
Fibrosis, the Bad Actor in Cardiorenal Syndromes: Mechanisms Involved



Abstract
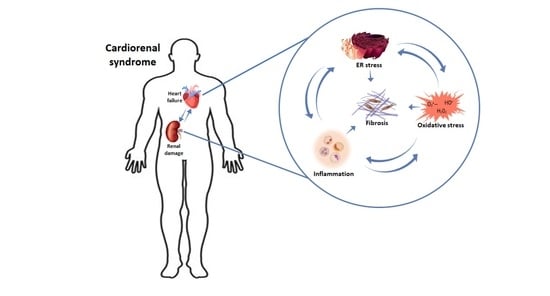
{kind=link}
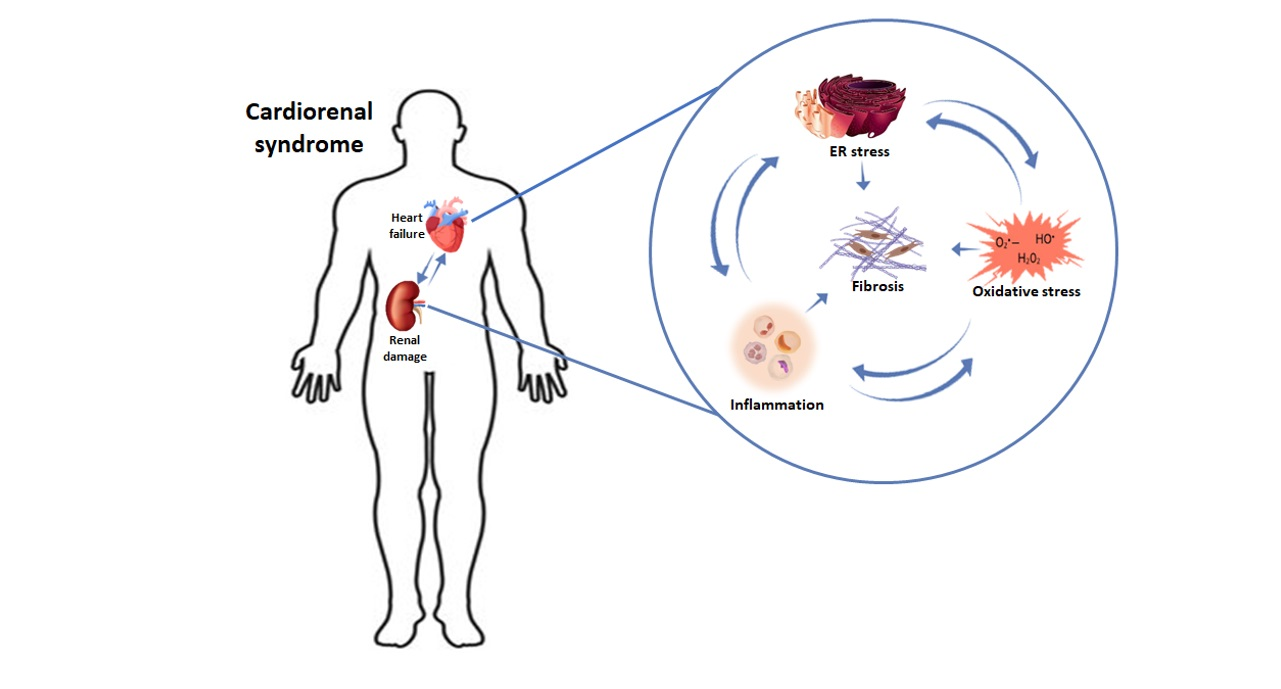
{kind=link}
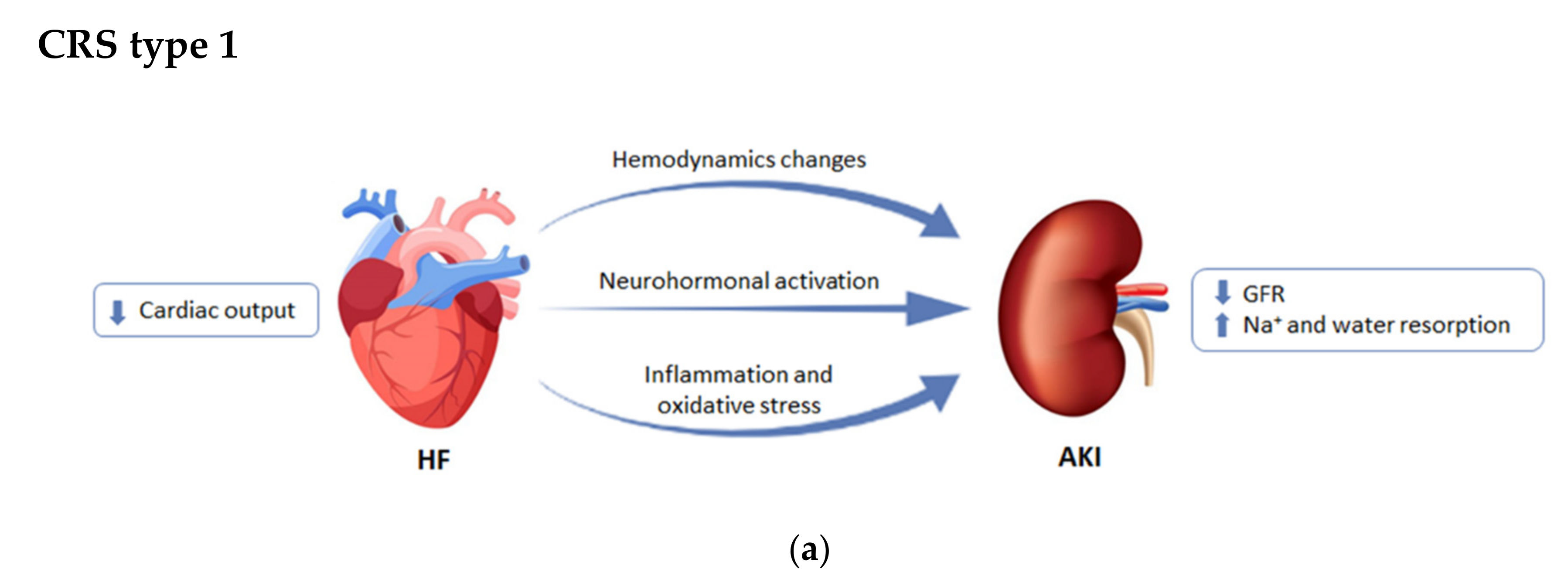
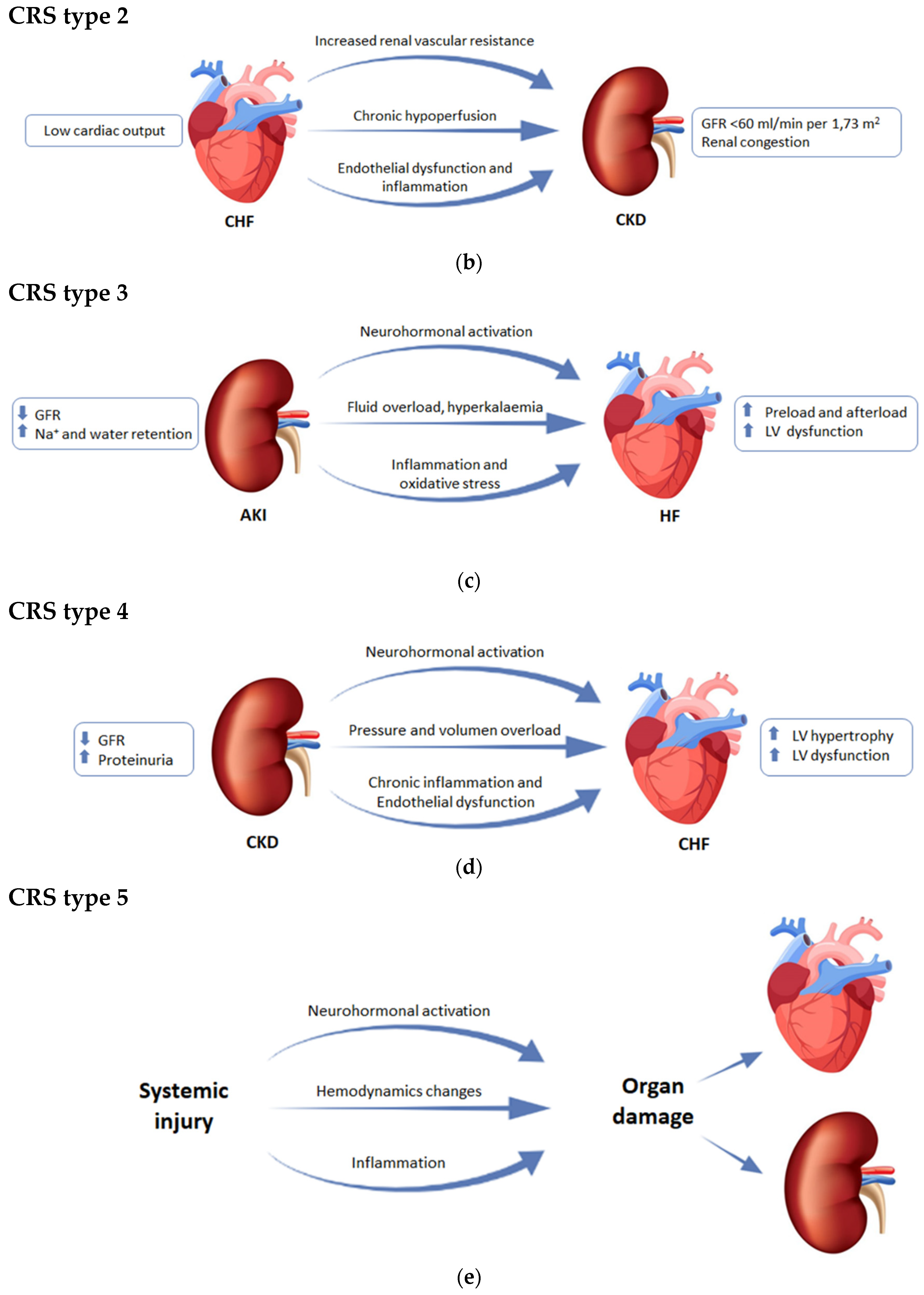{kind=link}
{kind=link}
1. Introduction
2. CRS Classification
2.1. CRS Type 1 or Acute Cardiorenal Syndrome
2.2. CRS Type 2 or Chronic Cardiorenal Syndrome
2.3. CRS Type 3 or Acute Reno-Cardiac Syndrome
2.4. CRS Type 4 or Chronic Reno-Cardiac Syndrome
2.5. CRS Type 5 or Secondary Cardiorenal Syndrome
3. Pathophysiology of CRS
3.1. Cardiac Alterations Associated with CKD
3.2. Renal Alteration Associated to HF
3.3. Fibrosis
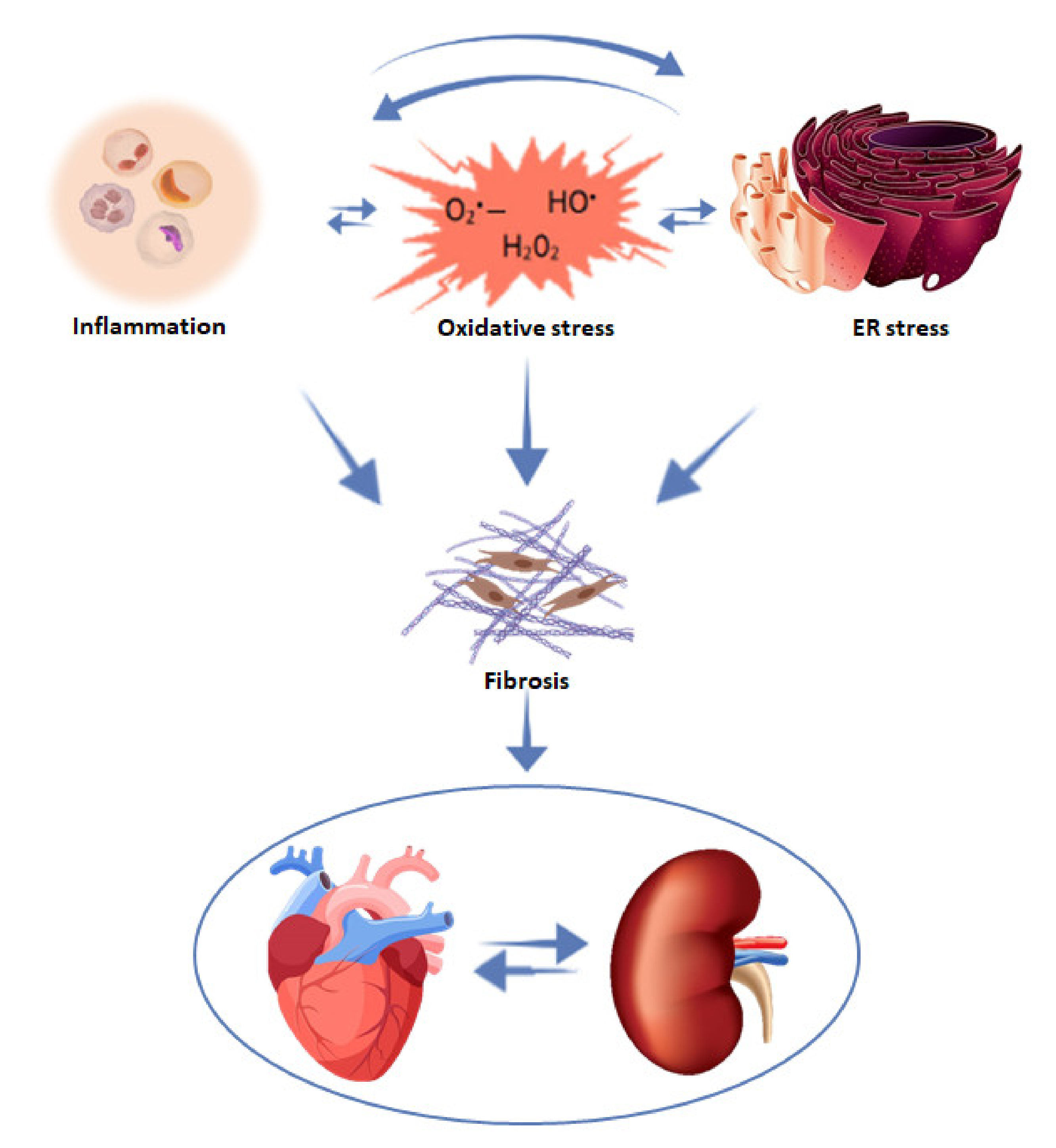
4. Mechanisms Involved in Fibrosis Progression
4.1. Inflammation
4.2. Oxidative Stress
4.3. Endoplasmic Reticulum Stress
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Bright, R. Cases and Observations Illustrative of Renal Disease, Accompanied with the Secretion of Albuminous Urine. Med. Chir. Rev. 1836, 25, 23–35. [Google Scholar]
- Zannad, F.; Rossignol, P. Cardiorenal Syndrome Revisited. Circulation 2018, 138, 929–944. [Google Scholar] [CrossRef]
- Cabandugama, P.K.; Gardner, M.J.; Sowers, J.R. The Renin Angiotensin Aldosterone System in Obesity and Hypertension. Med. Clin. North Am. 2017, 101, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Panas, R. Diabetes and Cardiorenal Syndrome: Understanding the “Triple Threat”. Hell. J. Cardiol. 2017, 58, 342–347. [Google Scholar] [CrossRef] [PubMed]
- McCullough, P.A.; Jurkovitz, C.T.; Pergola, P.E.; McGill, J.B.; Brown, W.W.; Collins, A.J.; Chen, S.-C.; Li, S.; Singh, A.; Norris, K.C.; et al. Independent Components of Chronic Kidney Disease as a Cardiovascular Risk State. Arch. Intern. Med. 2007, 167, 1122–1129. [Google Scholar] [CrossRef]
- Raina, R.; Nair, N.; Chakraborty, R.; Nemer, L.; Dasgupta, R.; Varian, K. An Update on the Pathophysiology and Treatment of Cardiorenal Syndrome. Cardiol. Res. 2020, 11, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Ronco, C.; McCullough, P.; Anker, S.D.; Anand, I.; Aspromonte, N.; Bagshaw, S.M.; Bellomo, R.; Berl, T.; Bobek, I.; Cruz, D.N.; et al. Cardio-Renal Syndromes: Report from the Consensus Conference of the Acute Dialysis Quality Initiative. Eur. Heart J. 2009, 31, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Heywood, J.T.; Fonarow, G.; Costanzo, M.R.; Mathur, V.S.; Wigneswaran, J.R.; Wynne, J. High Prevalence of Renal Dysfunction and Its Impact on Outcome in 118,465 Patients Hospitalized with Acute Decompensated Heart Failure: A Report from the ADHERE Database. J. Card. Fail. 2007, 13, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Ronco, C.; Cicoira, M.; McCullough, P.A. Cardiorenal Syndrome Type 1. J. Am. Coll. Cardiol. 2012, 60, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Prins, K.W.; Thenappan, T.; Markowitz, J.S.; Pritzker, M.R. Cardiorenal Syndrome Type 1: Renal Dysfunction in Acute Decom-Pensated Heart Failure. J. Clin. Outcomes Manag. 2015, 22, 443–454. [Google Scholar] [PubMed]
- Ronco, C.; Bellasi, A.; Di Lullo, L. Cardiorenal Syndrome: An Overview. Adv. Chronic Kidney Dis. 2018, 25, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Mullens, W.; Abrahams, Z.; Francis, G.S.; Sokos, G.; Taylor, D.O.; Starling, R.C.; Young, J.B.; Tang, W.W. Importance of Venous Congestion for Worsening of Renal Function in Advanced Decompensated Heart Failure. J. Am. Coll. Cardiol. 2009, 53, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Ames, M.K.; Atkins, C.E.; Pitt, B. The renin-angiotensin-aldosterone System and Its Suppression. J. Veter. Intern. Med. 2019, 33, 363–382. [Google Scholar] [CrossRef] [PubMed]
- Brewster, U.C.; Perazella, A.M. The Renin-Angiotensin-Aldosterone System and the Kidney: Effects on Kidney Disease. Am. J. Med. 2004, 116, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Virzì, G.M.; Clementi, A.; De Cal, M.; Brocca, A.; Day, S.; Pastori, S.; Bolin, C.; Vescovo, G.; Ronco, C. Oxidative Stress: Dual Pathway Induction in Cardiorenal Syndrome Type 1 Pathogenesis. Oxidative Med. Cell. Longev. 2015, 2015, 1–9. [Google Scholar] [CrossRef]
- Harrison, J.C.; Smart, S.D.G.; Besley, E.M.H.; Kelly, J.R.; Read, M.I.; Yao, Y.; Sammut, I. A Clinically Relevant Functional Model of Type-2 Cardio-Renal Syndrome with Paraventricular Changes Consequent to Chronic Ischaemic Heart Failure. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Rangaswami, J.; Bhalla, V.; Blair, J.E.; Chang, T.I.; Costa, S.; Lentine, K.L.; Lerma, E.; Mezue, K.; Molitch, M.; Mullens, W.; et al. Cardiorenal Syndrome: Classification, Pathophysiology, Diagnosis, and Treatment Strategies: A Scientific Statement from the American Heart Association. Circulation 2019, 139, e840–e878. [Google Scholar] [CrossRef]
- Stevens, P.E.; Levin, A. Kidney Disease: Improving Global Outcomes Chronic Kidney Disease Guideline Development Work Group Members. Evaluation and Management of Chronic Kidney Disease: Synopsis of the Kidney Disease: Improving Global Outcomes 2012 Clinical Practice Guideline. Ann. Intern. Med. 2013, 158, 825–830. [Google Scholar] [CrossRef]
- Damman, K.; Valente, M.A.; Voors, A.A.; O’Connor, C.M.; Van Veldhuisen, D.J.; Hillege, H.L. Renal impairment, worsening renal function, and outcome in patients with heart failure: An updated meta-analysis. Eur. Heart J. 2014, 35, 455–469. [Google Scholar] [CrossRef] [PubMed]
- De Vecchis, R.; Baldi, C. Cardiorenal Syndrome Type 2: From Diagnosis to optimal Management. Ther. Clin. Risk Manag. 2014, 10, 949–961. [Google Scholar] [CrossRef][Green Version]
- Di Lullo, L.; Reeves, P.B.; Bellasi, A.; Ronco, C. Cardiorenal Syndrome in Acute Kidney Injury. Semin. Nephrol. 2019, 39, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Bagshaw, S.M.; Hoste, E.; Braam, B.; Briguori, C.; Kellum, J.A.; McCullough, P.A.; Ronco, C. Cardiorenal Syndrome Type 3: Pathophysiologic and Epidemiologic Considerations. Contrib. Nephrol. 2013, 182, 137–157. [Google Scholar] [CrossRef]
- Kumar, U.; Wettersten, N.; Garimella, P.S. Cardiorenal Syndrome. Cardiol. Clin. 2019, 37, 251–265. [Google Scholar] [CrossRef] [PubMed]
- Uduman, J. Epidemiology of Cardiorenal Syndrome. Adv. Chronic Kidney Dis. 2018, 25, 391–399. [Google Scholar] [CrossRef]
- Mentzer, R.M.; Oz, M.C.; Sladen, R.N.; Graeve, A.H.; Hebeler, R.F.; Luber, J.M.; Smedira, N.G. Effects of Perioperative Nesiritide in Patients with Left Ventricular Dysfunction Undergoing Cardiac Surgery: The NAPA Trial. J. Am. Coll. Cardiol. 2007, 49, 716–726. [Google Scholar] [CrossRef]
- Di Lullo, L.; Bellasi, A.; Barbera, V.; Russo, D.; Russo, L.; Di Iorio, B.; Cozzolino, M.; Ronco, C. Pathophysiology of the Cardio-Renal Syndromes Types 1–5: An Uptodate. Indian Heart J. 2017, 69, 255–265. [Google Scholar] [CrossRef]
- Hillege, H.L.; Nitsch, D.; Pfeffer, M.A.; Swedberg, K.; McMurray, J.J.; Yusuf, S.; Granger, C.B.; Michelson, E.L.; Ostergren, J.; Cornel, J.; et al. Renal Function as a Predictor of Outcome in a Broad Spectrum of Patients with Heart Failure. Circulation 2006, 113, 671–678. [Google Scholar] [CrossRef]
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.-Y. Chronic Kidney Disease and the Risks of Death, Cardiovascular Events, and Hospitalization. N. Eng. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef]
- Suresh, H.; Arun, B.S.; Moger, V.; Swamy, M. Cardiorenal Syndrome Type 4: A Study of Cardiovascular Diseases in Chronic Kidney Disease. Indian Heart J. 2017, 69, 11–16. [Google Scholar] [CrossRef]
- Clementi, A.; Virzì, G.M.; Goh, C.Y.; Cruz, D.N.; Granata, A.; Vescovo, G.; Ronco, C. Cardiorenal Syndrome Type 4: A Review. Cardiorenal Med. 2013, 3, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.L.; Rabb, H.; Shaw, A.D.; Singbartl, K.; Ronco, C.; McCullough, P.A.; Kellum, J.A. Cardiorenal Syndrome Type 5: Clinical Presentation, Pathophysiology and Management Strategies from the Eleventh Consensus Conference of the Acute Dialysis Quality Initiative (ADQI). Contrib. Nephrol. 2013, 182, 174–194. [Google Scholar] [CrossRef]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.-L.; Rello, J.; Marshall, J.K.; Silva, E.; Anzueto, A.; Martin, C.D.; Moreno, R.; Lipman, J.; Gomersall, C.; Sakr, Y.; et al. International Study of the Prevalence and Outcomes of Infection in Intensive Care Units. JAMA 2009, 302, 2323–2329. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.L.; Program to Improve Care in Acute Renal Disease (PICARD) Study Group; Bouchard, J.; Soroko, S.B.; Ikizler, T.; Paganini, E.P.; Chertow, G.M.; Himmelfarb, J. Sepsis as a Cause and Consequence of Acute Kidney Injury: Program to Improve Care in Acute Renal Disease. Intensiv. Care Med. 2010, 37, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Ronco, C.; McCullough, P.A.; Anker, S.D.; Anand, I.; Aspromonte, N.; Bagshaw, S.M.; Bellomo, R.; Berl, T.; Bobek, I.; Cruz, D.N.; et al. Cardiorenal Syndromes: An Executive Summary from the Consensus Conference of the Acute Dialysis Quality Initiative (ADQI). Contrib. Nephrol. 2010, 165, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.; Grams, M.E.; Sang, Y.; Astor, B.C.; Blankestijn, P.J.; Brunskill, N.J.; Collins, J.F.; Kalra, P.A.; Kovesdy, C.P.; Levin, A.; et al. Risk Factors for Prognosis in Patients with Severely Decreased GFR. Kidney Int. Rep. 2018, 3, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, K.; Van Der Velde, M.; Astor, B.C.; Woodward, M.; Levey, A.S.; De Jong, P.E.; Coresh, J.; Gansevoort, R.T. Association of Estimated Glomerular Filtration Rate and Albuminuria with All-Cause and Cardiovascular Mortality in General Population Cohorts: A Collaborative Meta-Analysis. Lancet 2010, 375, 2073–2081. [Google Scholar] [CrossRef]
- Matsushita, K.; Coresh, J.; Sang, Y.; Chalmers, J.; Fox, C.; Guallar, E.; Jafar, T.; Jassal, S.K.; Landman, G.W.D.; Muntner, P.; et al. Estimated Glomerular Filtration Rate and Albuminuria for Prediction of Cardiovascular Outcomes: A Collaborative Meta-Analysis of Individual Participant Data. Lancet Diabetes Endocrinol. 2015, 3, 514–525. [Google Scholar] [CrossRef]
- James, M.T.; Grams, M.E.; Woodward, M.; Elley, C.R.; Green, J.A.; Wheeler, D.C.; de Jong, P.; Gansevoort, R.T.; Levey, A.S.; Warnock, D.G.; et al. A Meta-Analysis of the Association of Estimated GFR, Albuminuria, Diabetes Mellitus, and Hypertension with Acute Kidney Injury. Am. J. Kidney Dis. 2015, 66, 602–612. [Google Scholar] [CrossRef]
- Matsuo, H.; Dohi, K.; Machida, H.; Takeuchi, H.; Aoki, T.; Nishimura, H.; Yasutomi, M.; Senga, M.; Ichikawa, T.; Kakuta, K.; et al. Echocardiographic Assessment of Cardiac Structural and Functional Abnormalities in Patients with End-Stage Renal Disease Receiving Chronic Hemodialysis. Circ. J. 2018, 82, 586–595. [Google Scholar] [CrossRef]
- Otsuka, T.; Suzuki, M.; Yoshikawa, H.; Sugi, K. Left Ventricular Diastolic Dysfunction in the Early Stage of Chronic Kidney Disease. J. Cardiol. 2009, 54, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Dobre, M.; Roy, J.; Tao, K.; Anderson, A.H.; Bansal, N.; Chen, J.; Deo, R.; Drawz, P.; Feldman, H.I.; Hamm, L.L.; et al. Serum Bicarbonate and Structural and Functional Cardiac Abnormalities in Chronic Kidney Disease - A Report from the Chronic Renal Insufficiency Cohort Study. Am. J. Nephrol. 2016, 43, 411–420. [Google Scholar] [CrossRef]
- Shlipak, M.G.; Fried, L.F.; Cushman, M.; Manolio, T.A.; Peterson, D.; Stehman-Breen, C.; Bleyer, A.; Newman, A.B.; Siscovick, D.; Psaty, B. Cardiovascular Mortality Risk in Chronic Kidney Disease. JAMA 2005, 293, 1737–1745. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Hsu, C.-Y.; Li, Y.; Mishra, R.K.; Keane, M.; Rosas, S.E.; Dries, D.; Xie, D.; Chen, J.; He, J.; et al. Associations Between Kidney Function and Subclinical Cardiac Abnormalities in CKD. J. Am. Soc. Nephrol. 2012, 23, 1725–1734. [Google Scholar] [CrossRef] [PubMed]
- Taddei, S.; Nami, R.; Bruno, R.M.; Quatrini, I.; Nuti, R. Hypertension, Left Ventricular Hypertrophy and Chronic Kidney Disease. Heart Fail. Rev. 2010, 16, 615–620. [Google Scholar] [CrossRef]
- Matsumoto, M.; Io, H.; Furukawa, M.; Okumura, K.; Masuda, A.; Seto, T.; Takagi, M.; Sato, M.; Nagahama, L.; Omote, K.; et al. Risk Factors Associated with Increased Left Ventricular Mass Index in Chronic Kidney Disease Patients Evaluated Using Echocardiography. J. Nephrol. 2012, 25, 794–801. [Google Scholar] [CrossRef] [PubMed]
- Pluta, A.; Stróżecki, P.; Krintus, M.; Odrowaz-Sypniewska, G.; Manitius, J. Left Ventricular Remodeling and Arterial Remodeling in Patients with Chronic Kidney Disease Stage 1–3. Ren. Fail. 2015, 37, 1–6. [Google Scholar] [CrossRef]
- Matsushita, K.; Ballew, S.; Coresh, J. Influence of Chronic Kidney Disease on Cardiac Structure and Function. Curr. Hypertens. Rep. 2015, 17, 1–9. [Google Scholar] [CrossRef]
- Toida, T.; Toida, R.; Yamashita, R.; Komiya, N.; Uezono, S.; Komatsu, H.; Ishikawa, T.; Kitamura, K.; Sato, Y.; Fujimoto, S. Grading of Left Ventricular Diastolic Dysfunction with Preserved Systolic Function by the 2016 American Society of Echocardiography/European Association of Cardiovascular Imaging Recommendations Contributes to Predicting Cardiovascular Events in Hemodialysis Patients. Cardiorenal Med. 2019, 9, 190–200. [Google Scholar] [CrossRef]
- Escoli, R.; Carvalho, M.J.; Cabrita, A.; Rodrigues, A. Diastolic Dysfunction, an Underestimated New Challenge in Dialysis. Ther. Apher. Dial. 2019, 23, 108–117. [Google Scholar] [CrossRef]
- Cai, Q.-Z.; Lu, X.-Z.; Lu, Y.; Wang, A.Y.-M. Longitudinal Changes of Cardiac Structure and Function in CKD (CASCADE Study). J. Am. Soc. Nephrol. 2014, 25, 1599–1608. [Google Scholar] [CrossRef]
- Franczyk, B.; Gluba, A.; Olszewski, R.; Banach, M.; Rysz, J. Heart Function Disturbances in Chronic Kidney Disease – Echocardiographic Indices. Arch. Med Sci. 2014, 10, 1109–1116. [Google Scholar] [CrossRef] [PubMed]
- Vogel, M.W.; Slusser, J.P.; Hodge, D.O.; Chen, H.H. The Natural History of Preclinical Diastolic Dysfunction. Circ. Heart Fail. 2012, 5, 144–151. [Google Scholar] [CrossRef]
- Shah, S.; Kitzman, D.W.; Borlaug, B.; Van Heerebeek, L.; Zile, M.; Kass, D.A.; Paulus, W.J. Phenotype-Specific Treatment of Heart Failure with Preserved Ejection Fraction. Circulation 2016, 134, 73–90. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, I.A.; De Mutsert, R.; Rabelink, T.J.; Jukema, J.W.; De Roos, A.; Rosendaal, F.R.; Lamb, H.J.; De Vries, A.P. Associations Between Normal Range Albuminuria, Renal Function and Cardiovascular Function in a Population-Based Imaging Study. Atherosclerosis 2018, 272, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.M.; Lam, C.S.; Cheng, S.; Verma, A.; Desai, A.S.; Rocha, R.A.; Hilkert, R.; Izzo, J.; Oparil, S.; Pitt, B.; et al. The Relationship Between Renal Impairment and Left Ventricular Structure, Function, and ventricular–arterial Interaction in Hypertension. J. Hypertens. 2011, 29, 1829–1836. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, K.; Kwak, L.; Sang, Y.; Ballew, S.H.; Skali, H.; Shah, A.M.; Coresh, J.; Solomon, S. Kidney Disease Measures and Left Ventricular Structure and Function: The Atherosclerosis Risk in Communities Study. J. Am. Heart Assoc. 2017, 6, e006259. [Google Scholar] [CrossRef]
- Zhou, J.; Cui, X.; Jin, X.; Zhou, J.; Zhang, H.; Tang, B.; Fu, M.; Herlitz, H.; Cui, J.; Zhu, H.; et al. Association of Renal Biochemical Parameters with Left Ventricular Diastolic Dysfunction in a Community-Based Elderly Population in China: A Cross-Sectional Study. PLoS ONE 2014, 9, e88638. [Google Scholar] [CrossRef]
- Kang, E.; Ryu, H.; Kim, J.; Lee, J.; Lee, K.; Chae, D.; Sung, S.A.; Kim, S.W.; Ahn, C.; Oh, K. Association Between High-Sensitivity Cardiac Troponin T and Echocardiographic Parameters in Chronic Kidney Disease: Results from the KNOW-CKD Cohort Study. J. Am. Heart Assoc. 2019, 8, e013357. [Google Scholar] [CrossRef]
- McCullough, P.A.; Jefferies, J.L. Novel Markers and Therapies for Patients with Acute Heart Failure and Renal Dysfunction. Am. J. Med. 2015, 128, 312-e1–312.e22. [Google Scholar] [CrossRef]
- Savic-Radojevic, A.; Pljesa-Ercegovac, M.; Matic, M.; Simic, D.; Radovanovic, S.; Simic, T. Novel Biomarkers of Heart Failure. Adv. Clin. Chem. 2017, 79, 93–152. [Google Scholar] [CrossRef] [PubMed]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; González-Juanatey, J.R.; Harjola, V.-P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. J. Heart Fail. 2016, 18, 891–975. [Google Scholar] [CrossRef] [PubMed]
- Arslan, M.; Dedic, A.; Boersma, E.; A Dubois, E. Serial High-Sensitivity Cardiac Troponin T Measurements to Rule Out Acute Myocardial Infarction and a Single High Baseline Measurement for Swift Rule-In: A Systematic Review and Meta-Analysis. Eur. Heart J. Acute Cardiovasc. Care 2020, 9, 14–22. [Google Scholar] [CrossRef]
- Westermann, D.; Neumann, J.T.; Sörensen, N.A.; Blankenberg, D.W.J.T.N.N.A.S.S. High-Sensitivity Assays for Troponin in Patients with Cardiac Disease. Nat. Rev. Cardiol. 2017, 14, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Hill, N.R.; Fatoba, S.T.; Oke, J.L.; Hirst, J.; O’Callaghan, C.A.; Lasserson, D.; Hobbs, R. Global Prevalence of Chronic Kidney Disease—A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0158765. [Google Scholar] [CrossRef] [PubMed]
- Vogt, L.; Bangalore, S.; Fayyad, R.; Melamed, S.; Hovingh, G.K.; DeMicco, D.A.; Waters, D.D. Atorvastatin Has a Dose-Dependent Beneficial Effect on Kidney Function and Associated Cardiovascular Outcomes: Post Hoc Analysis of 6 Double-Blind Randomized Controlled Trials. J. Am. Heart Assoc. 2019, 8, e010827. [Google Scholar] [CrossRef]
- Dalal, R.; Bruss, Z.S.; Sehdev, J.S. Physiology, Renal Blood Flow and Filtration; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Borovac, J.A.; D’Amario, D.; Bozic, J.; Glavas, D. Sympathetic Nervous System Activation and Heart Failure: Current State of Evidence and the Pathophysiology in the Light of Novel Biomarkers. World J. Cardiol. 2020, 12, 373–408. [Google Scholar] [CrossRef]
- Dlugos, C.P.; Picciotto, C.; Lepa, C.; Krakow, M.; Stöber, A.; Eddy, M.-L.; Weide, T.; Jeibmann, A.; Krahn, M.; Van Marck, V.; et al. Nephrin Signaling Results in Integrin β1 Activation. J. Am. Soc. Nephrol. 2019, 30, 1006–1019. [Google Scholar] [CrossRef]
- Lichtnekert, J.; Kaverina, N.V.; Eng, D.G.; Gross, K.W.; Kutz, J.N.; Pippin, J.W.; Shankland, S.J. Renin-Angiotensin-Aldosterone System Inhibition Increases Podocyte Derivation from Cells of Renin Lineage. J. Am. Soc. Nephrol. 2016, 27, 3611–3627. [Google Scholar] [CrossRef]
- Stoll, D.; Yokota, R.; Aragão, D.S.; Casarini, D.E. Both Aldosterone and Spironolactone Can Modulate the Intracellular ACE/ANG II/AT1 and ACE2/ANG (1-7)/MAS Receptor Axes in Human Mesangial Cells. Physiol. Rep. 2019, 7, e14105. [Google Scholar] [CrossRef]
- Gómez, G.I.; Fernández, P.; Velarde, V.; Sáez, J.C. Angiotensin II-Induced Mesangial Cell Damage Is Preceded by Cell Membrane Permeabilization Due to Upregulation of Non-Selective Channels. Int. J. Mol. Sci. 2018, 19, 957. [Google Scholar] [CrossRef]
- Xu, Z.; Li, W.; Han, J.; Zou, C.; Huang, W.; Yu, W.; Shan, X.; Lum, H.; Li, X.; Liang, G. Angiotensin II Induces Kidney Inflammatory Injury and Fibrosis through Binding to Myeloid Differentiation Protein-2 (MD2). Sci. Rep. 2017, 7, srep44911. [Google Scholar] [CrossRef] [PubMed]
- Brankovic, M.; Akkerhuis, K.M.; Van Boven, N.; Manintveld, O.; Germans, T.; Brugts, J.; Caliskan, K.; Umans, V.; Constantinescu, A.; Kardys, I. Real-Life Use of Neurohormonal Antagonists and Loop Diuretics in Chronic Heart Failure: Analysis of Serial Biomarker Measurements and Clinical Outcome. Clin. Pharmacol. Ther. 2017, 104, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, D.; Singh, G. Effects of Single and Dual RAAS Blockade Therapy on Progressive Kidney Disease Transition to CKD in Rats. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2019, 393, 615–627. [Google Scholar] [CrossRef]
- Dörr, O.; Liebetrau, C.; Möllmann, H.; Gaede, L.; Troidl, C.; Wiebe, J.; Renker, M.; Bauer, T.; Hamm, C.; Nef, H. Long-Term Verification of Functional and Structural Renal Damage After Renal Sympathetic Denervation. Catheter. Cardiovasc. Interv. 2016, 87, 1298–1303. [Google Scholar] [CrossRef] [PubMed]
- Damman, K.; Tang, W.W.; Testani, J.M.; McMurray, J.J. Terminology and Definition of Changes Renal Function in Heart Failure. Eur. Heart J. 2014, 35, 3413–3416. [Google Scholar] [CrossRef] [PubMed]
- Damman, K.; Testani, J.M. The Kidney in Heart Failure: An Update. Eur. Heart J. 2015, 36, 1437–1444. [Google Scholar] [CrossRef]
- Norris, K.C.; Smoyer, K.E.; Rolland, C.; Van Der Vaart, J.; Grubb, E.B. Albuminuria, Serum Creatinine, and Estimated Glomerular Filtration Rate as Predictors of Cardio-Renal Outcomes in Patients with Type 2 Diabetes Mellitus and Kidney Disease: A Systematic Literature Review. BMC Nephrol. 2018, 19, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.E.; Solomon, S.D.; Gerstein, H.; Zetterstrand, S.; Olofsson, B.; Michelson, E.L.; Granger, C.B.; Swedberg, K.; A Pfeffer, M.; Yusuf, S.; et al. Albuminuria in Chronic Heart Failure: Prevalence and Prognostic Importance. Lancet 2009, 374, 543–550. [Google Scholar] [CrossRef]
- Yang, Y.; Kim, K.; Hwang, I.; Yim, T.; Do, W.; Kim, M.; Lee, S.; Jung, H.-Y.; Choi, J.-Y.; Park, S.-H.; et al. Cystatin C–Based Equation for Predicting the Glomerular Filtration Rate in Kidney Transplant Recipients. Transplant. Proc. 2017, 49, 1018–1022. [Google Scholar] [CrossRef]
- Wang, D.; Feng, J.-F.; Wang, A.-Q.; Yang, Y.-W.; Liu, Y.-S. Role of Cystatin C and Glomerular Filtration Rate in Diagnosis of Kidney Impairment in Hepatic Cirrhosis Patients. Medicine 2017, 96, e6949. [Google Scholar] [CrossRef] [PubMed]
- Richter, B.; Sulzgruber, P.; Koller, L.; Steininger, M.; El-Hamid, F.; Rothgerber, D.J.; Forster, S.; Goliasch, G.; Silbert, B.I.; Meyer, E.L.; et al. Blood Urea Nitrogen Has Additive Value Beyond Estimated Glomerular Filtration Rate for Prediction of Long-Term Mortality in Patients with Acute Myocardial Infarction. Eur. J. Intern. Med. 2019, 59, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Seki, M.; Nakayama, M.; Sakoh, T.; Yoshitomi, R.; Fukui, A.; Katafuchi, E.; Tsuda, S.; Nakano, T.; Tsuruya, K.; Kitazono, T. Blood Urea Nitrogen Is Independently Associated with Renal Outcomes in Japanese Patients with Stage 3–5 Chronic Kidney Disease: A Prospective Observational Study. BMC Nephrol. 2019, 20, 115. [Google Scholar] [CrossRef] [PubMed]
- Buonafine, M.; Martinez-Martinez, E.; Jaisser, F. More Than a Simple Biomarker: The Role of NGAL in Cardiovascular and Renal Diseases. Clin. Sci. 2018, 132, 909–923. [Google Scholar] [CrossRef] [PubMed]
- Merdler, I.; Rozenfeld, K.-L.; Zahler, D.; Shtark, M.; Goldiner, I.; Loewenstein, I.S.; Fortis, L.; Hochstadt, A.; Keren, G.; Banai, S.; et al. Neutrophil Gelatinase-Associated Lipocalin for the Early Prediction of Acute Kidney Injury in ST-Segment Elevation Myocardial Infarction Patients Treated with Primary Percutaneous Coronary Intervention. Cardiorenal Med. 2020, 10, 154–161. [Google Scholar] [CrossRef]
- Moresco, R.N.; Bochi, G.V.; Stein, C.S.; de Carvalho, J.A.M.; Cembranel, B.M.; Bollick, Y.S. Urinary Kidney Injury Molecule-1 in Renal Disease. Clin. Chim. Acta 2018, 487, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Maydan, O.; McDade, P.G.; Liu, Y.; Wu, X.-R.; Matsell, D.; Eddy, A.A. Uromodulin Deficiency Alters Tubular Injury and Interstitial Inflammation But Not Fibrosis in Experimental Obstructive Nephropathy. Physiol. Rep. 2018, 6, e13654. [Google Scholar] [CrossRef]
- Nogare, A.L.; Veronese, F.V.; Carpio, V.N.; Montenegro, R.M.; Pedroso, J.A.; Pegas, K.L.; Gonçalves, L.F.; Manfro, R.C. Kidney Injury Molecule-1 Expression in Human Kidney Transplants with Interstitial Fibrosis and Tubular Atrophy. BMC Nephrol. 2015, 16, 19. [Google Scholar] [CrossRef]
- Humphreys, B.D.; Xu, F.; Sabbisetti, V.; Grgic, I.; Naini, S.M.; Wang, N.; Chen, D.; Xiao, S.; Patel, D.; Henderson, J.M.; et al. Chronic Epithelial Kidney Injury Molecule-1 Expression Causes Murine Kidney Fibrosis. J. Clin. Investig. 2013, 123, 4023–4035. [Google Scholar] [CrossRef]
- Edelstein, C.L. Biomarkers of Acute Kidney Injury. Adv. Chronic Kidney Dis. 2008, 15, 222–234. [Google Scholar] [CrossRef]
- Parikh, C.R.; Abraham, E.; Ancukiewicz, M.; Edelstein, C.L. Urine IL-18 Is an Early Diagnostic Marker for Acute Kidney Injury and Predicts Mortality in the Intensive Care Unit. J. Am. Soc. Nephrol. 2005, 16, 3046–3052. [Google Scholar] [CrossRef]
- Szabó, Z.; Magga, J.; Alakoski, T.; Ulvila, J.; Piuhola, J.; Vainio, L.; Kivirikko, K.I.; Vuolteenaho, O.; Ruskoaho, H.; Lipson, K.; et al. Connective Tissue Growth Factor Inhibition Attenuates Left Ventricular Remodeling and Dysfunction in Pressure Overload–Induced Heart Failure. Hypertension 2014, 63, 1235–1240. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, C.; Hong, X.; Miao, J.; Liao, Y.; Hou, F.F.; Zhou, L.; Liu, Y. Wnt/β-Catenin Signaling Mediates Both Heart and Kidney Injury in Type 2 Cardiorenal Syndrome. Kidney Int. 2019, 95, 815–829. [Google Scholar] [CrossRef]
- Lekawanvijit, S.; Kompa, A.R.; Zhang, Y.; Wang, B.H.; Kelly, D.J.; Krum, H. Myocardial Infarction Impairs Renal Function, Induces Renal Interstitial Fibrosis, and Increases Renal KIM-1 Expression: Implications for Cardiorenal Syndrome. Am. J. Physiol. Circ. Physiol. 2012, 302, H1884–H1893. [Google Scholar] [CrossRef]
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis—A Common Pathway to Organ Injury and Failure. N. Engl. J. Med. 2015, 372, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.T.; Brilla, C.G. Factors Associated with Reactive and Reparative Fibrosis of the Myocardium. Cell. Mol. Alter. Fail. Human Heart 1992, 87 (Suppl. S1), 291–301. [Google Scholar] [CrossRef]
- Baum, J.; Duffy, H.S. Fibroblasts and Myofibroblasts: What Are We Talking About? J. Cardiovasc. Pharmacol. 2011, 57, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Mack, M.; Yanagita, M. Origin of Myofibroblasts and Cellular Events Triggering Fibrosis. Kidney Int. 2015, 87, 297–307. [Google Scholar] [CrossRef]
- Györfi, A.-H.; Matei, A.-E.; Distler, J.H. Targeting TGF-β Signaling for the Treatment of Fibrosis. Matrix Biol. 2018, 68–69, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Valiente-Alandi, I.; Potter, S.J.; Salvador, A.M.; Schafer, A.E.; Schips, T.; Carrillo-Salinas, F.J.; Gibson, A.M.; Nieman, M.L.; Perkins, C.; Sargent, M.A.; et al. Inhibiting Fibronectin Attenuates Fibrosis and Improves Cardiac Function in a Model of Heart Failure. Circulation 2018, 138, 1236–1252. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Ma, T.; Lian, X.; Gao, J.; Wang, W.; Weng, W.; Lu, X.; Sun, W.; Cheng, Y.; Fu, Y.; et al. Clopidogrel Reduces Fibronectin Accumulation and Improves Diabetes-Induced Renal Fibrosis. Int. J. Biol. Sci. 2019, 15, 239–252. [Google Scholar] [CrossRef]
- Karsdal, M.A.; Nielsen, S.H.; Leeming, D.J.; Langholm, L.L.; Nielsen, M.J.; Manon-Jensen, T.; Siebuhr, A.; Gudmann, N.S.; Ronnow, S.; Sand, J.M.; et al. The good and the bad collagens of fibrosis—Their role in signaling and organ function. Adv. Drug Deliv. Rev. 2017, 121, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Pathophysiology of Myocardial Infarction. Compr. Physiol. 2015, 5, 1841–1875. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Cardiac Fibrosis: Cell Biological Mechanisms, Molecular Pathways and Therapeutic Opportunities. Mol. Asp. Med. 2019, 65, 70–99. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. The Extracellular Matrix in Ischemic and Nonischemic Heart Failure. Circ. Res. 2019, 125, 117–146. [Google Scholar] [CrossRef]
- Ytrehus, K.; Hulot, J.-S.; Perrino, C.; Schiattarella, G.; Madonna, R. Perivascular Fibrosis and the Microvasculature of the Heart. Still Hidden Secrets of Pathophysiology? Vasc. Pharmacol. 2018, 107, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Talman, V.; Ruskoaho, H. Cardiac fibrosis in myocardial infarction—from repair and remodeling to regeneration. Cell Tissue Res. 2016, 365, 563–581. [Google Scholar] [CrossRef]
- Shinde, A.V.; Frangogiannis, N.G. Fibroblasts in Myocardial Infarction: A Role in Inflammation and Repair. J. Mol. Cell. Cardiol. 2014, 70, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Schilter, H.; Findlay, A.D.; Perryman, L.; Yow, T.T.; Moses, J.; Zahoor, A.; Turner, C.I.; Deodhar, M.; Foot, J.S.; Zhou, W.; et al. The Lysyl Oxidase Like 2/3 Enzymatic Inhibitor, PXS-5153A, Reduces Crosslinks and Ameliorates Fibrosis. J. Cell. Mol. Med. 2018, 23, 1759–1770. [Google Scholar] [CrossRef]
- Doi, M.; Kusachi, S.; Murakami, T.; Ninomiya, Y.; Murakami, M.; Nakahama, M.; Takeda, K.; Komatsubara, I.; Naito, I.; Tsuji, T. Time-Dependent Changes of Decorin in the Infarct Zone After Experimentally Induced Myocardial Infarction in Rats: Comparison with Biglycan. Pathol. Res. Pract. 2000, 196, 23–33. [Google Scholar] [CrossRef]
- Li, L.; Okada, H.; Takemura, G.; Kosai, K.-I.; Kanamori, H.; Esaki, M.; Takahashi, T.; Goto, K.; Tsujimoto, A.; Maruyama, R.; et al. Postinfarction Gene Therapy with Adenoviral Vector Expressing Decorin Mitigates Cardiac Remodeling and Dysfunction. Am. J. Physiol. Circ. Physiol. 2009, 297, H1504–H1513. [Google Scholar] [CrossRef]
- Nakahama, M.; Murakami, T.; Kusachi, S.; Naito, I.; Takeda, K.; Ohnishi, H.; Komatsubara, I.; Oka, T.; Ninomiya, Y.; Tsuji, T. Expression of Perlecan Proteoglycan in the Infarct Zone of Mouse Myocardial Infarction. J. Mol. Cell. Cardiol. 2000, 32, 1087–1100. [Google Scholar] [CrossRef] [PubMed]
- Sasse, P.; Malan, D.; Fleischmann, M.; Roell, W.; Gustafsson, E.; Bostani, T.; Fan, Y.; Kolbe, T.; Breitbach, M.; Addicks, K.; et al. Perlecan Is Critical for Heart Stability. Cardiovasc. Res. 2008, 80, 435–444. [Google Scholar] [CrossRef]
- Fu, X.; Khalil, H.; Kanisicak, O.; Boyer, J.G.; Vagnozzi, R.J.; Maliken, B.D.; Sargent, M.A.; Prasad, V.; Valiente-Alandi, I.; Blaxall, B.C.; et al. Specialized Fibroblast Differentiated States Underlie Scar Formation in the Infarcted Mouse Heart. J. Clin. Investig. 2018, 128, 2127–2143. [Google Scholar] [CrossRef]
- Woodiwiss, A.J.; Tsotetsi, O.J.; Sprott, S.; Lancaster, E.J.; Mela, T.; Chung, E.S.; Meyer, T.E.; Norton, G. Reduction in Myocardial Collagen Cross-Linking Parallels Left Ventricular Dilatation in Rat Models of Systolic Chamber Dysfunction. Circulation 2001, 103, 155–160. [Google Scholar] [CrossRef]
- Santamaria, J.G.; Villalba, M.; Busnadiego, O.; López-Olañeta, M.M.; Sandoval, P.; Snabel, J.; López-Cabrera, M.; Erler, J.; Hanemaaijer, R.; Lara-Pezzi, E.; et al. Matrix Cross-Linking Lysyl Oxidases Are Induced in Response to Myocardial Infarction and Promote Cardiac Dysfunction. Cardiovasc. Res. 2015, 109, 67–78. [Google Scholar] [CrossRef]
- Siebermair, J.; Suksaranjit, P.; McGann, C.J.; Peterson, K.A.; Kheirkhahan, M.; Baher, A.A.; Damal, K.; Wakili, R.; Marrouche, N.F.; Wilson, B.D. Atrial Fibrosis in non–atrial Fibrillation Individuals and Prediction of Atrial Fibrillation by Use of Late Gadolinium Enhancement Magnetic Resonance Imaging. J. Cardiovasc. Electrophysiol. 2019, 30, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.C.; Koutsogeorgis, I.D.; Grapsa, J.; Papadopoulos, C.; Katsivas, A.; Nihoyannopoulos, P. Left Atrium and the Imaging of Atrial Fibrosis: Catch It If You Can! Eur. J. Clin. Investig. 2014, 44, 872–881. [Google Scholar] [CrossRef]
- Yang, J.; Savvatis, K.; Kang, J.S.; Fan, P.; Zhong, H.; Schwartz, K.; Barry, V.; Mikels-Vigdal, A.; Karpinski, S.; Kornyeyev, D.; et al. Targeting LOXL2 for Cardiac Interstitial Fibrosis and Heart Failure Treatment. Nat. Commun. 2016, 7, 13710. [Google Scholar] [CrossRef] [PubMed]
- De Gaspari, M.; Toscano, G.; Bagozzi, L.; Metra, M.; Lombardi, C.; Rizzo, S.; Angelini, A.; Marra, M.P.; Gerosa, G.; Basso, C. Endomyocardial Fibrosis and Myocardial Infarction Leading to Diastolic and Systolic Dysfunction Requiring Transplantation. Cardiovasc. Pathol. Off. J. Soc. Cardiovasc. Pathol. 2019, 38, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Nucifora, G.; Muser, D.; Gianfagna, P.; Morocutti, G.; Proclemer, A. Systolic and Diastolic Myocardial Mechanics in Hypertrophic Cardiomyopathy and Their Link to the Extent of Hypertrophy, Replacement Fibrosis and Interstitial Fibrosis. Int. J. Cardiovasc. Imaging 2015, 31, 1603–1610. [Google Scholar] [CrossRef] [PubMed]
- Shinde, A.V.; Su, Y.; Palanski, B.A.; Fujikura, K.; Garcia, M.J.; Frangogiannis, N.G. Pharmacologic Inhibition of the Enzymatic Effects of Tissue Transglutaminase Reduces Cardiac Fibrosis and Attenuates Cardiomyocyte Hypertrophy Following Pressure Overload. J. Mol. Cell. Cardiol. 2018, 117, 36–48. [Google Scholar] [CrossRef] [PubMed]
- A Beltrami, C.; Finato, N.; Rocco, M.; A Feruglio, G.; Puricelli, C.; Cigola, E.; Quaini, F.; Sonnenblick, E.H.; Olivetti, G.; Anversa, P. Structural Basis of End-Stage Failure in Ischemic Cardiomyopathy in Humans. Circulation 1994, 89, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.N.; Ayyadurai, P.; Saad, M.; E Kosmas, C.; Dogar, M.U.; Patel, U.; Vittorio, T.J. Galectin-3 and Soluble ST2 As Complementary Tools to Cardiac MRI for Sudden Cardiac Death Risk Stratification in Heart Failure: A Review. JRSM Cardiovasc. Dis. 2020, 9. [Google Scholar] [CrossRef]
- Kim, E.K.; Chattranukulchai, P.; Klem, I. Cardiac Magnetic Resonance Scar Imaging for Sudden Cardiac Death Risk Stratification in Patients with Non-Ischemic Cardiomyopathy. Korean J. Radiol. 2015, 16, 683–695. [Google Scholar] [CrossRef]
- Kato, S.; Saito, N.; Kirigaya, H.; Gyotoku, D.; Iinuma, N.; Kusakawa, Y.; Iguchi, K.; Nakachi, T.; Fukui, K.; Futaki, M.; et al. Prognostic Significance of Quantitative Assessment of Focal Myocardial Fibrosis in Patients with Heart Failure with Preserved Ejection Fraction. Int. J. Cardiol. 2015, 191, 314–319. [Google Scholar] [CrossRef]
- King, J.B.; Azadani, P.N.; Suksaranjit, P.; Bress, A.P.; Witt, D.M.; Han, F.T.; Chelu, M.; Silver, M.A.; Biskupiak, J.; Wilson, B.D.; et al. Left Atrial Fibrosis and Risk of Cerebrovascular and Cardiovascular Events in Patients with Atrial Fibrillation. J. Am. Coll. Cardiol. 2017, 70, 1311–1321. [Google Scholar] [CrossRef] [PubMed]
- Peddakkulappagari, C.S.; Saifi, M.A.; Khurana, A.; Anchi, P.; Singh, M.; Godugu, C. Withaferin A Ameliorates Renal Injury Due to Its Potent Effect on Inflammatory Signaling. BioFactors 2019, 45, 750–762. [Google Scholar] [CrossRef]
- Tammaro, A.; Florquin, S.; Brok, M.; Claessen, N.; Butter, L.M.; Teske, G.J.D.; de Boer, O.; Vogl, T.; Leemans, J.C.; Dessing, M.C. S100A8/A9 Promotes Parenchymal Damage and Renal Fibrosis in Obstructive Nephropathy. Clin. Exp. Immunol. 2018, 193, 361–375. [Google Scholar] [CrossRef]
- Grande, M.T.; Sanchez-Laorden, B.; López-Blau, C.; De Frutos, C.A.; Boutet, A.; Arévalo, M.; Rowe, R.G.; Weiss, S.J.; López-Novoa, J.M.; Nieto, M.A. Snail1-Induced Partial Epithelial-to-Mesenchymal Transition Drives Renal Fibrosis in Mice and Can Be Targeted to Reverse Established Disease. Nat. Med. 2015, 21, 989–997. [Google Scholar] [CrossRef]
- Xie, K.; Bao, L.; Jiang, X.; Ye, Z.; Bing, J.; Dong, Y.; Gao, D.; Ji, X.; Jiang, T.; Li, J.; et al. The Association of Metabolic Syndrome Components and Chronic Kidney Disease in Patients with Hypertension. Lipids Health Dis. 2019, 18, 1–6. [Google Scholar] [CrossRef]
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K. Diabetic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2017, 12, 2032–2045. [Google Scholar] [CrossRef]
- Mordi, I.; Mordi, N.; Delles, C.; Tzemos, N. Endothelial Dysfunction in Human Essential Hypertension. J. Hypertens. 2016, 34, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Vanhoutte, P.M. Macro- and Microvascular Endothelial Dysfunction in Diabetes. J. Diabetes 2017, 9, 434–449. [Google Scholar] [CrossRef]
- Moriya, T.; Yamagishi, T.; Matsubara, M.; Ouchi, M. Serial Renal Biopsies in Normo- and Microalbuminuric Patients with Type 2 Diabetes Demonstrate That Loss of Renal Function Is Associated with a Reduction in Glomerular Filtration Surface Secondary to Mesangial Expansion. J. Diabetes Complicat. 2019, 33, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Heintz, B.; Stöcker, G.; Mrowka, C.; Rentz, U.; Melzer, H.; Stickeler, E.; Sieberth, H.-G.; Greiling, H.; Haubeck, H.-D. Decreased Glomerular Basement Membrane Heparan Sulfate Proteoglycan in Essential Hypertension. Hypertension 1995, 25, 399–407. [Google Scholar] [CrossRef]
- Salem, R.M.; Todd, J.N.; Sandholm, N.; Cole, J.B.; Chen, W.-M.; Andrews, D.; Pezzolesi, M.G.; McKeigue, P.M.; Hiraki, L.T.; Qiu, C.; et al. Genome-Wide Association Study of Diabetic Kidney Disease Highlights Biology Involved in Glomerular Basement Membrane Collagen. J. Am. Soc. Nephrol. 2019, 30, 2000–2016. [Google Scholar] [CrossRef] [PubMed]
- Tung, C.-W.; Hsu, Y.-C.; Shih, Y.-H.; Chang, P.-J.; Lin, C.-L. Glomerular Mesangial Cell and Podocyte Injuries in Diabetic Nephropathy. Nephrology 2018, 23, 32–37. [Google Scholar] [CrossRef]
- Puelles, V.G.; Cullen-McEwen, L.; Taylor, G.E.; Li, J.; Hughson, M.D.; Kerr, P.G.; Hoy, W.E.; Bertram, J. Human Podocyte Depletion in Association with Older Age and Hypertension. Am. J. Physiol. Physiol. 2016, 310, F656–F668. [Google Scholar] [CrossRef]
- Liu, Y. Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 2011, 7, 684–696. [Google Scholar] [CrossRef]
- Conlin, C.C.; Huang, Y.; Gordon, B.A.J.; Zhang, J.L. Quantitative Characterization of Glomerular Fibrosis with Magnetic Resonance Imaging: A Feasibility Study in a Rat Glomerulonephritis Model. Am. J. Physiol. Physiol. 2018, 314, F747–F752. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Lou, K.; Tatum, K.; Funk, J.; Wu, J.; Bartkowiak, T.; Kagan, D.; Lou, Y. Differentiating Glomerular Inflammation from Fibrosis in a Bone Marrow Chimera for Rat Anti-Glomerular Basement Membrane Glomerulonephritis. Am. J. Nephrol. 2015, 42, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Genovese, F.; A Manresa, A.; Leeming, D.J.; Karsdal, M.A.; Boor, P. The Extracellular Matrix in the Kidney: A Source of Novel Non-Invasive Biomarkers of Kidney Fibrosis? Fibrogenesis Tissue Repair 2014, 7, 4. [Google Scholar] [CrossRef]
- Bohle, A.; Mackensen-Haen, S.; Gise, H.V. Significance of Tubulointerstitial Changes in the Renal Cortex for the Excretory Function and Concentration Ability of the Kidney: A Morphometric Contribution. Am. J. Nephrol. 1987, 7, 421–433. [Google Scholar] [CrossRef]
- Tervaert, T.W.C.; Mooyaart, A.; Amann, K.; Cohen, A.H.; Cook, H.T.; Drachenberg, C.B.; Ferrario, F.; Fogo, A.B.; Haas, M.; De Heer, E.; et al. Pathologic Classification of Diabetic Nephropathy. J. Am. Soc. Nephrol. 2010, 21, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Boor, P.; Perkuhn, M.; Ms, M.W.; Zok, S.; Martin, W.; Ms, J.G.; Schoth, F.; Ostendorf, T.; Kuhl, C.; Floege, J. Diffusion-Weighted MRI Does Not Reflect Kidney Fibrosis in a Rat Model of Fibrosis. J. Magn. Reson. Imaging 2015, 42, 990–998. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, Z.; Liu, M.; Zhu, J.; Zhang, X.; Zhang, T.; Li, S.; Li, Y. Assessment of Renal Fibrosis in Chronic Kidney Disease Using Diffusion-Weighted MRI. Clin. Radiol. 2014, 69, 1117–1122. [Google Scholar] [CrossRef]
- Eadon, M.T.; Schwantes-An, T.-H.; Phillips, C.L.; Roberts, A.R.; Greene, C.V.; Hallab, A.; Hart, K.J.; Lipp, S.N.; Perez-Ledezma, C.; Omar, K.O.; et al. Kidney Histopathology and Prediction of Kidney Failure: A Retrospective Cohort Study. Am. J. Kidney Dis. 2020, 76, 350–360. [Google Scholar] [CrossRef]
- Belghasem, M.E.; A’Amar, O.; Roth, D.; Walker, J.; Arinze, N.; Richards, S.M.; Francis, J.M.; Salant, D.J.; Chitalia, V.C.; Bigio, I.J. Towards Minimally-Invasive, Quantitative Assessment of Chronic Kidney Disease Using Optical Spectroscopy. Sci. Rep. 2019, 9, 7168. [Google Scholar] [CrossRef] [PubMed]
- Nath, K.A. Tubulointerstitial Changes As a Major Determinant in the Progression of Renal Damage. Am. J. Kidney Dis. 1992, 20, 1–17. [Google Scholar] [CrossRef]
- Howie, A.J.; Ferreira, M.A.S.; Adu, D. Prognostic Value of Simple Measurement of Chronic Damage in Renal Biopsy Specimens. Nephrol. Dial. Transplant. 2001, 16, 1163–1169. [Google Scholar] [CrossRef]
- Kaissling, B.; LeHir, M.; Kriz, W. Renal Epithelial Injury and Fibrosis. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 931–939. [Google Scholar] [CrossRef]
- Rosenberg, A.; Kopp, J. Focal Segmental Glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2017, 12, 502–517. [Google Scholar] [CrossRef]
- Palatini, P. Glomerular Hyperfiltration: A Marker of Early Renal Damage in Pre-Diabetes and Pre-Hypertension. Nephrol. Dial. Transplant. 2012, 27, 1708–1714. [Google Scholar] [CrossRef] [PubMed]
- Kriz, W.; Lehir, M. Pathways to Nephron Loss Starting from Glomerular diseases—Insights from Animal Models. Kidney Int. 2005, 67, 404–419. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Li, Y.; Yue, S.; Liu, X.; Wang, L.; Xiong, M.; Wang, G.; Nie, S.; Xu, X. The Profiles of Biopsy-Proven Renal Tubulointerstitial Lesions in Patients with Glomerular Disease. Ann. Transl. Med. 2020, 8, 1066. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.M.K.; Nikolic-Paterson, D.J.; Lan, H.-Y. Macrophages: Versatile Players in Renal Inflammation and Fibrosis. Nat. Rev. Nephrol. 2019, 15, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.-H. Mesangial Cells and Renal Fibrosis. Adv. Exp. Med. Biol. 2019, 1165, 165–194. [Google Scholar] [CrossRef]
- Bülow, R.D.; Boor, P. Extracellular Matrix in Kidney Fibrosis: More Than Just a Scaffold. J. Histochem. Cytochem. 2019, 67, 643–661. [Google Scholar] [CrossRef]
- Müller, G.A.; Rodemann, H.P. Characterization of Human Renal Fibroblasts in Health and Disease: I. Immunophenotyping of Cultured Tubular Epithelial Cells and Fibroblasts Derived from Kidneys with Histologically Proven Interstitial Fibrosis. Am. J. Kidney Dis. 1991, 17, 680–683. [Google Scholar] [CrossRef]
- Rodemann, H.P.; Müller, G.A. Characterization of Human Renal Fibroblasts in Health and Disease: II. In Vitro Growth, Differentiation, and Collagen Synthesis of Fibroblasts from Kidneys with Interstitial Fibrosis. Am. J. Kidney Dis. 1991, 17, 684–686. [Google Scholar] [CrossRef]
- Koesters, R.; Kaissling, B.; LeHir, M.; Picard, N.; Theilig, F.; Gebhardt, R.; Glick, A.B.; Hähnel, B.; Hosser, H.; Gröne, H.-J.; et al. Tubular Overexpression of Transforming Growth Factor-β1 Induces Autophagy and Fibrosis But Not Mesenchymal Transition of Renal Epithelial Cells. Am. J. Pathol. 2010, 177, 632–643. [Google Scholar] [CrossRef] [PubMed]
- Kriz, W.; Hähnel, B.; Hosser, H.; Ostendorf, T.; Gaertner, S.; Kränzlin, B.; Gretz, N.; Shimizu, F.; Floege, J. Pathways to Recovery and Loss of Nephrons in Anti-Thy-1 Nephritis. J. Am. Soc. Nephrol. 2003, 14, 1904–1926. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Pollock, A.S.; Mahimkar, R.; Olson, J.L.; Lovett, D.H. Matrix Metalloproteinase 2 and Basement Membrane Integrity: A Unifying Mechanism for Progressive Renal Injury. FASEB J. 2006, 20, 1898–1900. [Google Scholar] [CrossRef] [PubMed]
- Nadasdy, T.; Laszik, Z.; E Blick, K.; Johnson, L.D.; Silva, F.G. Proliferative Activity of Intrinsic Cell Populations in the Normal Human Kidney. J. Am. Soc. Nephrol. 1994, 4, 2032–2039. [Google Scholar] [CrossRef]
- Humphreys, B.D.; Valerius, M.T.; Kobayashi, A.; Mugford, J.W.; Soeung, S.; Duffield, J.S.; McMahon, A.P.; Bonventre, J.V. Intrinsic Epithelial Cells Repair the Kidney After Injury. Cell Stem Cell 2008, 2, 284–291. [Google Scholar] [CrossRef]
- Yang, L.; Besschetnova, T.Y.; Brooks, C.R.; Shah, J.V.; Bonventre, J.V. Epithelial Cell Cycle Arrest in G2/M Mediates Kidney Fibrosis After Injury. Nat. Med. 2010, 16, 535–543. [Google Scholar] [CrossRef]
- Grgic, I.; Campanholle, G.; Bijol, V.; Wang, C.; Sabbisetti, V.S.; Ichimura, T.; Humphreys, B.D.; Bonventre, J.V. Targeted Proximal Tubule Injury Triggers Interstitial Fibrosis and Glomerulosclerosis. Kidney Int. 2012, 82, 172–183. [Google Scholar] [CrossRef]
- Xu, L.; Sharkey, D.; Cantley, L.G. Tubular GM-CSF Promotes Late MCP-1/CCR2-Mediated Fibrosis and Inflammation After Ischemia/Reperfusion Injury. J. Am. Soc. Nephrol. 2019, 30, 1825–1840. [Google Scholar] [CrossRef]
- Arfian, N.; Wahyudi, D.A.P.; Zulfatina, I.B.; Citta, A.N.; Anggorowati, N.; Multazam, A.; Romi, M.M.; Sari, D.C.R. Chlorogenic Acid Attenuates Kidney Ischemic/Reperfusion Injury via Reducing Inflammation, Tubular Injury, and Myofibroblast Formation. BioMed Res. Int. 2019, 2019, 1–10. [Google Scholar] [CrossRef]
- Jing, W.; Vaziri, N.D.; Nunes, A.C.F.; Suematsu, Y.; Farzaneh, T.; Khazaeli, M.; Moradi, H. LCZ696 (Sacubitril/Valsartan) Ameliorates Oxidative Stress, Inflammation, Fibrosis and Improves Renal Function Beyond Angiotensin Receptor Blockade in CKD. Am. J. Transl. Res. 2017, 9, 5473–5484. [Google Scholar]
- Fortrie, G.; De Geus, H.R.H.; Betjes, M.G.H. The Aftermath of Acute Kidney Injury: A Narrative Review of Long-Term Mortality and Renal Function. Crit. Care 2019, 23, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Stroo, I.; Stokman, G.; Teske, G.J.D.; Raven, A.; Butter, L.M.; Florquin, S.; Leemans, J.C. Chemokine Expression in Renal ischemia/Reperfusion Injury Is Most Profound During the Reparative Phase. Int. Immunol. 2010, 22, 433–442. [Google Scholar] [CrossRef]
- Ali, B.H.; Al-Salam, S.; Al Suleimani, Y.; Al Kalbani, J.; Al Bahlani, S.; Ashique, M.; Manoj, P.; Al Dhahli, B.; Al Abri, N.; Naser, H.T.; et al. Curcumin Ameliorates Kidney Function and Oxidative Stress in Experimental Chronic Kidney Disease. Basic Clin. Pharmacol. Toxicol. 2018, 122, 65–73. [Google Scholar] [CrossRef]
- Fabre, T.; Kared, H.; Friedman, S.L.; Shoukry, N.H. IL-17A Enhances the Expression of Profibrotic Genes through Upregulation of the TGF-β Receptor on Hepatic Stellate Cells in a JNK-Dependent Manner. J. Immunol. 2014, 193, 3925–3933. [Google Scholar] [CrossRef]
- Mack, M. Inflammation and Fibrosis. Matrix Biol. 2018, 68-69, 106–121. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGFβ Signalling in Context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Finnson, K.; Almadani, Y.; Philip, A. Non-Canonical (non-SMAD2/3) TGF-β Signaling in Fibrosis: Mechanisms and Targets. Semin. Cell Dev. Biol. 2020, 101, 115–122. [Google Scholar] [CrossRef]
- Sun, K.-H.; Chang, Y.; Reed, N.I.; Sheppard, D. α-Smooth Muscle Actin Is an Inconsistent Marker of Fibroblasts Responsible for Force-Dependent TGFβ Activation or Collagen Production across Multiple Models of Organ Fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 2016, 310, L824–L836. [Google Scholar] [CrossRef] [PubMed]
- Walker, E.J.; Heydet, D.; Veldre, T.; Ghildyal, R. Transcriptomic Changes During TGF-β-Mediated Differentiation of Airway Fibroblasts to Myofibroblasts. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef]
- Chen, W.; Dijke, P.T. Immunoregulation by Members of the TGFβ Superfamily. Nat. Rev. Immunol. 2016, 16, 723–740. [Google Scholar] [CrossRef] [PubMed]
- Gravning, J.; Ørn, S.; Kaasbøll, O.J.; Martinov, V.N.; Manhenke, C.; Dickstein, K.; Edvardsen, T.; Attramadal, H.; Ahmed, M.S. Myocardial Connective Tissue Growth Factor (CCN2/CTGF) Attenuates Left Ventricular Remodeling After Myocardial Infarction. PLoS ONE 2012, 7, e52120. [Google Scholar] [CrossRef]
- Mao, L.; Liu, L.; Zhang, T.; Wu, X.; Zhang, T.; Xu, Y. MKL1 Mediates TGF-β-induced CTGF Transcription to Promote Renal Fibrosis. J. Cell. Physiol. 2020, 235, 4790–4803. [Google Scholar] [CrossRef]
- Mori, T.; Kawara, S.; Shinozaki, M.; Hayashi, N.; Kakinuma, T.; Igarashi, A.; Takigawa, M.; Nakanishi, T.; Takehara, K. Role and in-Teraction of Connective Tissue Growth Factor with Transforming Growth Factor-Beta in Persistent Fibrosis: A Mouse Fibrosis Model. J. Cell Physiol. 1999, 181, 153–159. [Google Scholar] [CrossRef]
- (96) Frazier, K.; Williams, S.; Kothapalli, D.; Klapper, H.; Grotendorst, G.R. Stimulation of Fibroblast Cell Growth, Matrix Production, and Granulation Tissue Formation by Connective Tissue Growth Factor. J. Investig. Dermatol. 1996, 107, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, A.; Nashiro, K.; Kikuchi, K.; Sato, S.; Ihn, H.; Fujimoto, M.; Grotendorst, G.R.; Takehara, K. Connective Tissue Growth Factor Gene Expression in Tissue Sections from Localized Scleroderma, Keloid, and Other Fibrotic Skin Disorders. J. Investig. Dermatol. 1996, 106, 729–733. [Google Scholar] [CrossRef]
- Inanc, S.; Keleş, D.; Oktay, G. An Improved Collagen Zymography Approach for Evaluating the Collagenases MMP-1, MMP-8, and MMP-13. Biotechnology 2017, 63, 174–180. [Google Scholar] [CrossRef]
- Falconer, A.M.D.; Chan, C.M.; Gray, J.; Nagashima, I.; Holland, R.A.; Shimizu, H.; Pickford, A.R.; Rowan, A.D.; Wilkinson, D.J. Collagenolytic Matrix Metalloproteinases Antagonize Proteinase-Activated Receptor-2 Activation, Providing Insights into Extracellular Matrix Turnover. J. Biol. Chem. 2019, 294, 10266–10277. [Google Scholar] [CrossRef]
- Toth, M.; Sohail, A.; Fridman, R. Assessment of Gelatinases (MMP-2 and MMP-9) by Gelatin Zymography. In Metastasis Research Protocols; Humana Press: Totowa, NJ, USA, 2012; pp. 163–174. [Google Scholar]
- Vafashoar, F.; Mousavizadeh, K.; Poormoghim, H.; Tavasoli, A.; Shabestari, T.M.; JavadMoosavi, S.A.; Mojtabavi, N. Gelatinases Increase in Bleomycin-Induced Systemic Sclerosis Mouse Model. Iran. J. Allergy Asthma Immunol. 2019, 18, 182–189. [Google Scholar] [CrossRef]
- Butler, G.S.; Connor, A.R.; Sounni, N.E.; Eckhard, U.; Morrison, C.J.; Noel, A.; Overall, C.M. Degradomic and Yeast 2-Hybrid Inactive Catalytic Domain Substrate Trapping Identifies New Membrane-Type 1 Matrix Metalloproteinase (MMP14) Substrates: CCN3 (Nov) and CCN5 (WISP2). Matrix Biol. 2017, 59, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Mirastschijski, U.; Dinesh, N.; Baskaran, S.; Wedekind, D.; Gavrilovic, J.; Murray, M.Y.; Bevan, D.; Kelm, S. Novel Specific Human and Mouse stromelysin-1 (MMP-3) and stromelysin-2 (MMP-10) Antibodies for Biochemical and Immunohistochemical Analyses. Wound Repair Regen. 2019, 27, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Giannandrea, M.; Parks, W.C. Diverse Functions of Matrix Metalloproteinases During Fibrosis. Dis. Model. Mech. 2014, 7, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Gao, M.; Li, J.; Sun, J.; Wu, R.; Han, D.; Tan, J.; Wang, J.; Wang, B.; Zhang, L.; et al. MMP-9-positive Neutrophils Are Essential for Establishing Profibrotic Microenvironment in the Obstructed Kidney of UUO Mice. Acta Physiol. 2019, 227, e13317. [Google Scholar] [CrossRef]
- Zhao, Y.; Qiao, X.; Tan, T.K.; Zhao, H.; Zhang, Y.; Liu, L.; Zhang, J.; Wang, L.; Cao, Q.; Wang, Y.; et al. Matrix Metalloproteinase 9-Dependent Notch Signaling Contributes to Kidney Fibrosis through Peritubular Endothelial-Mesenchymal Transition. Nephrol. Dial. Transplant. 2016, 32, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Chiao, Y.A.; Ramirez, T.A.; Zamilpa, R.; Okoronkwo, S.M.; Dai, Q.; Zhang, J.; Jin, Y.-F.; Lindsey, M.L. Matrix Metalloproteinase-9 Deletion Attenuates Myocardial Fibrosis and Diastolic Dysfunction in Ageing Mice. Cardiovasc. Res. 2012, 96, 444–455. [Google Scholar] [CrossRef]
- Altieri, P.; Brunelli, C.; Garibaldi, S.; Nicolino, A.; Ubaldi, S.; Spallarossa, P.; Olivotti, L.; Rossettin, P.; Barsotti, A.; Ghigliotti, G. Metalloproteinases 2 and 9 Are Increased in Plasma of Patients with Heart Failure. Eur. J. Clin. Investig. 2003, 33, 648–656. [Google Scholar] [CrossRef]
- Takamiya, Y.; Fukami, K.; Yamagishi, S.-I.; Kaida, Y.; Nakayama, Y.; Obara, N.; Iwatani, R.; Ando, R.; Koike, K.; Matsui, T.; et al. Experimental Diabetic Nephropathy Is Accelerated in Matrix Metalloproteinase-2 Knockout Mice. Nephrol. Dial. Transplant. 2012, 28, 55–62. [Google Scholar] [CrossRef]
- Jao, T.-M.; Nangaku, M.; Wu, C.-H.; Sugahara, M.; Saito, H.; Maekawa, H.; Ishimoto, Y.; Aoe, M.; Inoue, T.; Tanaka, T.; et al. ATF6α Downregulation of PPARα Promotes Lipotoxicity-Induced Tubulointerstitial Fibrosis. Kidney Int. 2019, 95, 577–589. [Google Scholar] [CrossRef]
- Jiménez-González, S.; Marín-Royo, G.; Jurado-López, R.; Bartolomé, M.V.; Miranda, A.R.; Luaces, M.; Islas, F.; Nieto, M.L.; Martínez-Martínez, E.; Cachofeiro, V. The Crosstalk Between Cardiac Lipotoxicity and Mitochondrial Oxidative Stress in the Cardiac Alterations in Diet-Induced Obesity in Rats. Cells 2020, 9, 451. [Google Scholar] [CrossRef]
- Delgado-Valero, B.; de la Fuente-Chávez, L.; Romero-Miranda, A.; Bartolomé, M.V.; Ramchandani, B.; Islas, F.; Luaces, M.; Cachofeiro, V.; Martínez-Martínez, E. Role of Endoplasmic Reticulum Stress in Renal Damage After Myocardial Infarction. Clin. Sci. 2021, 135, 143–159. [Google Scholar] [CrossRef]
- Nogueira, A.; Pires, M.J.; Oliveira, P.A. Pathophysiological Mechanisms of Renal Fibrosis: A Review of Animal Models and Therapeutic Strategies. Vivo 2017, 31, 1–22. [Google Scholar] [CrossRef]
- Sun, H.-J. Current Opinion for Hypertension in Renal Fibrosis. Adv. Exp. Med. Biol. 2019, 1165, 37–47. [Google Scholar] [CrossRef]
- Rubinstein, J.; Sanford, D. Treatment of Cardiorenal Syndrome. Cardiol. Clin. 2019, 37, 267–273. [Google Scholar] [CrossRef]
- Wang, L.; Tian, X.; Cao, Y.; Ma, X.; Shang, L.; Li, H.; Zhang, X.; Deng, F.; Li, S.; Guo, T.; et al. Cardiac Shock Wave Therapy Improves Ventricular Function by Relieving Fibrosis Through PI3K/Akt Signaling Pathway: Evidence from a Rat Model of Post-Infarction Heart Failure. Front. Cardiovasc. Med. 2021, 8, 693875. [Google Scholar] [CrossRef] [PubMed]
- Zindel, J.; Kubes, P. DAMPs, PAMPs, and LAMPs in Immunity and Sterile Inflammation. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 493–518. [Google Scholar] [CrossRef] [PubMed]
- Abdulkhaleq, L.A.; Assi, M.A.; Abdullah, R.; Zamri-Saad, M.; Taufiq-Yap, Y.H.; Hezmee, M.N.M. The Crucial Roles of Inflammatory Mediators in Inflammation: A Review. Veter. World 2018, 11, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.; Kupper, T. Old Meets New: The Interaction Between Innate and Adaptive Immunity. J. Investig. Dermatol. 2005, 125, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic Inflammation in the Etiology of Disease across the Life Span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- Odegaard, A.O.; Goff, D.R., Jr.; Sanchez, O.A.; Goff, D.C.; Reiner, A.P.; Gross, M.D. Oxidative Stress, Inflammation, Endothelial Dysfunction and Incidence of Type 2 Diabetes. Cardiovasc. Diabetol. 2016, 15, 1–12. [Google Scholar] [CrossRef]
- Kanazawa, I.; Tanaka, S.; Sugimoto, T. The Association Between Osteocalcin and Chronic Inflammation in Patients with Type 2 Diabetes Mellitus. Calcif. Tissue Int. 2018, 103, 599–605. [Google Scholar] [CrossRef]
- Xiao, L.; Harrison, D.G. Inflammation in Hypertension. Can. J. Cardiol. 2020, 36, 635–647. [Google Scholar] [CrossRef]
- Rios, F.; Zou, Z.-G.; Harvey, A.P.; Harvey, K.Y.; Nosalski, R.; Anyfanti, P.; Camargo, L.L.; Lacchini, S.; Ryazanov, A.G.; Ryazanova, L.; et al. Chanzyme TRPM7 Protects Against Cardiovascular Inflammation and Fibrosis. Cardiovasc. Res. 2020, 116, 721–735. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Mihai, S.; Codrici, E.; Popescu, I.D.; Enciu, A.-M.; Albulescu, L.; Necula, L.G.; Mambet, C.; Anton, G.; Tanase, C. Inflammation-Related Mechanisms in Chronic Kidney Disease Prediction, Progression, and Outcome. J. Immunol. Res. 2018, 2018, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Torres, S.; Fabersani, E.; Marquez, A.; Gauffin-Cano, P. Adipose Tissue Inflammation and Metabolic Syndrome. The Proactive Role of Probiotics. Eur. J. Nutr. 2018, 58, 27–43. [Google Scholar] [CrossRef] [PubMed]
- Pierce, G.F.; A Mustoe, T.; Lingelbach, J.; Masakowski, V.R.; Griffin, G.L.; Senior, R.M.; Deuel, T.F. Platelet-Derived Growth Factor and Transforming Growth Factor-Beta Enhance Tissue Repair Activities by Unique Mechanisms. J. Cell Biol. 1989, 109, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Rossaint, J.; Margraf, A.; Zarbock, A. Role of Platelets in Leukocyte Recruitment and Resolution of Inflammation. Front. Immunol. 2018, 9, 2712. [Google Scholar] [CrossRef] [PubMed]
- Lichtman, M.K.; Otero-Vinas, M.; Falanga, V. Transforming Growth Factor Beta (TGF-β) Isoforms in Wound Healing and Fibrosis. Wound Repair Regen. 2015, 24, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Amdur, R.L.; Feldman, H.I.; Gupta, J.; Yang, W.; Kanetsky, P.; Shlipak, M.; Rahman, M.; Lash, J.P.; Townsend, R.R.; Ojo, A.; et al. Inflammation and Progression of CKD: The CRIC Study. Clin. J. Am. Soc. Nephrol. 2016, 11, 1546–1556. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, J.H.; Abraham, A.G.; Xu, Y.; Schelling, J.R.; Feldman, H.I.; Sabbisetti, V.S.; Gonzalez, M.C.; Coca, S.; Schrauben, S.J.; Waikar, S.S.; et al. Plasma Biomarkers of Tubular Injury and Inflammation Are Associated with CKD Progression in Children. J. Am. Soc. Nephrol. 2020, 31, 1067–1077. [Google Scholar] [CrossRef]
- Wen, Y.; Lu, X.; Ren, J.; Privratsky, J.R.; Yang, B.; Rudemiller, N.P.; Zhang, J.; Griffiths, R.; Jain, M.K.; Nedospasov, S.A.; et al. KLF4 in Macrophages Attenuates TNFα-Mediated Kidney Injury and Fibrosis. J. Am. Soc. Nephrol. 2019, 30, 1925–1938. [Google Scholar] [CrossRef]
- Brandt, S.; Ballhause, T.M.; Bernhardt, A.; Becker, A.; Salaru, D.; Le-Deffge, H.M.; Fehr, A.; Fu, Y.; Philipsen, L.; Djudjaj, S.; et al. Fibrosis and Immune Cell Infiltration Are Separate Events Regulated by Cell-Specific Receptor Notch3 Expression. J. Am. Soc. Nephrol. 2020, 31, 2589–2608. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-Y.; Jiang, H.; Pan, J.; Huang, X.-R.; Wang, Y.-C.; Huang, H.-F.; To, K.-F.; Nikolic-Paterson, D.J.; Lan, H.-Y.; Chen, J.-H. Macrophage-to-Myofibroblast Transition Contributes to Interstitial Fibrosis in Chronic Renal Allograft Injury. J. Am. Soc. Nephrol. 2017, 28, 2053–2067. [Google Scholar] [CrossRef] [PubMed]
- Kormann, R.; Kavvadas, P.; Placier, S.; Vandermeersch, S.; Dorison, A.; Dussaule, J.-C.; Chadjichristos, C.E.; Prakoura, N.; Chatziantoniou, C. Periostin Promotes Cell Proliferation and Macrophage Polarization to Drive Repair After AKI. J. Am. Soc. Nephrol. 2020, 31, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Simões, F.C.; Cahill, T.J.; Kenyon, A.; Gavriouchkina, D.; Vieira, J.M.; Sun, X.; Pezzolla, D.; Ravaud, C.; Masmanian, E.; Weinberger, M.; et al. Macrophages Directly Contribute Collagen to Scar Formation During Zebrafish Heart Regeneration and Mouse Heart Repair. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef]
- Shen, B.; Liu, X.; Fan, Y.; Qiu, J. Macrophages Regulate Renal Fibrosis Through Modulating TGFβ Superfamily Signaling. Inflammation 2014, 37, 2076–2084. [Google Scholar] [CrossRef] [PubMed]
- Wright, T.M. Cytokines in Acute and Chronic Inflammation. Front. Biosci. 1997, 2, d12–d26. [Google Scholar] [CrossRef] [PubMed]
- Panico, K.; Abrahão, M.V.; Sonoda, M.T.; Muzi-Filho, H.; Vieyra, A.; Carneiro-Ramos, M.S. Cardiac Inflammation After Ischemia-Reperfusion of the Kidney: Role of the Sympathetic Nervous System and the Renin-Angiotensin System. Cell. Physiol. Biochem. 2019, 53, 587–605. [Google Scholar] [CrossRef]
- Virzì, G.M.; Breglia, A.; Castellani, C.; Ankawi, G.; Bolin, C.; De Cal, M.; Cianci, V.; Angelini, A.; Vescovo, G.; Ronco, C. Lipopolysaccharide in Systemic Circulation Induces Activation of Inflammatory Response and Oxidative Stress in Cardiorenal Syndrome Type 1. J. Nephrol. 2019, 32, 803–810. [Google Scholar] [CrossRef]
- Virzì, G.M.; Breglia, A.; Brocca, A.; De Cal, M.; Bolin, C.; Vescovo, G.; Ronco, C. Levels of Proinflammatory Cytokines, Oxidative Stress, and Tissue Damage Markers in Patients with Acute Heart Failure with and without Cardiorenal Syndrome Type 1. Cardiorenal Med. 2018, 8, 321–331. [Google Scholar] [CrossRef]
- Colombo, P.C.; Ganda, A.; Lin, J.; Onat, D.; Harxhi, A.; Iyasere, J.E.; Uriel, N.; Cotter, G. Inflammatory Activation: Cardiac, Renal, and Cardio-Renal Interactions in Patients with the Cardiorenal Syndrome. Heart Fail. Rev. 2012, 17, 177–190. [Google Scholar] [CrossRef]
- Li, R.; Mi, X.; Yang, S.; Yang, Y.; Zhang, S.; Hui, R.; Chen, Y.; Zhang, W. Long-Term Stimulation of Angiotensin II Induced Endothelial Senescence and Dysfunction. Exp. Gerontol. 2019, 119, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Han, J.; Zhang, H.; Xu, J.; Jiang, L.; Ge, W. Kaempferol Prevents Against Ang II-Induced Cardiac Remodeling Through Attenuating Ang II-Induced Inflammation and Oxidative Stress. J. Cardiovasc. Pharmacol. 2019, 74, 326–335. [Google Scholar] [CrossRef]
- Lang, P.-P.; Bai, J.; Zhang, Y.-L.; Yang, X.-L.; Xia, Y.-L.; Lin, Q.-Y.; Li, H.-H. Blockade of Intercellular Adhesion Molecule-1 Prevents Angiotensin II-Induced Hypertension and Vascular Dysfunction. Lab. Investig. 2019, 100, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Kalra, D.; Sivasubramanian, N.; Mann, D.L. Angiotensin II Induces Tumor Necrosis Factor Biosynthesis in the Adult Mammalian Heart Through a Protein Kinase C–Dependent Pathway. Circulation 2002, 105, 2198–2205. [Google Scholar] [CrossRef]
- Ozawa, Y.; Kobori, H.; Suzaki, Y.; Navar, L.G. Sustained Renal Interstitial Macrophage Infiltration Following Chronic Angiotensin II Infusions. Am. J. Physiol. Physiol. 2007, 292, F330–F339. [Google Scholar] [CrossRef]
- Frenay, A.-R.S.; Yazdani, S.; Boersema, M.; Van Der Graaf, A.M.; Waanders, F.; Born, J.V.D.; Navis, G.J.; Van Goor, H. Incomplete Restoration of Angiotensin II - Induced Renal Extracellular Matrix Deposition and Inflammation Despite Complete Functional Recovery in Rats. PLoS ONE 2015, 10, e0129732. [Google Scholar] [CrossRef]
- Zhang, J.-D.; Patel, M.B.; Griffiths, R.; Dolber, P.C.; Ruiz, P.; Sparks, M.A.; Stegbauer, J.; Jin, H.; Gomez, J.A.; Buckley, A.F.; et al. Type 1 Angiotensin Receptors on Macrophages Ameliorate IL-1 receptor–mediated Kidney Fibrosis. J. Clin. Investig. 2014, 124, 2198–2203. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Xiong, M.; Chen, C.; Du, L.; Liu, Z.; Shi, Y.; Zhang, M.; Gong, J.; Song, X.; Xiang, R.; et al. Legumain, an Asparaginyl Endopeptidase, Mediates the Effect of M2 Macrophages on Attenuating Renal Interstitial Fibrosis in Obstructive Nephropathy. Kidney Int. 2018, 94, 91–101. [Google Scholar] [CrossRef]
- Witherel, C.E.; Abebayehu, D.; Barker, T.H.; Spiller, K.L. Macrophage and Fibroblast Interactions in Biomaterial-Mediated Fibrosis. Adv. Health Mater. 2019, 8, e1801451. [Google Scholar] [CrossRef]
- Colombo, P.C.; Onat, D.; Harxhi, A.; Demmer, R.T.; Hayashi, Y.; Jelic, S.; LeJemtel, T.H.; Bucciarelli, L.; Kebschull, M.; Papapanou, P.N.; et al. Peripheral Venous Congestion Causes Inflammation, Neurohormonal, and Endothelial Cell Activation. Eur. Heart J. 2013, 35, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Colombo, P.C.; Rastogi, S.; Onat, D.; Zacà, V.; Gupta, R.C.; Jorde, U.P.; Sabbah, H.N. Activation of Endothelial Cells in Conduit Veins of Dogs with Heart Failure and Veins of Normal Dogs After Vascular Stretch by Acute Volume Loading. J. Card. Fail. 2009, 15, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Berguetti, T.S.; Quintaes, L.S.P.; Pereira, T.H.; Robaina, M.C.; Cruz, A.L.S.; Maia, R.C.; De Souza, P.S.; Hancio, T. TNF-α Modulates P-Glycoprotein Expression and Contributes to Cellular Proliferation via Extracellular Vesicles. Cells 2019, 8, 500. [Google Scholar] [CrossRef]
- Chen, T.; Zhang, X.; Zhu, G.; Liu, H.; Chen, J.; Wang, Y.; He, X. Quercetin Inhibits TNF-α Induced HUVECs Apoptosis and Inflammation via Downregulating NF-KB and AP-1 Signaling Pathway in Vitro. Medicine 2020, 99, e22241. [Google Scholar] [CrossRef]
- Zelová, H.; Hošek, J. TNF-α Signalling and Inflammation: Interactions Between Old Acquaintances. Inflamm. Res. 2013, 62, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Sziksz, E.; Pap, D.; Lippai, R.; Béres, N.J.; Fekete, A.; Szabó, A.J.; Vannay, Á. Fibrosis Related Inflammatory Mediators: Role of the IL-10 Cytokine Family. Mediat. Inflamm. 2015, 2015, 1–15. [Google Scholar] [CrossRef]
- Wynn, T.A. Cellular and Molecular Mechanisms of Fibrosis. J. Pathol. 2007, 214, 199–210. [Google Scholar] [CrossRef]
- Shao, D.; Suresh, R.; Vakil, V.; Gomer, R.; Pilling, D. Pivotal Advance: Th-1 Cytokines Inhibit, and Th-2 Cytokines Promote Fibrocyte Differentiation. J. Leukoc. Biol. 2008, 83, 1323–1333. [Google Scholar] [CrossRef] [PubMed]
- Le Floc’H, A.; Allinne, J.; Nagashima, K.; Scott, G.; Birchard, D.; Asrat, S.; Bai, Y.; Lim, W.K.; Martin, J.; Huang, T.; et al. Dual Blockade of IL-4 and IL-13 with Dupilumab, an IL-4Rα Antibody, Is Required to Broadly Inhibit Type 2 Inflammation. Allergy 2019, 75, 1188–1204. [Google Scholar] [CrossRef]
- Reiman, R.M.; Thompson, R.W.; Feng, C.; Hari, D.; Knight, R.; Cheever, A.W.; Rosenberg, H.F.; Wynn, T.A. Interleukin-5 (IL-5) Augments the Progression of Liver Fibrosis by Regulating IL-13 Activity. Infect. Immun. 2006, 74, 1471–1479. [Google Scholar] [CrossRef] [PubMed]
- Pesce, J.; Kaviratne, M.; Ramalingam, T.R.; Thompson, R.W.; Urban, J.; Cheever, A.W.; Young, D.A.; Collins, M.; Grusby, M.J.; Wynn, T.A. The IL-21 Receptor Augments Th2 Effector Function and Alternative Macrophage Activation. J. Clin. Investig. 2006, 116, 2044–2055. [Google Scholar] [CrossRef]
- Korn, T.; Bettelli, E.; Gao, W.; Awasthi, A.; Jäger, A.; Strom, T.B.; Oukka, M.; Kuchroo, V.K. IL-21 Initiates an Alternative Pathway to Induce Proinflammatory TH17 Cells. Nat. Cell Biol. 2007, 448, 484–487. [Google Scholar] [CrossRef]
- Nurieva, R.; Yang, X.O.; Martinez, G.; Zhang, Y.; Panopoulos, A.; Ma, L.; Schluns, K.; Tian, Q.; Watowich, S.S.; Jetten, A.; et al. Essential Autocrine Regulation by IL-21 in the Generation of Inflammatory T Cells. Nat. Cell Biol. 2007, 448, 480–483. [Google Scholar] [CrossRef]
- Lei, L.; Zhao, C.; Qin, F.; He, Z.Y.; Wang, X.; Zhong, X.N. Th17 Cells and IL-17 Promote the Skin and Lung Inflammation and Fibrosis Process in a Bleomycin-Induced Murine Model of Systemic Sclerosis. Clin. Exp. Rheumatol. 2016, 34 (Suppl. S100), 14–22. [Google Scholar]
- Wu, L.; Ong, S.; Talor, M.V.; Barin, J.G.; Baldeviano, G.C.; Kass, D.A.; Bedja, D.; Zhang, H.; Sheikh, A.; Margolick, J.B.; et al. Cardiac Fibroblasts Mediate IL-17A–driven Inflammatory Dilated Cardiomyopathy. J. Exp. Med. 2014, 211, 1449–1464. [Google Scholar] [CrossRef] [PubMed]
- Sommerfeld, S.D.; Cherry, C.; Schwab, R.M.; Chung, L.; Maestas, D.R., Jr.; Laffont, P.; Stein, J.E.; Tam, A.; Ganguly, S.; Housseau, F.; et al. Interleukin-36γ–producing Macrophages Drive IL-17–mediated Fibrosis. Sci. Immunol. 2019, 4, eaax4783. [Google Scholar] [CrossRef]
- Ramani, K.; Tan, R.J.; Zhou, D.; Coleman, B.M.; Jawale, C.V.; Liu, Y.; Biswas, P.S. IL-17 Receptor Signaling Negatively Regulates the Development of Tubulointerstitial Fibrosis in the Kidney. Mediat. Inflamm. 2018, 2018, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Sharma, S.; Zhu, L.X.; Keane, M.P.; Luo, J.; Zhang, L.; Burdick, M.D.; Lin, Y.Q.; Dohadwala, M.; Gardner, B.; et al. IL-7 Inhibits Fibroblast TGF-β Production and Signaling in Pulmonary Fibrosis. J. Clin. Investig. 2002, 109, 931–937. [Google Scholar] [CrossRef]
- Demols, A.; Van Laethem, J.-L.; Quertinmont, E.; Degraef, C.; Delhaye, M.; Geerts, A.; Devière, J. Endogenous Interleukin-10 Modulates Fibrosis and Regeneration in Experimental Chronic Pancreatitis. Am. J. Physiol. Liver Physiol. 2002, 282, G1105–G1112. [Google Scholar] [CrossRef] [PubMed]
- Shamskhou, E.A.; Kratochvil, M.J.; Orcholski, M.E.; Nagy, N.; Kaber, G.; Steen, E.; Balaji, S.; Yuan, K.; Keswani, S.; Danielson, B.; et al. Hydrogel-Based Delivery of Il-10 Improves Treatment of Bleomycin-Induced Lung Fibrosis in Mice. Biomaterials 2019, 203, 52–62. [Google Scholar] [CrossRef]
- Guan, Q.; Weiss, C.R.; Wang, S.; Qing, G.; Yang, X.; Warrington, R.J.; Bernstein, C.N.; Peng, Z. Reversing Ongoing Chronic Intestinal Inflammation and Fibrosis by Sustained Block of IL-12 and IL-23 Using a Vaccine in Mice. Inflamm. Bowel Dis. 2018, 24, 1941–1952. [Google Scholar] [CrossRef]
- Keane, M.P.; Belperio, J.A.; Burdick, M.D.; Strieter, R.M. IL-12 Attenuates Bleomycin-Induced Pulmonary Fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 2001, 281, L92–L97. [Google Scholar] [CrossRef] [PubMed]
- Weidenbusch, M.; Song, S.; Iwakura, T.; Shi, C.; Rodler, S.; Kobold, S.; Mulay, S.R.; Honarpisheh, M.M.; Anders, H.-J. IL-22 Sustains Epithelial Integrity in Progressive Kidney Remodeling and Fibrosis. Physiol. Rep. 2018, 6, e13817. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, Y.; Fan, J.; Zhang, X.; Luan, J.; Bian, Q.; Ding, T.; Wang, Y.; Wang, Z.; Song, P.; et al. Interleukin-22 Ameliorated Renal Injury and Fibrosis in Diabetic Nephropathy through Inhibition of NLRP3 Inflammasome Activation. Cell Death Dis. 2017, 8, e2937. [Google Scholar] [CrossRef]
- Lee, J.-W.; Oh, J.E.; Rhee, K.-J.; Yoo, B.-S.; Eom, Y.W.; Park, S.W.; Lee, J.H.; Son, J.-W.; Youn, Y.J.; Ahn, M.-S.; et al. Co-Treatment with Interferon-γ and 1-Methyl Tryptophan Ameliorates Cardiac Fibrosis through Cardiac Myofibroblasts Apoptosis. Mol. Cell. Biochem. 2019, 458, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Poosti, F.; Bansal, R.; Yazdani, S.; Prakash, J.; Post, E.; Klok, P.; Born, J.V.D.; de Borst, M.H.; van Goor, H.; Poelstra, K.; et al. Selective Delivery of IFN-γ to Renal Interstitial Myofibroblasts: A Novel Strategy for the Treatment of Renal Fibrosis. FASEB J. 2015, 29, 1029–1042. [Google Scholar] [CrossRef]
- Liechty, K.W.; Kim, H.B.; Adzick, N.; Crombleholme, T.M. Fetal Wound Repair Results in Scar Formation in Interleukin-10–deficient Mice in a Syngeneic Murine Model of Scarless Fetal Wound Repair. J. Pediatr. Surg. 2000, 35, 866–873. [Google Scholar] [CrossRef]
- Lin, W.-R.; Lim, S.-N.; Yen, T.-H.; Alison, M.R. The Influence of Bone Marrow-Secreted IL-10 in a Mouse Model of Cerulein-Induced Pancreatic Fibrosis. BioMed Res. Int. 2016, 2016, 1–11. [Google Scholar] [CrossRef]
- Gupta, J.; Mitra, N.; Kanetsky, P.A.; Devaney, J.; Wing, M.R.; Reilly, M.; Shah, V.O.; Balakrishnan, V.S.; Guzman, N.J.; Girndt, M.; et al. Association Between Albuminuria, Kidney Function, and Inflammatory Biomarker Profile in CKD in CRIC. Clin. J. Am. Soc. Nephrol. 2012, 7, 1938–1946. [Google Scholar] [CrossRef]
- Li, R.; Guo, Y.; Zhang, Y.; Zhang, X.; Zhu, L.; Yan, T. Salidroside Ameliorates Renal Interstitial Fibrosis by Inhibiting the TLR4/NF-κB and MAPK Signaling Pathways. Int. J. Mol. Sci. 2019, 20, 1103. [Google Scholar] [CrossRef]
- Edeling, M.; Ragi, G.; Huang, S.; Pavenstädt, H.; Susztak, K. Developmental Signalling Pathways in Renal Fibrosis: The Roles of Notch, Wnt and Hedgehog. Nat. Rev. Nephrol. 2016, 12, 426–439. [Google Scholar] [CrossRef]
- Cho, E.; Kim, M.; Ko, Y.S.; Lee, H.Y.; Song, M.; Kim, H.-K.; Cho, W.-Y.; Jo, S.-K. Role of Inflammation in the Pathogenesis of Cardiorenal Syndrome in a Rat Myocardial Infarction Model. Nephrol. Dial. Transplant. 2013, 28, 2766–2778. [Google Scholar] [CrossRef]
- Yhee, J.-Y.; Yu, C.-H.; Kim, J.-H.; Sur, J.-H. Effects of T Lymphocytes, Interleukin-1, and Interleukin-6 on Renal Fibrosis in Canine End-Stage Renal Disease. J. Veter. Diagn. Investig. 2008, 20, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Van Linthout, S.; Miteva, K.; Tschöpe, C. Crosstalk Between Fibroblasts and Inflammatory Cells. Cardiovasc. Res. 2014, 102, 258–269. [Google Scholar] [CrossRef] [PubMed]
- Black, L.M.; Lever, J.M.; Agarwal, A. Renal Inflammation and Fibrosis: A Double-Edged Sword. J. Histochem. Cytochem. 2019, 67, 663–681. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.-M. Inflammatory Mediators and Renal Fibrosis. Adv. Exp. Med. Biol. 2019, 1165, 381–406. [Google Scholar] [CrossRef]
- Iwano, M.; Neilson, E.G. Mechanisms of Tubulointerstitial Fibrosis. Curr. Opin. Nephrol. Hypertens. 2004, 13, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, S.D.; Frangogiannis, N.G. The Biological Basis for Cardiac Repair After Myocardial Infarction. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef]
- Sun, K.; Li, Y.-Y.; Jin, J. A Double-Edged Sword of Immuno-Microenvironment in Cardiac Homeostasis and Injury Repair. Signal Transduct. Target. Ther. 2021, 6, 1–16. [Google Scholar] [CrossRef]
- Fadok, V.A.; Bratton, D.L.; Konowal, A.; Freed, P.W.; Westcott, J.Y.; Henson, P.M. Macrophages That Have Ingested Apoptotic Cells In Vitro Inhibit Proinflammatory Cytokine Production through autocrine/Paracrine Mechanisms Involving TGF-Beta, PGE2, and PAF. J. Clin. Investig. 1998, 101, 890–898. [Google Scholar] [CrossRef] [PubMed]
- Song, E.; Ouyang, N.; Hörbelt, M.; Antus, B.; Wang, M.; Exton, M.S. Influence of Alternatively and Classically Activated Macrophages on Fibrogenic Activities of Human Fibroblasts. Cell. Immunol. 2000, 204, 19–28. [Google Scholar] [CrossRef]
- Valgimigli, M.; Ceconi, C.; Malagutti, P.; Merli, E.; Soukhomovskaia, O.; Francolini, G.; Cicchitelli, G.; Olivares, A.; Parrinello, G.; Percoco, G.; et al. Tumor Necrosis Factor-α Receptor 1 Is a Major Predictor of Mortality and New-Onset Heart Failure in Patients with Acute Myocardial Infarction. Circulation 2005, 111, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, N.; Wada, H.; Kanda, T.; Niwa, T.; Yamada, Y.; Saito, K.; Fujiwara, H.; Sekikawa, K.; Seishima, M. Improved Myocardial ischemia/Reperfusion Injury in Mice Lacking Tumor Necrosis Factor-α. J. Am. Coll. Cardiol. 2002, 39, 1229–1235. [Google Scholar] [CrossRef]
- Kurrelmeyer, K.M.; Michael, L.H.; Baumgarten, G.; Taffet, G.E.; Peschon, J.J.; Sivasubramanian, N.; Entman, M.L.; Mann, D.L. Endogenous Tumor Necrosis Factor Protects the Adult Cardiac Myocyte Against Ischemic-Induced Apoptosis in a Murine Model of Acute Myocardial Infarction. Proc. Natl. Acad. Sci. USA 2000, 97, 5456–5461. [Google Scholar] [CrossRef]
- Huang, M.; Li, J.-Y. Physiological Regulation of Reactive Oxygen Species in Organisms Based on Their Physicochemical Properties. Acta Physiol. 2020, 228, e13351. [Google Scholar] [CrossRef] [PubMed]
- Pisoschi, A.M.; Pop, A. The Role of Antioxidants in the Chemistry of Oxidative Stress: A Review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef]
- Lau, N.; Pluth, M.D. Reactive Sulfur Species (RSS): Persulfides, Polysulfides, Potential, and Problems. Curr. Opin. Chem. Biol. 2019, 49, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Hu, J.; Liu, S. Reactive Oxygen, Nitrogen, and Sulfur Species (RONSS)-Responsive Polymersomes for Triggered Drug Release. Macromol. Rapid Commun. 2017, 38, 10–1002. [Google Scholar] [CrossRef]
- Luo, Z.; Xu, X.; Sho, T.; Zhang, J.; Xu, W.; Yao, J.; Xu, J. ROS-Induced Autophagy Regulates Porcine Trophectoderm Cell Apoptosis, Proliferation, and Differentiation. Am. J. Physiol. Physiol. 2019, 316, C198–C209. [Google Scholar] [CrossRef]
- Pei, J.; Wang, F.; Pei, S.; Bai, R.; Cong, X.; Nie, Y.; Chen, X. Hydrogen Sulfide Promotes Cardiomyocyte Proliferation and Heart Regeneration via ROS Scavenging. Oxidative Med. Cell. Longev. 2020, 2020, 1–11. [Google Scholar] [CrossRef]
- Zhang, G.; He, J.; Ye, X.; Zhu, J.; Hu, X.; Shen, M.; Ma, Y.; Mao, Z.; Song, H.; Chen, F. β-Thujaplicin Induces Autophagic Cell Death, Apoptosis, and Cell Cycle Arrest through ROS-Mediated Akt and p38/ERK MAPK Signaling in Human Hepatocellular Carcinoma. Cell Death Dis. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Kang, R.; Li, R.; Dai, P.; Li, Z.; Li, Y.; Li, C. Deoxynivalenol Induced Apoptosis and Inflammation of IPEC-J2 Cells by Promoting ROS Production. Environ. Pollut. 2019, 251, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, C.L.; Davies, M.J. Detection, Identification, and Quantification of Oxidative Protein Modifications. J. Biol. Chem. 2019, 294, 19683–19708. [Google Scholar] [CrossRef] [PubMed]
- Kowalska, M.; Piekut, T.; Prendecki, M.; Sodel, A.; Kozubski, W.; Dorszewska, J. Mitochondrial and Nuclear DNA Oxidative Damage in Physiological and Pathological Aging. DNA Cell Biol. 2020, 39, 1410–1420. [Google Scholar] [CrossRef]
- Salehi, F.; Behboudi, H.; Kavoosi, G.; Ardestani, S.K. Oxidative DNA Damage Induced by ROS-Modulating Agents with the Ability to Target DNA: A Comparison of the Biological Characteristics of Citrus Pectin and Apple Pectin. Sci. Rep. 2018, 8, 1–16. [Google Scholar] [CrossRef]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Tsikas, D. Assessment of Lipid Peroxidation by Measuring Malondialdehyde (MDA) and Relatives in Biological Samples: Analytical and Biological Challenges. Anal. Biochem. 2017, 524, 13–30. [Google Scholar] [CrossRef]
- Gallo, G.; Sprovierio, P.; Martino, G. 4-Hydroxynonenal and Oxidative Stress in Several Organelles and Its Damaging Effects on Cell Functions. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2020, 71, 10–26402. [Google Scholar]
- Schrader, M.; Fahimi, H. Peroxisomes and Oxidative Stress. Biochim. Biophys. Acta Bioenerg. 2006, 1763, 1755–1766. [Google Scholar] [CrossRef]
- Antonenkov, V.D.; Grunau, S.; Ohlmeier, S.; Hiltunen, K. Peroxisomes Are Oxidative Organelles. Antioxid. Redox Signal 2010, 13, 525–537. [Google Scholar] [CrossRef]
- Zeeshan, H.M.A.; Lee, G.H.; Kim, H.-R.; Chae, H.-J. Endoplasmic Reticulum Stress and Associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free Radicals: Properties, Sources, Targets, and Their Implication in Various Diseases. Ind. J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef]
- Oyewole, A.O.; Birch-Machin, M.A. Mitochondria-targeted Antioxidants. FASEB J. 2015, 29, 4766–4771. [Google Scholar] [CrossRef]
- Iacovino, L.G.; Manzella, N.; Resta, J.; Vanoni, M.A.; Rotilio, L.; Pisani, L.; Edmondson, D.E.; Parini, A.; Mattevi, A.; Mialet-Perez, J.; et al. Rational Redesign of Monoamine Oxidase A into a Dehydrogenase to Probe ROS in Cardiac Aging. ACS Chem. Biol. 2020, 15, 1795–1800. [Google Scholar] [CrossRef]
- Kaludercic, N.; Mialet-Perez, J.; Paolocci, N.; Parini, A.; Di Lisa, F. Monoamine Oxidases As Sources of Oxidants in the Heart. J. Mol. Cell. Cardiol. 2014, 73, 34–42. [Google Scholar] [CrossRef]
- Dey, S.; Sidor, A.; O’Rourke, B. Compartment-Specific Control of Reactive Oxygen Species Scavenging by Antioxidant Pathway Enzymes. J. Biol. Chem. 2016, 291, 11185–11197. [Google Scholar] [CrossRef]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef] [PubMed]
- Dinh, Q.N.; Drummond, G.; Sobey, C.G.; Chrissobolis, S. Roles of Inflammation, Oxidative Stress, and Vascular Dysfunction in Hypertension. BioMed Res. Int. 2014, 2014, 1–11. [Google Scholar] [CrossRef]
- Chiba, T.; Peasley, K.D.; Cargill, K.R.; Maringer, K.V.; Bharathi, S.S.; Mukherjee, E.; Zhang, Y.; Holtz, A.; Basisty, N.; Yagobian, S.D.; et al. Sirtuin 5 Regulates Proximal Tubule Fatty Acid Oxidation to Protect Against AKI. J. Am. Soc. Nephrol. 2019, 30, 2384–2398. [Google Scholar] [CrossRef]
- Papinska, A.M.; Rodgers, K.E. Long-Term Administration of Angiotensin (1–7) to db/Db Mice Reduces Oxidative Stress Damage in the Kidneys and Prevents Renal Dysfunction. Oxidative Med. Cell. Longev. 2018, 2018, 1–10. [Google Scholar] [CrossRef]
- Kumar, S.; Wang, G.; Zheng, N.; Cheng, W.; Ouyang, K.; Lin, H.; Liao, Y.; Liu, J. HIMF (Hypoxia-Induced Mitogenic Factor)-IL (Interleukin)-6 Signaling Mediates Cardiomyocyte-Fibroblast Crosstalk to Promote Cardiac Hypertrophy and Fibrosis. Hypertension 2019, 73, 1058–1070. [Google Scholar] [CrossRef]
- He, T.; Guan, X.; Wang, S.; Xiao, T.; Yang, K.; Xu, X.; Wang, J.; Zhao, J. Resveratrol Prevents High Glucose-Induced epithelial–mesenchymal Transition in Renal Tubular Epithelial Cells by Inhibiting NADPH oxidase/ROS/ERK Pathway. Mol. Cell. Endocrinol. 2015, 402, 13–20. [Google Scholar] [CrossRef]
- Carthy, J.M. TGFβ Signaling and the Control of Myofibroblast Differentiation: Implications for Chronic Inflammatory Disorders. J. Cell. Physiol. 2017, 233, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-A.; Kim, M.-J.; Park, S.-Y.; Kim, J.-S.; Lee, S.-J.; Woo, H.A.; Kim, D.-K.; Nam, J.-S.; Sheen, Y.Y. EW-7197 Inhibits Hepatic, Renal, and Pulmonary Fibrosis by Blocking TGF-β/Smad and ROS Signaling. Cell. Mol. Life Sci. 2014, 72, 2023–2039. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yuan, X.; Li, W.; Cao, Q.; Shu, Y. Aspirin-Triggered Resolvin D1 Inhibits TGF-β1-Induced EMT through the Inhibition of the MTOR Pathway by Reducing the Expression of PKM2 and Is Closely Linked to Oxidative Stress. Int. J. Mol. Med. 2016, 38, 1235–1242. [Google Scholar] [CrossRef]
- De Bleser, P.J.; Xu, G.; Rombouts, K.; Rogiers, V.; Geerts, A. Glutathione Levels Discriminate Between Oxidative Stress and Transforming Growth Factor-β Signaling in Activated Rat Hepatic Stellate Cells. J. Biol. Chem. 1999, 274, 33881–33887. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, S.; Kuroda, J.; Ago, T.; Zhai, P.; Ikeda, Y.; Oka, S.; Fong, G.-H.; Tian, R.; Sadoshima, J. Broad Suppression of NADPH Oxidase Activity Exacerbates Ischemia/Reperfusion Injury Through Inadvertent Downregulation of Hypoxia-Inducible Factor-1α and Upregulation of Peroxisome Proliferator–activated Receptor-α. Circ. Res. 2013, 112, 1135–1149. [Google Scholar] [CrossRef]
- Verzola, D.; Ratto, E.; Villaggio, B.; Parodi, E.L.; Pontremoli, R.; Garibotto, G.; Viazzi, F. Uric Acid Promotes Apoptosis in Human Proximal Tubule Cells by Oxidative Stress and the Activation of NADPH Oxidase NOX 4. PLoS ONE 2014, 9, e115210. [Google Scholar] [CrossRef] [PubMed]
- Buvelot, H.; Jaquet, V.; Krause, K.-H. Mammalian NADPH Oxidases. Methods Mol. Biol. 2019, 1982, 17–36. [Google Scholar] [CrossRef]
- Lan, T.; Kisseleva, T.; Brenner, D.A. Deficiency of NOX1 or NOX4 Prevents Liver Inflammation and Fibrosis in Mice through Inhibition of Hepatic Stellate Cell Activation. PLoS ONE 2015, 10, e0129743. [Google Scholar] [CrossRef]
- Muñoz, M.; López-Oliva, M.E.; Rodríguez, C.; Martínez, M.P.; Sáenz-Medina, J.; Sánchez, A.; Climent, B.; Benedito, S.; García-Sacristán, A.; Rivera, L.; et al. Differential Contribution of Nox1, Nox2 and Nox4 to Kidney Vascular Oxidative Stress and Endothelial Dysfunction in Obesity. Redox Biol. 2020, 28, 101330. [Google Scholar] [CrossRef] [PubMed]
- Rajaram, R.D.; Dissard, R.; Faivre, A.; Ino, F.; Delitsikou, V.; Jaquet, V.; Cagarelli, T.; Lindenmeyer, M.; Jansen-Duerr, P.; Cohen, C.; et al. Tubular NOX4 Expression Decreases in Chronic Kidney Disease But Does Not Modify Fibrosis Evolution. Redox Biol. 2019, 26, 101234. [Google Scholar] [CrossRef]
- Braunersreuther, V.; Montecucco, F.; Ashri, M.; Pelli, G.; Galan, K.; Frias, M.; Burger, F.; Quinderé, A.L.G.; Montessuit, C.; Krause, K.-H.; et al. Role of NADPH Oxidase Isoforms NOX1, NOX2 and NOX4 in Myocardial ischemia/Reperfusion Injury. J. Mol. Cell. Cardiol. 2013, 64, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Yang, C.; Shao, L.; Zhu, H.; Wang, Y.; Huang, X.; Wang, S.; Hong, L. Targeting NOX 4 by Petunidin Improves anoxia/Reoxygenation-Induced Myocardium Injury. Eur. J. Pharmacol. 2020, 888, 173414. [Google Scholar] [CrossRef]
- Bondi, C.D.; Manickam, N.; Lee, D.Y.; Block, K.; Gorin, Y.; Abboud, H.E.; Barnes, J.L. NAD(P)H Oxidase Mediates TGF-β1–Induced Activation of Kidney Myofibroblasts. J. Am. Soc. Nephrol. 2009, 21, 93–102. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Xiong, J.; Nie, L.; Yu, Y.; Guan, X.; Xu, X.; Xiao, T.; Yang, K.; Liu, L.; Zhang, D.; et al. Resveratrol Inhibits Renal Interstitial Fibrosis in Diabetic Nephropathy by Regulating AMPK/NOX4/ROS Pathway. J. Mol. Med. 2016, 94, 1359–1371. [Google Scholar] [CrossRef]
- Breglia, A.; Virzì, G.M.; Pastori, S.; Brocca, A.; De Cal, M.; Bolin, C.; Vescovo, G.; Ronco, C. Determinants of Monocyte Apoptosis in Cardiorenal Syndrome Type 1. Cardiorenal Med. 2018, 8, 208–216. [Google Scholar] [CrossRef]
- Caio-Silva, W.; Dias, D.D.S.; JunHo, C.V.C.; Panico, K.; Neres-Santos, R.S.; Pelegrino, M.T.; Pieretti, J.C.; Seabra, A.B.; De Angelis, K.; Carneiro-Ramos, M.S. Characterization of the Oxidative Stress in Renal Ischemia/Reperfusion-Induced Cardiorenal Syndrome Type 3. BioMed Res. Int. 2020, 2020, 1–11. [Google Scholar] [CrossRef]
- Fox, B.M.; Gil, H.-W.; Kirkbride-Romeo, L.; Bagchi, R.; Wennersten, S.; Haefner, K.R.; Skrypnyk, N.I.; Brown, C.N.; Soranno, D.E.; Gist, K.M.; et al. Metabolomics Assessment Reveals Oxidative Stress and Altered Energy Production in the Heart After Ischemic Acute Kidney Injury in Mice. Kidney Int. 2019, 95, 590–610. [Google Scholar] [CrossRef]
- Guo, H.; Xu, D.; Kuroki, M.; Lu, Z.; Xu, X.; Geurts, A.; Osborn, J.W.; Chen, Y. Kidney Failure, Arterial Hypertension and Left Ventricular Hypertrophy in Rats with Loss of Function Mutation of SOD3. Free. Radic. Biol. Med. 2020, 152, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Oslowski, C.M.; Urano, F. Measuring ER Stress and the Unfolded Protein Response Using Mammalian Tissue Culture System. Methods Enzymol. 2011, 490, 71–92. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Villanueva, J.F.; Díaz-Molina, R.; García-González, V. Protein Folding and Mechanisms of Proteostasis. Int. J. Mol. Sci. 2015, 16, 17193–17230. [Google Scholar] [CrossRef]
- Read, A.; Schröder, M. The Unfolded Protein Response: An Overview. Biology 2021, 10, 384. [Google Scholar] [CrossRef]
- Sanyal, A.; Zbornik, E.A.; Watson, B.G.; Christoffer, C.; Ma, J.; Kihara, D.; Mattoo, S. Kinetic and Structural Parameters Governing Fic-Mediated adenylylation/AMPylation of the Hsp70 Chaperone, BiP/GRP78. Cell Stress Chaperon 2021, 1–18. [Google Scholar] [CrossRef]
- Song, S.; Tan, J.; Miao, Y.; Li, M.; Zhang, Q. Crosstalk of Autophagy and Apoptosis: Involvement of the Dual Role of Autophagy under ER Stress. J. Cell. Physiol. 2017, 232, 2977–2984. [Google Scholar] [CrossRef] [PubMed]
- Lam, M.; A Marsters, S.; Ashkenazi, A.; Walter, P. Misfolded Proteins Bind and Activate Death Receptor 5 to Trigger Apoptosis During Unresolved Endoplasmic Reticulum Stress. eLife 2020, 9, e52291. [Google Scholar] [CrossRef]
- Shrestha, N.; De Franco, E.; Arvan, P.; Cnop, M. Pathological β-Cell Endoplasmic Reticulum Stress in Type 2 Diabetes: Current Evidence. Front. Endocrinol. 2021, 12, 650158. [Google Scholar] [CrossRef]
- Parks, S.Z.; Gao, T.; Awuapura, N.J.; Ayathamattam, J.; Chabosseau, P.L.; Kalvakolanu, D.V.; Valdivia, H.H.; Rutter, G.A.; Leclerc, I. The Ca 2+ -binding Protein Sorcin Stimulates Transcriptional Activity of the Unfolded Protein Response Mediator ATF6. FEBS Lett. 2021, 10. [Google Scholar] [CrossRef]
- Baba, B.; Caliskan, M.; Boyuk, G.; Hacisevki, A. Chemical Chaperone PBA Attenuates ER Stress and Upregulates SOCS3 Expression as a Regulator of Leptin Signaling. Biochemistry 2021, 86, 480–488. [Google Scholar] [CrossRef]
- Yang, Y.; Zhou, Q.; Gao, A.; Chen, L.; Li, L. Endoplasmic Reticulum Stress and Focused Drug Discovery in Cardiovascular Disease. Clin. Chim. Acta 2020, 504, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Ajoolabady, A.; Wang, S.; Kroemer, G.; Klionsky, D.J.; Uversky, V.N.; Sowers, J.R.; Aslkhodapasandhokmabad, H.; Bi, Y.; Ge, J.; Ren, J. ER Stress in Cardiometabolic Diseases: From Molecular Mechanisms to Therapeutics. Endocr. Rev. 2021. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.-H.; Zheng, C.-M.; Chou, C.-L.; Chen, Y.-J.; Lee, Y.-H.; Lin, Y.-F.; Chiu, H.-W. Therapeutic Effect of Endothelin-Converting Enzyme Inhibitor on Chronic Kidney Disease through the Inhibition of Endoplasmic Reticulum Stress and the NLRP3 Inflammasome. Biomedicine 2021, 9, 398. [Google Scholar] [CrossRef]
- Lins, B.B.; Casare, F.A.M.; Fontenele, F.F.; Gonçalves, G.L.; Oliveira-Souza, M. Long-Term Angiotensin II Infusion Induces Oxidative and Endoplasmic Reticulum Stress and Modulates Na+ Transporters Through the Nephron. Front. Physiol. 2021, 12, 642752. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Wu, P.; Li, Y.; Shen, Y.; Xin, P.; Li, S.; Wang, Z.; Dai, X.; Zhu, W.; Wei, M. Myocardial Infarction Worsens Glomerular Injury and Microalbuminuria in Rats with Pre-Existing Renal Impairment Accompanied by the Activation of ER Stress and Inflammation. Mol. Biol. Rep. 2014, 41, 7911–7921. [Google Scholar] [CrossRef]
- Dickhout, J.G.; Carlisle, R.E.; Austin, R.C. Interrelationship Between Cardiac Hypertrophy, Heart Failure, and Chronic Kidney Disease. Circ. Res. 2011, 108, 629–642. [Google Scholar] [CrossRef]
- Olivares-Silva, F.; Espitia-Corredor, J.; Letelier, A.; Vivar, R.; Parra-Flores, P.; Olmedo, I.; Montenegro, J.; Pardo-Jiménez, V.; Díaz-Araya, G. TGF-β1 Decreases CHOP Expression and Prevents Cardiac Fibroblast Apoptosis Induced by Endoplasmic Reticulum Stress. Toxicol. Vitr. 2020, 70, 105041. [Google Scholar] [CrossRef]
- Yamamoto, T.; Endo, J.; Kataoka, M.; Matsuhashi, T.; Katsumata, Y.; Shirakawa, K.; Isobe, S.; Moriyama, H.; Goto, S.; Shimanaka, Y.; et al. Palmitate Induces Cardiomyocyte Death via Inositol Requiring Enzyme-1 (IRE1)-Mediated Signaling Independent of X-Box Binding Protein 1 (XBP1). Biochem. Biophys. Res. Commun. 2020, 526, 122–127. [Google Scholar] [CrossRef]
- Shu, S.; Zhu, J.; Liu, Z.; Tang, C.; Cai, J.; Dong, Z. Endoplasmic Reticulum Stress Is Activated in Post-Ischemic Kidneys to Promote Chronic Kidney Disease. EBioMedicine 2018, 37, 269–280. [Google Scholar] [CrossRef]
- Huang, D.; Yan, M.-L.; Chen, K.-K.; Sun, R.; Dong, Z.-F.; Wu, P.-L.; Li, S.; Zhu, G.-S.; Ma, S.-X.; Pan, Y.-S.; et al. Cardiac-Specific Overexpression of Silent Information Regulator 1 Protects Against Heart and Kidney Deterioration in Cardiorenal Syndrome via Inhibition of Endoplasmic Reticulum Stress. Cell. Physiol. Biochem. 2018, 46, 9–22. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Y.; Ding, W.; Wang, Y. Mito-TEMPO Alleviates Renal Fibrosis by Reducing Inflammation, Mitochondrial Dysfunction, and Endoplasmic Reticulum Stress. Oxidative Med. Cell. Longev. 2018, 2018, 1–13. [Google Scholar] [CrossRef]
- (66) Luo, T.; Kim, J.K.; Chen, B.; Abdel-Latif, A.; Kitakaze, M.; Yan, L. Attenuation of ER Stress Prevents Post-Infarction-Induced Cardiac Rupture and Remodeling by Modulating Both Cardiac Apoptosis and Fibrosis. Chem. Interact. 2015, 225, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Xiao, W.; Lee, K.; Salem, F.; Wen, J.; He, L.; Zhang, J.; Fei, Y.; Cheng, D.; Bao, H.; et al. Inhibition of Reticulon-1A–Mediated Endoplasmic Reticulum Stress in Early AKI Attenuates Renal Fibrosis Development. J. Am. Soc. Nephrol. 2017, 28, 2007–2021. [Google Scholar] [CrossRef]
- Han, J.; Pang, X.; Shi, X.; Zhang, Y.; Peng, Z.; Xing, Y. Ginkgo Biloba Extract EGB761 Ameliorates the Extracellular Matrix Accumulation and Mesenchymal Transformation of Renal Tubules in Diabetic Kidney Disease by Inhibiting Endoplasmic Reticulum Stress. BioMed Res. Int. 2021, 2021, 1–11. [Google Scholar] [CrossRef]
- Qu, J.; Li, M.; Li, D.; Xin, Y.; Li, J.; Lei, S.; Wu, W.; Liu, X. Stimulation of Sigma-1 Receptor Protects Against Cardiac Fibrosis by Alleviating IRE1 Pathway and Autophagy Impairment. Oxidative Med. Cell. Longev. 2021, 2021, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-T.; Jhao, P.-Y.; Hung, C.-T.; Wu, Y.-F.; Lin, S.-J.; Chiang, W.-C.; Lin, S.-L.; Yang, K.-C. Endoplasmic Reticulum Protein TXNDC5 Promotes Renal Fibrosis by Enforcing TGF-β Signaling in Kidney Fibroblasts. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Liu, S.-H.; Yang, C.-C.; Chan, D.-C.; Wu, C.-T.; Chen, L.-P.; Huang, J.-W.; Hung, K.-Y.; Chiang, C.-K. Chemical Chaperon 4-Phenylbutyrate Protects Against the Endoplasmic Reticulum Stress-Mediated Renal Fibrosis in Vivo and in Vitro. Oncotarget 2016, 7, 22116–22127. [Google Scholar] [CrossRef]
- Park, M.-J.; Oh, K.-S.; Nho, J.-H.; Kim, G.-Y.; Kim, D.-I. Asymmetric Dimethylarginine (ADMA) Treatment Induces Apoptosis in Cultured Rat Mesangial Cells via Endoplasmic Reticulum Stress Activation. Cell Biol. Int. 2016, 40, 662–670. [Google Scholar] [CrossRef]
- Matsuzaki, S.; Hiratsuka, T.; Taniguchi, M.; Shingaki, K.; Kubo, T.; Kiya, K.; Fujiwara, T.; Kanazawa, S.; Kanematsu, R.; Maeda, T.; et al. Physiological ER Stress Mediates the Differentiation of Fibroblasts. PLoS ONE 2015, 10, e0123578. [Google Scholar] [CrossRef] [PubMed]
- Shih, Y.-C.; Chen, C.-L.; Zhang, Y.; Mellor, R.L.; Kanter, E.M.; Fang, Y.; Wang, H.-C.; Hung, C.-T.; Nong, J.-Y.; Chen, H.-J.; et al. Endoplasmic Reticulum Protein TXNDC5 Augments Myocardial Fibrosis by Facilitating Extracellular Matrix Protein Folding and Redox-Sensitive Cardiac Fibroblast Activation. Circ. Res. 2018, 122, 1052–1068. [Google Scholar] [CrossRef]
- Pallet, N.; Bouvier, N.; Bendjallabah, A.; Rabant, M.; Flinois, J.P.; Hertig, A.; Legendre, C.; Beaune, P.; Thervet, E.; Anglicheau, D. Cyclosporine-Induced Endoplasmic Reticulum Stress Triggers Tubular Phenotypic Changes and Death. Arab. Archaeol. Epigr. 2008, 8, 2283–2296. [Google Scholar] [CrossRef]
- Li, Y.; Weng, X.; Wang, P.; He, Z.; Cheng, S.; Wang, D.; Li, X.; Cheng, G.; Li, T. 4-Phenylbutyrate Exerts Stage-Specific Effects on Cardiac Differentiation via HDAC Inhibition. PLoS ONE 2021, 16, e0250267. [Google Scholar] [CrossRef]
- Basseri, S.; Lhoták, Š.; Sharma, A.M.; Austin, R.C. The Chemical Chaperone 4-Phenylbutyrate Inhibits Adipogenesis by Modulating the Unfolded Protein Response. J. Lipid Res. 2009, 50, 2486–2501. [Google Scholar] [CrossRef]
- Akita, S.; Suzuki, K.; Yoshimoto, H.; Ohtsuru, A.; Hirano, A.; Yamashita, S. Cellular Mechanism Underlying Highly-Active or Antiretroviral Therapy-Induced Lipodystrophy: Atazanavir, a Protease Inhibitor, Compromises Adipogenic Conversion of Adipose-Derived Stem/Progenitor Cells through Accelerating ER Stress-Mediated Cell Death in Differentiating Adipocytes. Int. J. Mol. Sci. 2021, 22, 2114. [Google Scholar] [CrossRef]
- Shaffer, A.; Shelef, M.; Iwakoshi, N.N.; Lee, A.-H.; Qian, S.-B.; Zhao, H.; Yu, X.; Yang, L.; Tan, B.K.; Rosenwald, A.; et al. XBP1, Downstream of Blimp-1, Expands the Secretory Apparatus and Other Organelles, and Increases Protein Synthesis in Plasma Cell Differentiation. Immunity 2004, 21, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Gaudette, B.T.; Jones, D.D.; Bortnick, A.; Argon, Y.; Allman, D. MTORC1 Coordinates an Immediate Unfolded Protein Response-Related Transcriptome in Activated B Cells Preceding Antibody Secretion. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Meijer, B.J.; Smit, W.L.; Koelink, P.J.; Westendorp, B.F.; de Boer, R.J.; van der Meer, J.H.M.; Vermeulen, J.L.M.; Paton, J.C.; Paton, A.W.; Qin, J.; et al. Endoplasmic Reticulum Stress Regulates the Intestinal Stem Cell State through CtBP2. Sci. Rep. 2021, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.-I.; Yamaguchi, T.; Kaji, H.; Kanazawa, I.; Sugimoto, T. Advanced Glycation End Products Suppress Osteoblastic Differentiation of Stromal Cells by Activating Endoplasmic Reticulum Stress. Biochem. Biophys. Res. Commun. 2013, 438, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.Y.; Kim, H.S. α-Syntrophin Alleviates ER Stress to Maintain Protein Homeostasis During Myoblast Differentiation. FEBS Lett. 2021, 595, 1656–1670. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delgado-Valero, B.; Cachofeiro, V.; Martínez-Martínez, E. Fibrosis, the Bad Actor in Cardiorenal Syndromes: Mechanisms Involved. Cells 2021, 10, 1824. https://doi.org/10.3390/cells10071824
Delgado-Valero B, Cachofeiro V, Martínez-Martínez E. Fibrosis, the Bad Actor in Cardiorenal Syndromes: Mechanisms Involved. Cells. 2021; 10(7):1824. https://doi.org/10.3390/cells10071824
Chicago/Turabian StyleDelgado-Valero, Beatriz, Victoria Cachofeiro, and Ernesto Martínez-Martínez. 2021. "Fibrosis, the Bad Actor in Cardiorenal Syndromes: Mechanisms Involved" Cells 10, no. 7: 1824. https://doi.org/10.3390/cells10071824
APA StyleDelgado-Valero, B., Cachofeiro, V., & Martínez-Martínez, E. (2021). Fibrosis, the Bad Actor in Cardiorenal Syndromes: Mechanisms Involved. Cells, 10(7), 1824. https://doi.org/10.3390/cells10071824