Kuroiwa, Y.; Nishioka, K. Novel coronavirus disease 2019 (COVID-19) and cytokine storms for more effective treatments from an inflammatory pathophysiology. J. Clin. Med. 2021, 10, 801. [Google Scholar] [CrossRef]


{kind=link}
| Injuries | Pathogenic Mechanisms | Refs. |
|---|---|---|
| Alveolar endothelial injury | Endothelial barrier disruption induces intrapulmonary shunt, hypoxia, intravascular coagulation and the release of pro-inflammatory factors. | [1,5,35] |
| ACE2-decline-induced pulmonary injury | ACE2 deficiency leads to enhanced and protracted tissues, and vessel exposure to Ang II, which then enhances thrombosis and cell proliferation, increases tissue permeability, cytokine production and inflammation. | [25,26,27,28,29,30,31,32,33,34] |
| Loss of hypoxic pulmonary vasoconstriction (HPV) | HPV directs blood perfusion from badly-ventilated to well-ventilated alveoli to optimize gas exchange. | [36,37,38] |
| General pulmonary vasoconstriction | Lead to pulmonary hypertension and a risk of right-heart failure subsequently. | [39,40,41] |
| Severe ventilation-perfusion mismatch | Induce hypoxemia in the non-injured fraction or/and cause hyper-perfusion of the small injured fraction. | [42,43] |
| Intravascular coagulation and microthrombi formation | Lead to increased wasted ventilation and less efficient carbon dioxide removal. | [44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62] |
| Diffuse alveolar damage (DAD) | Lead to hypoxia at the edematous alveoli. | [63,64,65] |
| Alveolar edema | Lead to great decline in oxygen diffusion over the blood-air barrier (hypoxia); Hypoxia in turn inhibits oedema fluid clearance. | [5,66] |
| Impaired alveolar surfactant production | Increase alveolar surface tension and hamper alveolar fluid resorption. | [67,68,69,70,71] |
| Drugs or Treatments | Therapeutic Mechanisms | Therapeutic Effects | Refs. |
|---|---|---|---|
| Oxygen inhalation | Oxygenation enhancement | Alleviate hypoxia; however, high-flow oxygen lead to pulmonary toxic effects. | [35] |
| Mechanical ventilation | Oxygenation enhancement | Alleviate hypoxia; however, mechanical ventilation may cause mucus to be blown deep into the small airways, which then aggravates alveolar hypoxia. | [9] |
| ECMO | Oxygenation enhancement | Alleviate hypoxia; however, patients receiving ECMO still showed a high mortality rate. | [9] |
| Inhaled nitric oxide | Decrease pulmonary vascular resistance | Improve ventilation-perfusion ratio; however, may cause further arterial desaturation (hypoxia). | [35,47,48] |
| Aspirin | Anticoagulation | Reduce ICU cases, but show no apparent association with the fatality. | [49,50,51] |
| Heparin | Anticoagulation; block virus entry; increase antibody titres | Reduce the risk of in-hospital mortality and decrease the occurrence of severe cases; however, could not completely prevent occurrence of severe cases. | [52,53,54,55,56,57,58,59,60,61,62] |
| Exogenous pulmonary surfactant | Alleviate alveolar edema | Reduce the mortality of infants with neonatal RDS; however, clinical outcomes for COVID-19 patients need further investigation. | [67,68,69,70,71] |
| Ethanol–oxygen vapor therapy | Alleviate alveolar edema | May reduce occurrence of severe cases and the mortality rate (need clinical verification). | [72,73,74,75] |
| Conservative fluid management | Alleviate alveolar edema | May reduce occurrence of severe cases and the mortality rate (need clinical verification). | [76,77] |
| SpO2 Limits | Corresponding Disease Stage | Refs. | |
|---|---|---|---|
| SpO2 Lower Limit for Oxygen Therapy | 90% | In the SpO2 <90% group, the median occurrence time of lowest SpO2 was only one day after admission, indicating a very late stage. | [87] |
| 94.7% | Stage IV: bilateral diffuse inhibitions, over half of the lung areas involved, occasionally extended to the entire lung and defined as the white lung. | [88] | |
| 95% | 78.0% of the patients with SpO2 ≤95% would develop into severe diseases (late stages). | [89] | |
| 93% | Resting state SpO2 ≤93% indicates a severe condition (late stages). | [92] | |
| SpO2 Upper Limit for Oxygen Therapy | 96% | For most patients receiving oxygen therapy. | [94,95] |
| 88–92% | For patients at risk of hypercapnic respiratory failure. | [94] | |
| 88–92% | For patients with chronic type II respiratory failure. | [92] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, S.; Jiang, S.-C.; Zhang, Z.-W.; Fu, Y.-F.; Hu, J.; Li, Z.-L. The Role of Alveolar Edema in COVID-19. Cells 2021, 10, 1897. https://doi.org/10.3390/cells10081897
Yuan S, Jiang S-C, Zhang Z-W, Fu Y-F, Hu J, Li Z-L. The Role of Alveolar Edema in COVID-19. Cells. 2021; 10(8):1897. https://doi.org/10.3390/cells10081897
Chicago/Turabian StyleYuan, Shu, Si-Cong Jiang, Zhong-Wei Zhang, Yu-Fan Fu, Jing Hu, and Zi-Lin Li. 2021. "The Role of Alveolar Edema in COVID-19" Cells 10, no. 8: 1897. https://doi.org/10.3390/cells10081897
APA StyleYuan, S., Jiang, S.-C., Zhang, Z.-W., Fu, Y.-F., Hu, J., & Li, Z.-L. (2021). The Role of Alveolar Edema in COVID-19. Cells, 10(8), 1897. https://doi.org/10.3390/cells10081897